
Abstract
Background:
Bacteriophage diversity of Klebsiella-infecting phages isolated on Klebsiella spp. other than Klebsiella pneumoniae remains relatively underexplored.
Materials and Methods:
A novel bacteriophage RSU-F4K5 infecting a Klebsiella michiganensis strain representing ST191 was isolated and characterized. The phage was subjected to Whole Genome Sequencing (WGS), genome de novo assembly, genome functional annotation, and comparative genomics. Wet-lab characterization included Transmission Electron Microscopy (TEM), lysis dynamics turbidimetry, adsorption assay, and a one-step growth curve.
Results:
K. michiganensis phage RSU-F4K5 is a novel lytic siphophage with a complete genome of 48,657 bp (47.71% G + C) predicted to encode 103 proteins and demonstrating <70% intergenomic similarity to other publicly available phage genomes. Phage RSU-F4K5 failed to infect local Klebsiella spp. isolates other than its isolation host.
Conclusions:
The host-specificity demonstrated by RSU-F4K5 makes it highly unlikely to be useful for therapeutical applications. However, the isolation and characterization of phage RSU-F4K5 adds to the global knowledge of the phage diversity of Klebsiella spp.
Introduction
Klebsiella spp. are non-motile gram-negative, encapsulated rods from the family Enterobacteriaceae. 1 Members of this genus are widely distributed in various terrestrial, aquatic, and host-associated environments (e.g., soils, waters, sewage, plants, and animals). 2 Although Klebsiella spp. are normally found on healthy human mucosal surfaces where they commonly colonize (e.g., nose, throat, and intestinal tract), 3 representatives of the genus are recognized as opportunistic pathogens with most infections associated with hospitalization. 4
Klebsiella michiganensis, initially recognized as a distinct species based on an isolate originating from a toothbrush holder, is considered a part of the Klebsiella oxytoca species complex, alongside several other Klebsiella spp.5,6 To date, strains of K. michiganensis have been described in association with bloodstream infections,7,8 diarrhea, 9 and even severe sepsis in an infant, 10 implying that the species can be considered a rising nosocomial pathogen. Despite the likely underreporting due to the ease of misidentification as K. oxytoca, 11 like with many other bacterial pathogens, antibiotic-resistant strains of K. michiganensis also begin to emerge, hinting at the necessity of alternative options for their control and treatment.12–17
Phage therapy (PT; a deliberate use of viruses of bacteria-bacteriophages to control or eradicate target bacterial pathogen during patient treatment18,19) represents a promising solution for managing drug-resistant Klebsiella spp. infections. The efficacy of PT has already been demonstrated in connection with Klebsiella isolates via reduced bacterial loads and disruption of biofilm formation leading to more favorable outcomes in prosthetic joint infections,20,21 pneumonia, 22 and recurrent urinary tract infections, 23 among others. 24 However, most of the Klebsiella phage isolation, characterization, and PT efforts were aimed at the most notorious representative of the genus—Klebsiella pneumoniae, with the diversity of bacteriophages infecting other Klebsiella spp. remaining relatively underexplored and warranting an interest in further investigation. 25
Currently, public biological sequence repositories list more than 1200 Klebsiella bacteriophage complete genomes, with K. michiganensis specifically indicated as the host in the associated metadata for only 20 of these submissions (based on GenBank search conducted on January 10, 2025, see Table 1 for details). To expand the known cultured phage diversity associated with K. michiganensis, we isolated a novel siphophage RSU-F4K5 which was specific to a hospital wastewater-associated bacterial strain RSU-K5.
The Known Klebsiella michiganensis-Infecting Bacteriophages That Have Their Complete Genome Sequences Publicly Available
Methods
Phage isolation and propagation
Phage presented as “RSU-F4K5” was isolated from the filtrate of Pauls Stradins Clinical University Hospital wastewater (15 mL centrifuged at 6000 g for 15 min and passed through a 0.45 μm syringe filter). Briefly, 100 µL of the filtrate was plated with the 100 µL of the previously isolated RSU-K5 host overnight culture using a double-layer agar (DLA) overlay method (1.5% tryptic soy agar [TSA] as the bottom layer and 0.7% TSA as the top layer). After incubation for 18 h at 35°C, a single plaque was collected and transferred to 1 mL Luria Broth (LB), which was then used for serial dilution with a dilution factor step of 10, using these dilutions for DLA overlays. The procedure was repeated for three more passages to obtain an individual phage isolate, which was then propagated.
Propagation of the phage was performed through six near-confluent lysis DLA overlay plates. The top soft agar layers were collected in a tube and vigorously vortexed with the addition of ∼3.5 mL of liquid LB medium per plate. After an hour-long incubation at RT and subsequent centrifugation at 7000 g for 20 min, the supernatant was decanted and filtered through a 0.45 µm filter. The filtrate was titrated and aliquoted for long-term storage at −80°C, while the remainder was stored at +4°C as a stock for follow-up experiments. Routine experiments have also been carried out with the agarized LB (1.5% and 0.7% agar for bottom and top agar layers, respectively).
