
Abstract
Drug repurposing is of interest for therapeutics innovation in many human diseases including coronavirus disease 2019 (COVID-19). Methodological innovations in drug repurposing are currently being empowered by convergence of omics systems science and digital transformation of life sciences. This expert review article offers a systematic summary of the application of artificial intelligence (AI), particularly machine learning (ML), to drug repurposing and classifies and introduces the common clustering, dimensionality reduction, and other methods. We highlight, as a present-day high-profile example, the involvement of AI/ML-based drug discovery in the COVID-19 pandemic and discuss the collection and sharing of diverse data types, and the possible futures awaiting drug repurposing in an era of AI/ML and digital technologies. The article provides new insights on convergence of multi-omics and AI-based drug repurposing. We conclude with reflections on the various pathways to expedite innovation in drug development through drug repurposing for prompt responses to the current COVID-19 pandemic and future ecological crises in the 21st century.
Introduction
Drug repositioning is a method of developing new targets for existing drugs, which can significantly reduce time, cost, and other wastes as the targets are compounds that have already been tested for safety and pharmacokinetics (Jourdan et al, 2020; Rapicavoli et al, 2022). Drug repositioning relies on two main scientific bases that some diseases share common biological pathways and that a drug may have multiple targets that may be effective to different diseases (Jourdan et al, 2020). Within this context, and with the advent of the Big Data era, the generation of vast amounts of biological and chemical information has provided the scientific community with new opportunities to link drugs to diseases (March-Vila et al, 2017).
Since December 2019, coronavirus disease 2019 (COVID-19) has been recognized as a worldwide public health emergency (Hui et al, 2020) and declared as a global pandemic by the World Health Organization (WHO) in 2020. Although there is a massive vaccination campaign underway, emerging variants limit the efficacy of the campaign (Sibilio et al, 2021). Therefore, the search for new drugs that can treat patients remains important and urgent, and drug repositioning, with its time and cost advantages, certainly makes it highly visible.
This article highlights the emerging intersection with artificial intelligence (AI) and machine learning (ML) from a multi-omics perspective, and summarizes and discusses drug repositioning approaches that have been applied to COVID-19.
SARS Coronavirus-2: Pathogenesis and Focused Treatments
Coronaviruses are enveloped RNA viruses that are widely distributed in mammals, including humans, as well as birds, that cause respiratory, intestinal, hepatic, and neurological diseases (Zhu et al, 2020). Human coronaviruses (HCoVs) are positive-sense single-stranded RNA viruses and 30,000 bp long (Pirone et al, 2020). Before December 2019, six coronaviruses are known to cause human disease, of which, SARS coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV) are associated with fatal disease (Cui et al, 2019; Zhu et al, 2020).
Infection with the novel coronavirus SARS-CoV-2 belongs to the sarbecovirus subgenus of the Coronaviridae family (Zhu et al, 2020), caused a cluster of severe respiratory illnesses, which can lead to acute respiratory failure and even death (Pirone et al, 2020). WHO reported that there had been 516,922,683 cumulative cases and 6,259,945 cumulative deaths as of May 13, 2022.
Clinical deterioration in patients with severe COVID-19 disease is usually rapid, and a large part of the severe disease course is due to a cytokine storm leading to a massive inflammatory response, which leads to multi-organ failure or even death, which is thought to be possibly related to immune checkpoint activation and immune system failure (Behrens and Koretzky, 2017; Kim et al, 2021; Sibilio et al, 2021). At the same time, this massive immune response has set the stage for testing several immunomodulatory agents simultaneously with antiviral drugs (Sibilio et al, 2021), and several specific immunomodulatory agents, anti-cytokines such as interleukin-1 (IL-1) and IL-6 receptor antagonists, are considered to have potential for the treatment of cytokine storms (Rizk et al, 2020).
Multi-Omics and AI in Drug Repositioning
Avalanche of omics data
With the continuous development of new technologies for the determination of multi-omics data, the possibility of obtaining high-dimensional histological data quickly and efficiently is offered. Because a disease often emerges as a complex interaction between multiple genetic variants (Hirschhorn and Daly, 2005), a single layer of “omics” usually provides only limited insight into the biological mechanisms of the disease, with DNA, RNA, proteins, and metabolites often acting in complementary roles to join some biological function (Sun and Hu, 2016). Therefore, the integrated analysis of multi-omics data is of great importance in the study of complex biological processes and disease mechanisms.