Host range determination
The host range was determined by plating 10 µL of phage RSU-F4K5 stock (∼7.5 × 1010 PFU/mL) and its serial dilutions on lawns of different local Klebsiella spp. strains. The spots were applied before the formation of the lawn in a DLA overlay with the top layer seeded only with the host. The strains tested included a dozen genomically uncharacterized strains from the K. pneumoniae species complex isolated at different times from local hospitals or wastewater, as well as one of each genetically characterized K. pneumoniae isolates representing ST14/K2, ST147/K64, ST215/K5, ST219/KL114, and ST512/KL107. Furthermore, lytic activity of RSU-F4K5 was tested against four local strains allegedly representing K. oxytoca species complex. The formation of lysis zones or plaques was assessed after ∼18 h incubation at 35°C.
Details of the bacterial strains used for phage RSU-F4K5 host range testing are provided in the Supplementary Table S1.
Transmission electron microscopy
Transmission electron microscopy was performed as previously described, 26 the only modification was that the phage RSU-F4K5 lysate filtrate was used for negative staining with uranyl acetate.
Adsorption assay
Phage adsorption assay was performed using the protocol described by Svanberga 26 with slight modifications. The RSU-F4K5 phage was added to a flask with LB and 9.9 mL of exponentially growing bacteria at a 0.001 multiplicity of infection (MOI) and mixed. The flask was incubated at RT with shaking at 100 rpm (∼0.14 g). Afterward, 100 µL aliquots were collected immediately after infection, and then 1, 2, 3, 4, 5, 10, 15, 20, 25, and 30 min post-infection, diluted in 900 µL of LB and centrifuged at 9000 g for 2 min. The supernatant containing unadsorbed phages was diluted and plated in triplicate DLA assays. The plaques were counted after overnight incubation at 35°C. The whole experiment was performed in triplicate.
One-step growth curve
The one-step growth curve was done according to the protocol described previously 27 with slight modifications. The host culture at the exponential phase (10 mL, OD600 = 0.2) was infected with the RSU-F4K5 phage at an MOI of 0.001, and the flask was gently swirled by hand. After 5-min adsorption, the flask was centrifuged for 3 min at 9000 g to remove unadsorbed phages. The pelleted cells were resuspended in 10 mL of LB and incubated at 35°C with shaking at 150 rpm (∼0.32 g). Approximately 17 min post-infection, the samples were collected at 6-min intervals for 60 min, serially diluted, and plated in triplicate. The experiment was repeated independently twice.
The burst size was calculated as the titer of the phage RSU-F4K5 (PFU/mL) at the datapoint corresponding to the end of the rise period divided by the average titer of the datapoints representing latent period.
Lysis curve
To determine the bacteriolytic activity of RSU-F4K5 in a liquid environment, the host RSU-K5 culture was grown in LB to the exponential phase (OD600 ∼0.2), transferred to a 96-well plate (200 µL per well) and infected with the RSU-F4K5 phage at various MOIs (0.0001, 0.001, 0.01, 0.1, 1, 10, 100). There were six technical replicates (individual wells) for each condition tested per plate. The plate was incubated at 35°C for 24 h in a Varioskan LUX plate reader (Thermo Fisher Scientific, Waltham, MA, USA), shaking at 60 rpm (∼0.05 g) between hourly readings. The experiment was repeated three times.
DNA extraction
For phage DNA extraction, the lysate was filtered through a 0.45 µm filter and 300 µL were subjected to proteinase K (1 µL of 18.7 mg/mL; Thermo Fisher Scientific) and SDS (Sigma-Aldrich, 0.5% final concentration) treatment (1 h at 56°C) with occasional vortexing. Next, instructions of the Genomic DNA Clean & Concentrator-10 Kit (Zymo Research, Irvine, CA, USA) were followed. The same workflow was applied to extract the host DNA, starting from an overnight culture.
Whole-genome sequencing of the phage RSU-F4K5
Phage NGS library was prepared from 200 ng of dsDNA (quantified using Qubit [Invitrogen, Waltham, MA, USA]). First, dsDNA was sonicated using Covaris S220 (Covaris, Woburn, MA, USA) to an average fragment length of 550 bp. Afterward, the TruSeq DNA Nano Low-Throughput Library Prep Kit (Illumina, San Diego, CA, USA) protocol was followed using adapter #22 from the TruSeq DNA Single Indexes Set B (Illumina). The resultant library was checked using an Agilent 2100 bioanalyzer (Agilent, Santa Clara, CA, USA) with a High-Sensitivity DNA Kit (Agilent) and a Qubit fluorometer (Invitrogen) dsDNA high-sensitivity quantification assay (Invitrogen). The library was then pooled with 17 other differently barcoded libraries prepared using the same kit (equal amounts of each) and sequenced using the 500-cycle MiSeq Reagent Kit v2 nano (Illumina) and MiSeq sequencer (Illumina) in 250 bp paired-end conformation.