Figure 1 briefly represents the different layers of the multi-omics data. For the overview of omics modalities, background, and origin, please refer to the study by Manzoni et al (2018).
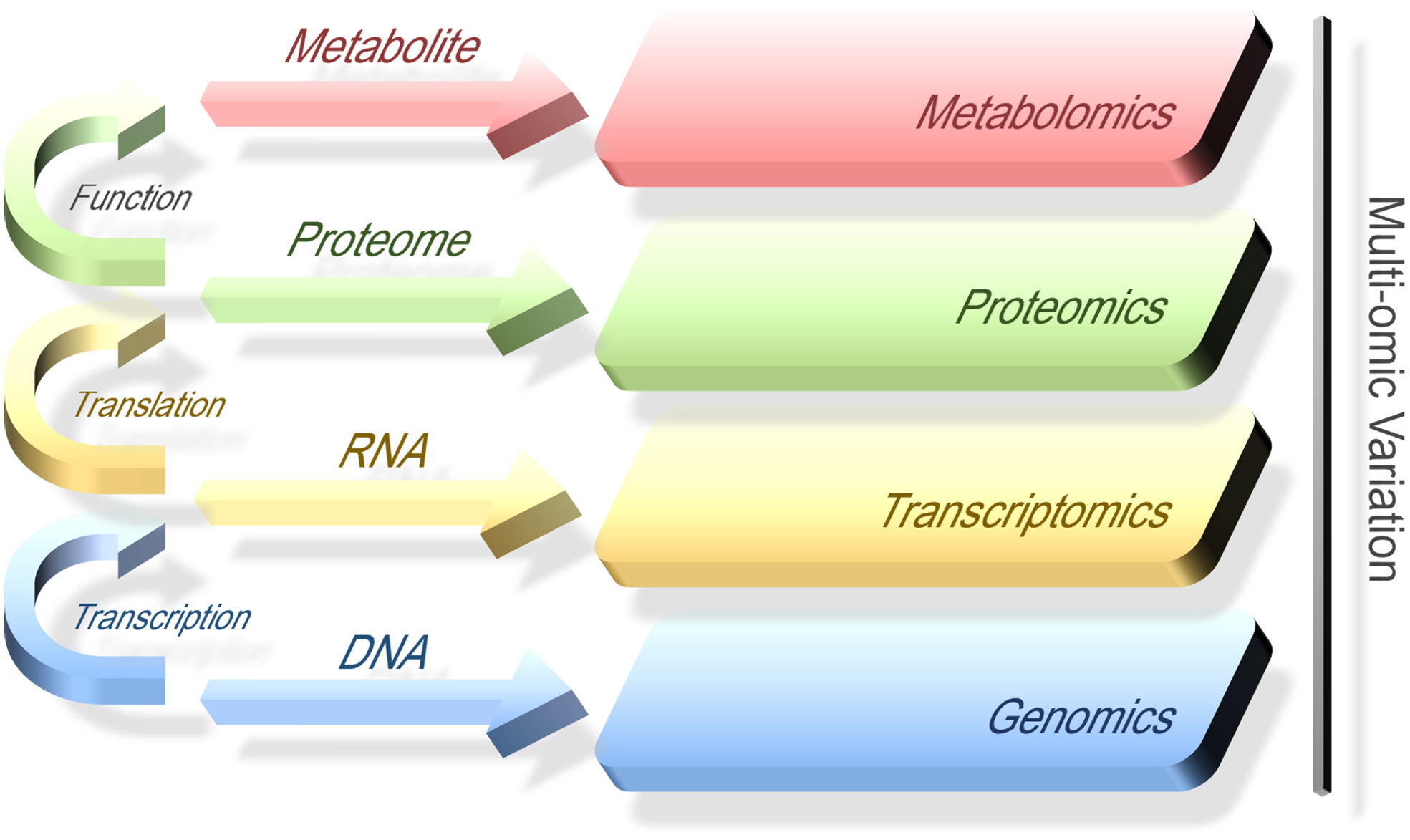
Different layers of multi-omics data. The straight arrows in the middle indicate the organismal molecules that constitute the different layers of the so-called omics cascade, and the curved arrows on the left side imply the biological process between different types of molecules.