Whole-genome sequencing of the strain RSU-K5
The host DNA library was prepared following the transposon-based QIAseq FX DNA Library UDI Kit (QIAGEN, Hilden, Germany) instructions, starting from 1 µg of input dsDNA. The library was pooled with other individually barcoded libraries and sequenced using the 500-cycle MiSeq Reagent Kit v2 (Illumina) on a MiSeq sequencer (Illumina) in a 250 bp paired-end conformation to get ∼0.35 gigabases of data.
De novo assembly and functional genome annotation
The “raw” demultiplexed phage read dataset was filtered and trimmed using BBduk (v.38.69 28 ). Reads with regions matching adapters or the phiX174 genome (used as a sequencing control) were removed. The remaining reads had their termini trimmed based on the individual base qualities (until Phred score >20 base is encountered), and only reads >50 bp post-trimming were retained. These trimmed and filtered reads (Sequencing Read Archive (SRA) accession: SRR32526756) were then subjected to de novo assembly using Unicycler (v.0.4.8 29 ). The de novo assembled genome of the phage was auto-annotated using Pharokka (v.1.7.1 30 ). Auto-annotation results were further validated and manually refined wherever necessary as previously described. 31 Phage genome molecule termini type elucidation/packaging strategy prediction was attempted via PhageTerm (v.1.0.12 32 ) and TerL phylogeny-based approach using an extended dataset of Merrill and colleagues, 33 similarly to as described in Ref. 34 .
The host strain genome was de novo assembled using Unicycler (v.0.4.8 29 ) with demultiplexed and trimmed reads as input. Before the assembly, the reads were trimmed using fastp (v.0.20.0 35 ) as follows: automatic detection and error correction from paired-end data enabled, Phred base quality threshold >20, the last trailing base of the reads removed, followed by low-quality base removal from the tails, discarding reads <50 bp afterward. The resulting assembly (GCA_046503775.1) was annotated using NCBI Prokaryotic Genome Annotation Pipeline (PGAP) (v.6.9 36 ). Host de novo assembly was subject to a complete Kleborate analysis under the default parameters for the K. oxytoca complex isolates (v.3.1.0, 37 “kosc” preset).
Metabuli (v.1.0.8 38 ) was used with GTDB (v.214.1 39 ) to evaluate the proportions of the host and phage contents in the trimmed read datasets of phage and host libraries under otherwise default settings.
Evolutionary relationships with other phages
The complete genome sequence of phage RSU-F4K5 was subjected to Nucleotide Basic Local Alignment Search Tool (BLASTN) against the non-redundant nucleotide database limited to Caudoviricetes (taxid:2731619), excluding uncultured/environmental sample sequences, using otherwise default megablast parameters. The top 30 highest-scoring hits were downloaded for a web Virus Intergenomic Distance Calculator (VIRIDIC) 40 analysis together with the complete genome sequence of RSU-F4K5.
Phylogenies were reconstructed for selected phage RSU-F4K5 proteins assumed to be functionally independent—the major capsid protein (MCP; XNP02778.1), terminase large subunit (TerL; XNP02736.1), helicase (XNP02814.1), and Mu Gam-like end protection protein (XNP02816.1).
First, a Protein Basic Local Alignment Search Tool (BLASTP) search against the non-redundant protein sequences was performed under default settings. All the hits to proteins of comparable length encoded by other cultured phage or bacterial genomes with a query coverage of at least 70% demonstrating an identity of at least 85% were selected and downloaded regardless of their annotations.
The respective analogous protein amino-acid-sequence-containing datasets were aligned using MAFFT (v.7.525 41 ) and the resulting multiple sequence alignments were subjected to ML phylogeny reconstructions in IQ-TREE (v. 2.0.6 42 ), inferring the best-fitting substitution model using the built-in ModelFinder, 43 allowing for polytomies and evaluating branch supports using 1000 ultrafast bootstrap 44 replicates.
The resulting trees were midpoint rooted and visualized in FigTree (v.1.4.4 45 ). Inkscape (v.1.0.1 46 ) was then used to combine and annotate the trees.
Results
Characteristics of K. michiganensis bacteriophage RSU-F4K5
The plaques phage RSU-F4K5 forms on the lawn of its isolation host strain are ∼1 mm in diameter, with a surrounding halo reaching a diameter of ∼5 mm in 18 h (Fig. 1A). Transmission electron microscopy revealed that RSU-F4K5 virions have a siphophage morphology: an icosahedral capsid with a diameter of 63.0 ± 2.5 nm attached to a 123.8 ± 4.7 nm long and 12.6 ± 2.2 nm wide non-contractile tail with a visually furcated baseplate featured at the distal end (Fig. 1B).