Through gene expression profiling, multi-omics data can be easily used for drug repurposing. The Connectivity Map (CMap) is a gene expression profiling database based on interventional gene expression proposed by Lamb et al (2006); it is mainly used to reveal functional associations between small-molecule compounds, genes, and diseases. CMap and Library of Integrated Network-Based Cellular Signatures (LINCS) as its extension are considered to be the key concepts behind various drug repurposing studies (Jarada et al, 2020). Specifically, the CMap database contains mainly gene expression profiles of different cell lines acted by a large number of perturbagens (small-molecule compounds, overexpressed genes, etc.).
By comparing reference data, cellular gene expression profiles are found with high similarity and thus investigate whether cells have some kind of connection to different inductions. For use, the list of up/downregulated differentially expressed genes obtained from the experimental analysis is compared with the database reference data set by CMap; the enrichment direction and intensity of each reference is scored according to the enrichment of differentially expressed genes in the reference gene expression profile to determine the degree of enrichment with the query feature, that is, the similarity of differentially expressed genes to the reference gene expression profile, and ranked.
Amemiya et al (2019) proposed a computational drug repositioning approach to perform an integrated multi-omics analysis based on transcriptomic, proteomic, and interactomic data to detect drug candidates for dengue hemorrhagic fever. In this study, signature genes were identified by integrating the Gene Expression Omnibus (GEO) data set, drug candidates were identified by CMap search, disease specific pathways were detected using Gene Set Enrichment Analysis (GSEA) approach for transcriptomic and proteomic data, and finally, a human–dengue virus protein–protein interaction (PPI) network was constructed (Amemiya et al, 2019).
Convergence of multi-omics and AI/ML applications toward drug repurposing
The biggest challenge in drug repositioning is to customize or optimize methods to develop promising, affordable, and efficient drug repositioning pipelines for complex diseases (Jin and Wong, 2014; Zeng et al, 2020), so screening methods for drug reuse are particularly important. Depending on the classification, the approaches involved in drug repositioning can be classified as drug-oriented, target-oriented, and disease or therapy-oriented according to the information available related to quality and quantity (Sahoo et al, 2021), or as network-based, ligand-based, chemogenomic and ML, ligand-based approaches, and so on, according to the means (March-Vila et al, 2017).
With the advent of high-throughput technologies, more and more data need to be explored and used by computational analysis and mining tools, and to achieve systematic or comprehensive repurposing, various computer-based approaches are gradually becoming mainstream (Rapicavoli et al, 2022). In silico drug repositioning is a hypothesis-driven approach that can translate available omics data through the collection of disease phenotypes and targets, such as genome-wide association analyses or gene expression response profiles, pathway mappings, compound structures, as well as data related to drug modes of action, into predictions of druggable targets, ideally, top provide a list of Food and Drug Administration (FDA)-approved drug candidates with potential modulatory/inhibitory functions (Mottini et al, 2021; Pushpakom et al, 2019).
Our focus here is on AI and ML methods that leverage publicly available databases and information sources. DSP-1181 was the first repurposed drug to enter clinical trials discovered through an AI approach, and the time from initial screening to the end of preclinical testing was reduced from the 4 years that would have been required to <12 months (Farghali et al, 2021). AI-based approaches have enabled a more nuanced and iterative process to rapidly identify potentially bioactive compounds from millions of drug candidates in a short period, which has revolutionized the drug development process and facilitated the realization of precision medicine (Boniolo et al, 2021; Nayarisseri et al, 2021).
AI has also led to the creation of many reverse vaccinology (RV) virtual frameworks that are often categorized as rule-based filtering models. ML enables the creation of models that can learn and generalize patterns within existing data and are able to make inferences from previously unseen data. With the advent of deep learning (DL), the learning process can also include the automatic extraction of features from raw data (Prasad and Kumar, 2021; Sarker, 2021).
As an example of fusing multi-omics and AI/ML applications, in our recent study, we proposed a new approach to drug repurposing involving two-stage prediction and ML with applications to inclusion body myositis, polymyositis, and dermatomyositis (Cong et al, 2022). First, diseases are clustered by gene expression, with the thought that similar patterns of altered gene expression imply critical pathways shared in different disease conditions. Second, drug efficacy is determined based on the ability to reverse altered gene expression, and the results are clustered to identify repurposing targets.
Since the number of clusters cannot be well determined by using Uniform Manifold Approximation and Projection for Dimension Reduction (UMAP) alone, we introduced the k-means method in combination with it to obtain effective grouping information while maintaining good clustering performance. As a result, disease-specific gene expression and 22 drugs for repurposing were identified. The details are presented in Figure 2. To date, many computational approaches to drug repositioning using ML technology continue to be proposed and improved, especially in the context of COVID-19 of global concern.
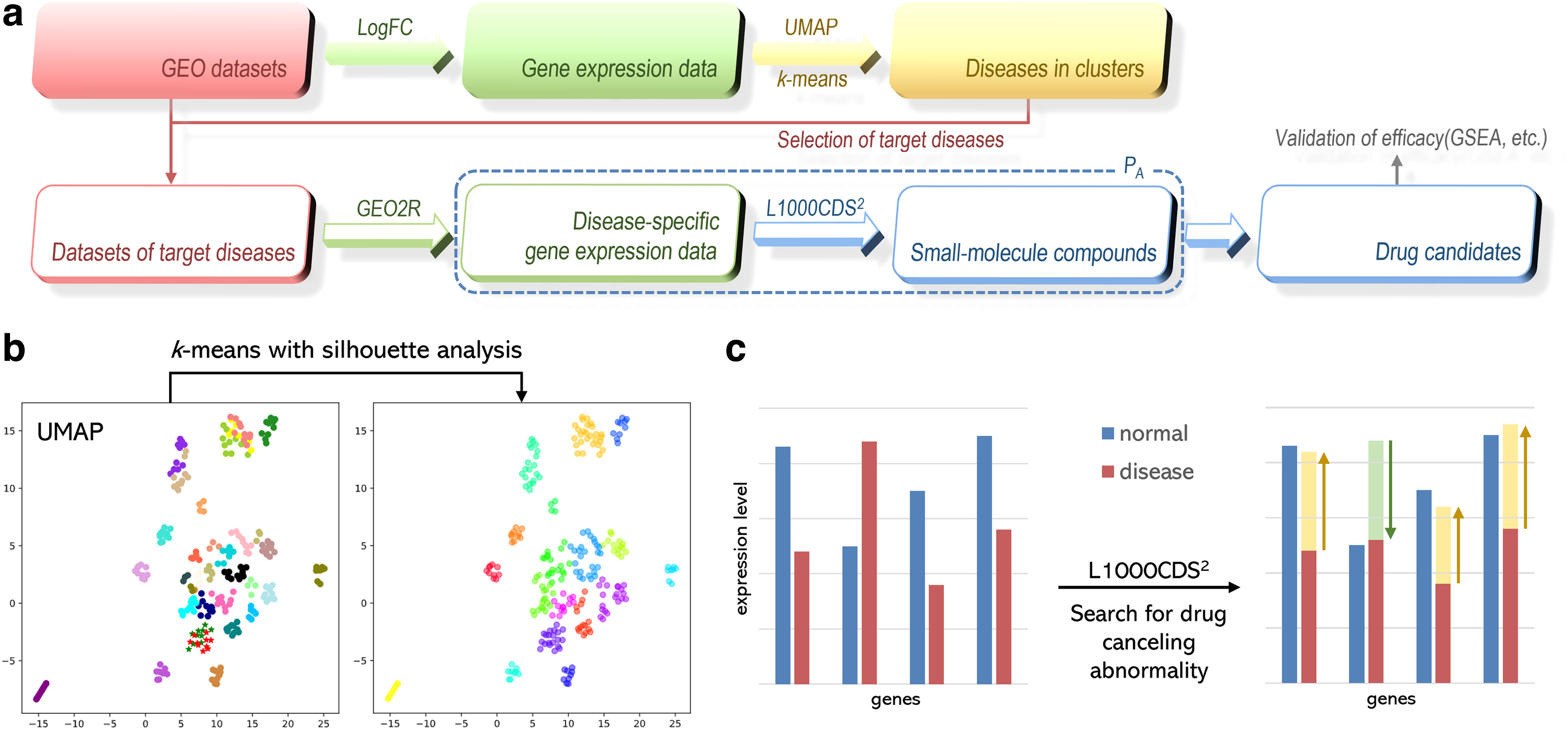
A new approach to drug repurposing with two-stage prediction, ML, and unsupervised clustering of gene expression. (
AI-Based Drug Repositioning as a Strategy for Identifying New Therapeutic Agents for COVID-19
Understanding the genetic regulatory code that controls gene expression is an important topic in the field of molecular biology and will provide us with the means to cure diseases. However, because the biological sequence space is too large to explore, experimental studies are limited to a single regulatory component in the context of single reporter genes (Zrimec et al, 2020). Therefore, various methods based on AI and DL have been introduced.