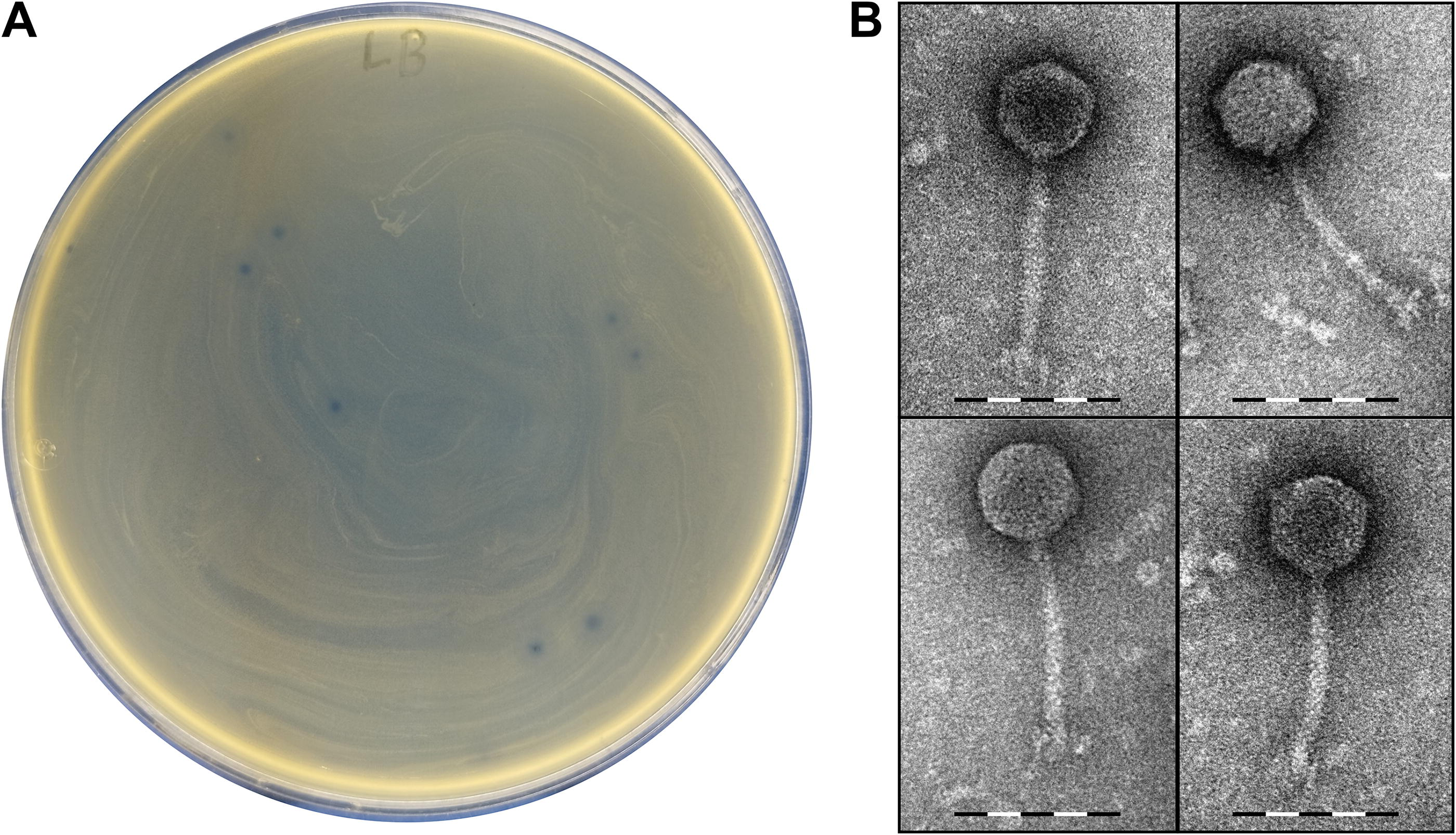
Morphology of the Klebsiella michiganensis bacteriophage RSU-F4K5.
No plaque formation was observed in a spot test when phage RSU-F4K5 was challenged against multiple strains of K. pneumoniae available in the laboratory (including the representatives of ST14/K2, ST147/K64, ST215/K5, ST219/KL114, and ST512/KL107 based on Kleborate/Kaptive 65 prediction using their short-read data) and some other bona fide K. oxytoca strains (based on matrix-assisted laser desorption ionization-time of flight mass spectrometry [MALDI-TOF MS] known to have limited resolution within the K. oxytoca species complex; previously isolated from wastewaters or patients as part of surveillance efforts unrelated to this study and shared with us by colleagues) that remain without genomic identification as of yet.
Notably, strain RSU-K5 was initially also acquired by us from the hospital as a strain of K. oxytoca based on a MALDI-TOF MS identification.
Upon infection of a log-phase host culture in liquid LB medium, ∼26% of the infective virions adsorb to the host cells during the very first minute post-infection, and less than 10% of the free virions remain in the medium after the first 5 min of co-incubation (Fig. 2A). One-step growth curve experiments revealed that the average burst size of RSU-F4K5 on its isolation host strain K. michiganensis RSU-K5 was on average 66 ± 10 virions, which were released into the surrounding medium after a ∼35-min-long latent period followed by an 18 min rise (Fig. 2B). However, pronounced lysis was observed in the liquid medium only when the phage RSU-F4K5 was added to its host strain at MOI as high as 100. But even then, after a few hours, regrowth of the host occurred (Fig. 2C).
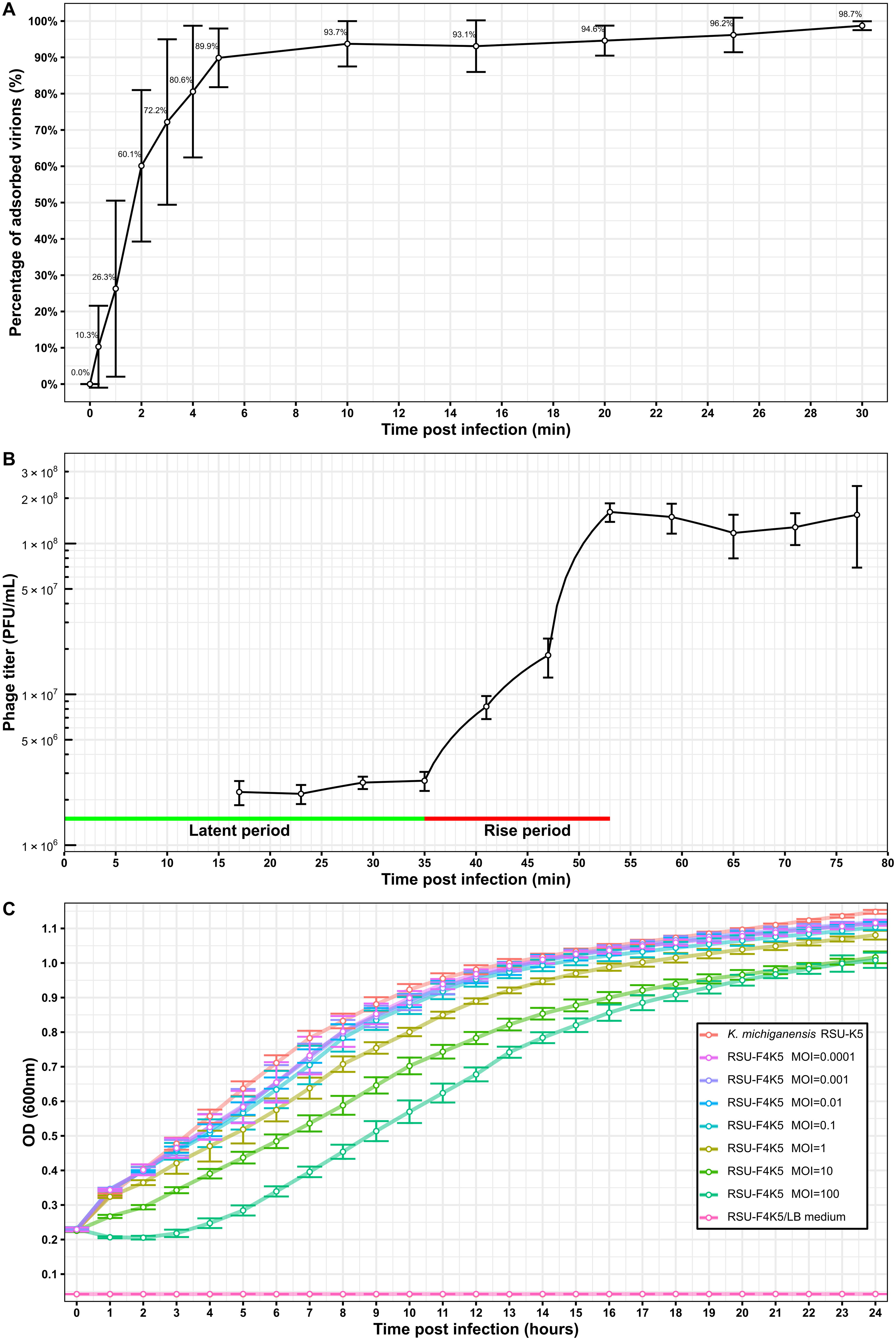
Interactions of the K. michiganensis bacteriophage RSU-F4K5 and its isolation host strain RSU-K5.
The genome of K. michiganensis strain RSU-K5
Demultiplexed raw read dataset corresponding to the host strain genomic DNA library comprised 958,103 read pairs yielding ∼350 Mbp. After trimming, 859,033 read pairs corresponding to ∼319.5 Mbp of the data remained (SRA accession: SRR31808208) and were used for de novo assembly. Classification of the trimmed library reads revealed that 99.81% of the reads belong to bacteria, and 74.46%, specifically to K. michiganensis.
The de novo assembly of the RSU-K5 genome has resulted in 51 contigs up to 887,701 bp in length and had an N50 of 401,685 bp (assembly accession: GCA_046503775.1). Functional annotation via National Center for Biotechnology Information (NCBI) PGAP identified 5840 features comprising 5623 coding genes, 132 pseudogenes, and 85 RNA genes. Based on a Kleborate analysis, strain RSU-K5 was determined to represent a rare K. michiganensis sequence type 191. 47
The genome of K. michiganensis bacteriophage RSU-F4K5
Raw demultiplexed read dataset of K. michiganensis bacteriophage RSU-F4K5 contained 68,468 reads of which 64,316 (∼94%) could be mapped onto the 48,567 bp-long de novo assembled genome using bwa mem (v.0.7.17-r1188 48 ), resulting in an average sequencing depth of 324x, as determined by samtools (v.1.20 49 ). The linear dsDNA genome of phage RSU-F4K5 had a G + C% of 47.71% and was predicted to encode up to 103 proteins, 64 of which remain functionally uncharacterized (so-called “hypothetical proteins”). No obvious temperate lifestyle, virulence, or antimicrobial resistance genes were identified in the genome of RSU-F4K5 (Supplementary Table S2).
Interestingly, when trimmed read classification was attempted (29,154 read pairs corresponding to ∼13.7 Mbp of data; SRA accession: SRR32526756), 54.10% were classified as “bacterial”; however, only 3.87%-as belonging to specifically K. michiganensis (the isolation and propagation host species of phage RSU-F4K5), while 45.90% were deemed unclassified.
No obvious fixed genome termini were identified using PhageTerm. TerL sequence phylogenetic tree reconstruction within the context of phages with experimentally determined packaging strategies demonstrated reliable clustering of RSU-F4K5 TerL with Sf6 TerL clade sequences representing headful packaging without a preferred packaging series initiation site 50 (Supplementary Fig. S1). These observations indicate that it is highly plausible that phage RSU-F4K5 employs a headful genome packaging strategy resulting in a population of virions with circularly permuted terminally redundant genomes.
Evolutionary relationships to other bacteriophages
When the complete genome nucleotide sequence of the siphophage RSU-F4K5 was queried against the non-redundant nucleotide sequences publicly available using BLASTN, similarity to several other Klebsiella-infecting bacteriophages was revealed. However, neither of the hits demonstrated a query coverage of more than 71%, with the Klebsiella phage 6991 serving as the closest cultured relative known to date (VIRIDIC-calculated intergenomic similarity of 63.9%), followed by phages VLCpiS13c and VLCpiS13b (Fig. 3). The genomes of these bacteriophages were revealed to be colinear and general annotated Open Reading Frame (ORF) synteny was observed (Fig. 4). However, multiple products encoded mostly by short ORFs of unknown function found in RSU-F4K5 were absent from the proteomes of the related phages being compared. Yet, the most notable differences in RSU-F4K5 included a distinct lysis protein region, the presence of a transposase, as well as dissimilarity of the putative tail fiber protein (Fig. 4).
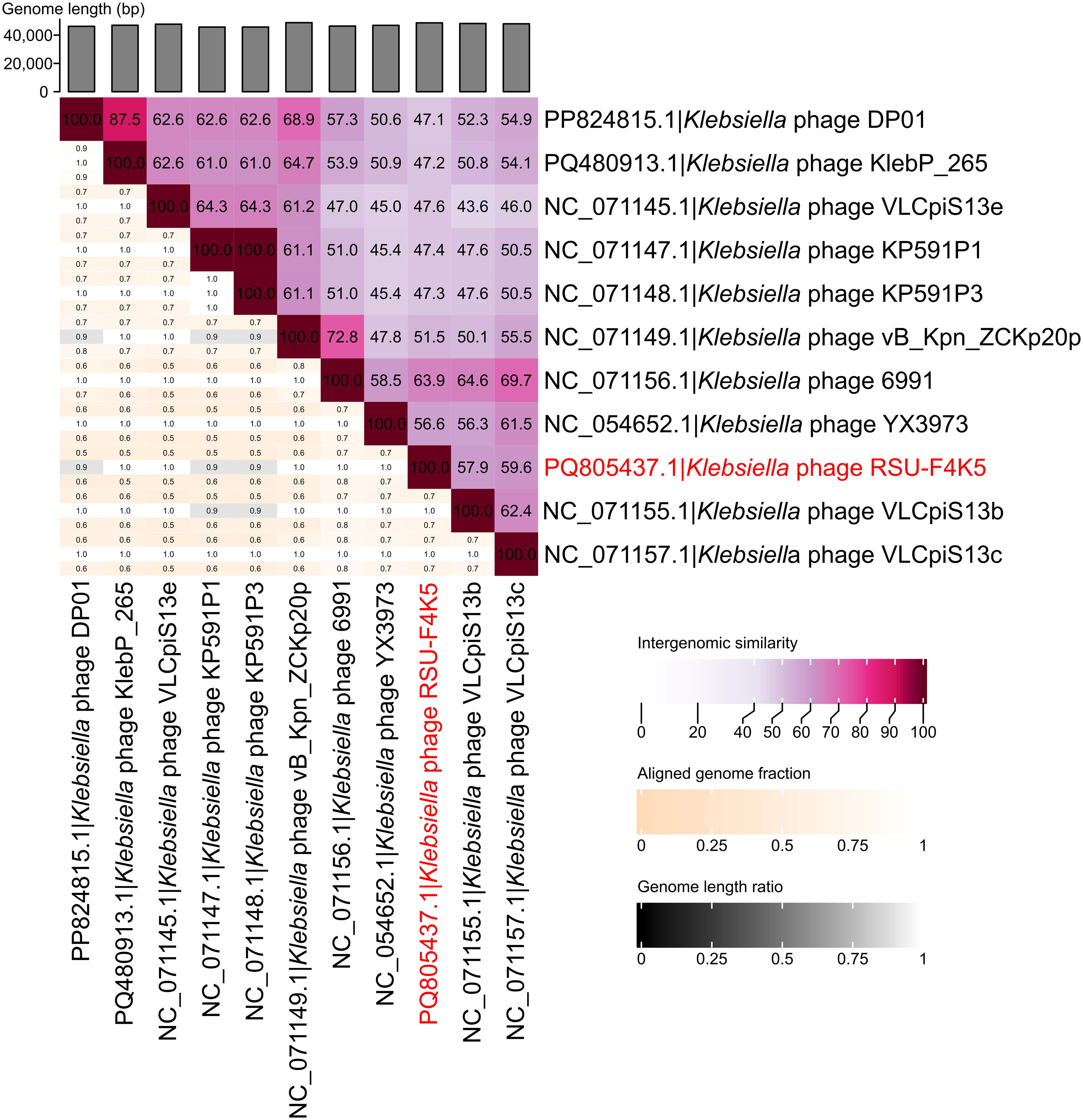
Similarities of the K. michiganensis bacteriophage RSU-F4K5 to other completely sequenced cultured bacteriophages. A VIRIDIC heatmap showing the pairwise intergenomic similarities of Klebsiella phage RSU-F4K5 and the 10 most similar bacteriophages.
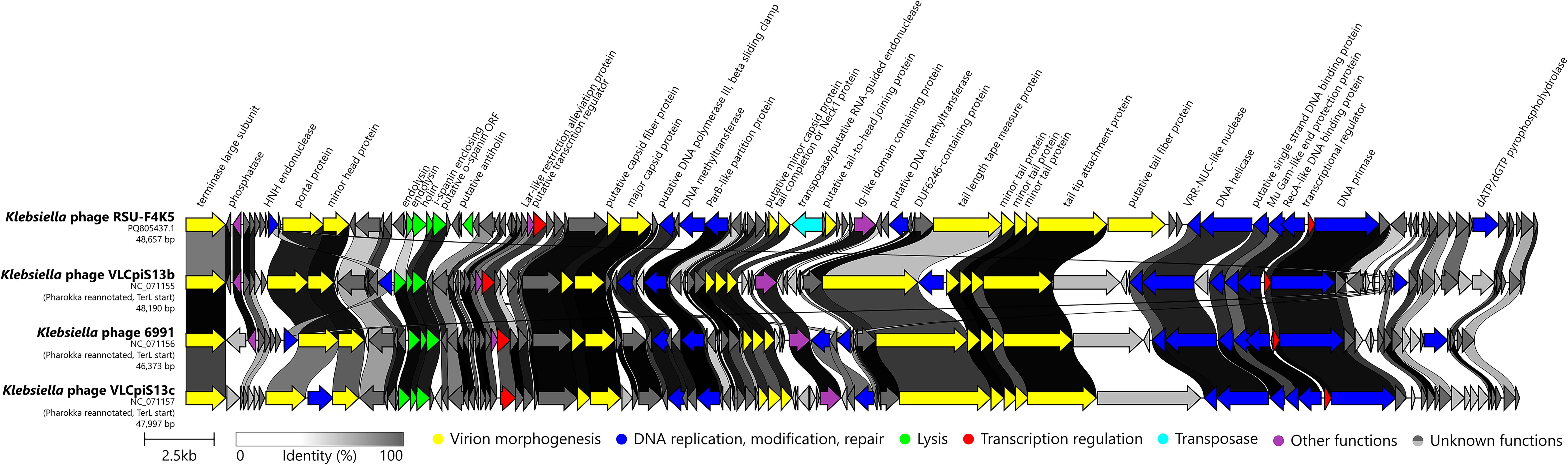
Comparison of genome organizations and proteome contents of the Klebsiella phage RSU-F4K5 and the three most intergenomically similar bacteriophages. The genomes are drawn to scale, genomes other than RSU-F4K5 were reannotated and reorganized to begin with the TerL-encoding ORF; the scale bar indicates 2500 base pairs. Arrows representing ORFs point in the direction of the transcription and are color-coded based on the function of their putative product according to the legend. The slanted labels above the arrows representing the phage RSU-F4K5 ORFs indicate the predicted function for the given ORF putative product if it could be functionally annotated. The ribbons connect homologous proteins of the phages and are colored according to their global identity. Pharokka (v.1.7.1 30 ) was used for reannotation of the related phage sequences and the figure was generated using Clinker (v.0.0.29 64 ) with further manual figure touch up in Inkscape (v.1.0.1 46 ).
Selected individual protein phylogeny reconstructions expectedly demonstrated that their counterparts from other Klebsiella-infecting phages are the most similar homologs. In the case of each, MCP (Fig. 5A), TerL (Fig. 5B), helicase (Fig. 5C), and Mu Gam-like protein (Fig. 5D), closely related homologs derived from genomic regions of bacteria such as strains of K. pneumoniae, Escherichia coli, and Salmonella enterica were noted.

Phylogenies of selected bona fide functionally-unrelated K. michiganensis phage RSU-F4K5 proteins. Midpoint-rooted maximum likelihood trees of the selected phage RSU-F4K5 protein amino acid sequences and similar sequences found in the proteomes of other cultured phages or bacteria.
Discussion
Based on the currently accepted intergenomic similarity-based criteria, phage RSU-F4K5 can be proposed as an isolate representing both a novel phage species and genus. 40 We hope that its baseline characterization provided in this study can serve as a point of reference for similar phages to be isolated in the future.
Although the phage RSU-F4K5 seems interesting from the point of view of bacteriophage diversity, based on the results presented within this study, we doubt that it will prove useful for practical purposes. Although the reliability of the observed narrow host range phage RSU-F4K5 demonstrated is limited by the ready unavailability of a wider diversity of Klebsiella spp. strains to offer it as potential hosts, its lack of ability to efficiently lyse its isolation host, even at the MOI as high as 100, seems also lackluster.
The reasons behind the regrowth of RSU-K5 cell population even when outnumbered by the phage RSU-F4K5 hundredfold remain a mystery. It would be interesting to check whether this has to do with merely a rather quick selection of resistant strains, to cognate receptor on which the phage cannot adsorb as efficiently (if at all), 51 or more intriguing potential aspects of phage-host interactions come into play—such as host cell persistence, 52 possible quorum-sensing effects, 53 or the activation of a hypothetical anti-RSU-F4K5 bacterial immune response, 54 to name a few options. 55
While bacteriophages are generally quite specific regarding their host ranges, the breadth of which might be limited to several strains of a single bacterial species, 56 “broad host-range” phages infecting multiple species within a host genus or even across genera are not rare. Recently, an article presented evidence that multiple distinct tailed bacteriophages isolated on various Klebsiella spp. were shown to be able to productively infect at least a single K. michiganensis strain despite the host diversity within the genus Klebsiella. 57
Undoubtedly, such bacteriophages that can infect multiple different species of bacteria are of great therapeutic utility and should be sought for potential practical use in generalized preparations that aim to target as much epidemiologically relevant targets as possible. 58 However, the ability of such phages to productively infect a particular representative of a species other than its isolation host can usually perceived just as a positive off-target “side effect” when an urgent preparation personalized for a particular patient to potentially undergo PT is pursued, sometimes even requiring further adaptation of a phage for a particular pathogen strain. 59 At the same time, one may expect that utilizing less commonplace bacteriophage isolation hosts might allow for a greater chance of recovering representatives of a yet under-sampled bacteriophage diversity in culture, especially that of highly specific phages.
Future experiments with an expanded host strain panel might reveal the true extent of the RSU-F4K5 host range, showcasing whether RSU-F4K5 is actually adapted to a completely different host species with its ability to lyse K. michiganensis strain RSU-K5 being merely a coincidence.
Wet-lab data on such isolates, and, more generally, on novel genomically characterized phage-host pairs, even if they might not be of particular immediate healthcare priority, have great potential to serve as a “feed” for future algorithms to be developed for prediction of intricacies concerning different phage-host interaction aspects, which are currently often very biased toward the most popular phage isolation hosts and of very limited utility elsewhere.
While the characterization of phages such as RSU-F4K5 is understandably incremental, we believe that each novel cultured phage matters. The focus on less popular phage isolation hosts and their phage diversity is expected to greatly aid in the development of the tools to not only better match isolated phages with the potential target strains but also to help in the characterization of novel bacteriophages that are yet to be isolated in culture.
Footnotes
Acknowledgments
The authors would like to acknowledge the support of Mrs. Renate Ruta Apse for continuous help in procuring the necessary materials. The authors are also thankful to Dr. Alisa Kazarina for her generous proposal of sharing a sequencing run. The authors are grateful to the National Microbiology Reference Laboratory of the Latvian Centre of Infectology for sharing genomic data associated with some of the K. pneumoniae strains used in this study.
Authors’ Contributions
N.B. and N.Z. wrote the initial version of the article. Each co-author of this article (N.B., D.R., J.J., K.S., K.R., J.K., and N.Z.) has contributed to this study by participating in a combination of one or more of the following activities: designing the study, acquiring samples and resources, performing experimental work, analyzing and interpreting the data, preparing visualizations. All the co-authors participated in preparing the final version of the article and agreed to its submission for peer review.
Ethical Statement
This research did not require an independent ethics committee approval because no human participants, animals, or genetically modified organisms requiring ethical oversight were involved. The subjects of this study do not fall under ethical review requirements.
Data Availability
The genomes of the phage and its host strain were submitted to GenBank and are publicly available from International Nucleotide Sequence Database Collaboration (INSDC) databases under the accession numbers PQ805437 and JBKFFE000000000, respectively, as a part of a larger relevant overarching BioProject (PRJNA1199463). Other data relevant to the analyses and the results presented are provided in the main text or supplementary materials associated with this work.
Author Disclosure Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding Information
This work was supported by the Nr. RSU-PAG-2024/1-0023 grant “Innovative strategies for limiting the antibacterial resistance of Klebsiella spp.: utilizing the synergy between phages and antibiotics (ReScUe-Kleb)” awarded to J.K.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.