Classification and regression
Linear regression is the simplest model, usually used to solve continuous numerical prediction problems, which uses regression analysis of mathematical statistics to determine the interdependence between variables. Logistic regression adds a sigmoid function mapping to linear regression and is often used to solve classification problems for estimating the likelihood of something (Cox, 1958).
Buza et al, (2020) proposed a new method called MOLIERE for drug–target interaction (DTI) prediction and compared their results with the bipartite local model (BLM), previously proposed by Yamanishi et al, (2008), that is popular in DTI by predicting the target protein of a given drug to reason about the drug that targets a given protein (Bleakley and Yamanishi, 2009; Buza et al, 2020; Yamanishi et al, 2008). In this study, a framework called asymmetric loss models was instantiated using linear regression, adding the use of weighted profile (WP) to BLM, which was considered to have better performance relative to the original BLM and WP (Buza et al, 2020).
Gottlieb et al (2011) proposed an algorithm for predicting drug–disease associations using various data sources called PREdicting Drug IndiCaTion (PREDICT). The algorithm uses drug–drug and disease–disease similarity measures as classification features, and they apply a logistic regression classifier to distinguish between true and false drug–disease associations and ultimately predict new associations.
Neural network (NN)-based approaches are data-driven methods that can learn potential feature representations directly from labeled training data, and a review article addresses NN-based biomedical classification methods in detail (Jarada et al, 2020).
Deep neural networks (DNNs) increase the number of layers based on NN, optimizes the model structure and training methods, and improves the computational power to better cope with large amounts of data (Sze et al, 2017).
Unlike traditional ML, both convolutional neural networks (CNNs) and recurrent neural network (RNNs) can autonomously learn features and autonomously optimize the weights of each layer to obtain the set of values that best represent the features in the training network. CNNs are mostly used in computer vision and image processing applications, and CNN is a network model that does not consider sequential data. And RNNs are models that consider sequence data, mostly used for language model and video data processing applications (Krizhevsky et al, 2017; Lecun et al, 1998; Olurotimi, 1994).
Based on CNNs, the computational framework named convolutional neural network for coexpression (CNNC) was proposed, which provides the ability to perform genetic relationship inference in a supervised manner and was shown to be superior to previous approaches in inferring interactions, causality, and function assignment (Yuan and Bar-Joseph, 2019).
It is worth mentioning that different kinds of NNs can be combined to build hybrid models for classification. As synthetic screening experiments are considered to be limited in their ability to study the overall relationship between different parts of gene regulatory structures and co-regulation, deep convolutional neural network (DCNN) was used to predict gene expression levels from natural DNA sequences (Agarwal and Shendure, 2020; Zrimec et al, 2020).
Support-vector network (SVM) is a two-group classification model, whose basic model is defined as a linear classifier with maximum interval on the feature space, and whose learning strategy is interval maximization, which can eventually be translated into the solution of a convex quadratic programming problem (Cortes et al, 1995).
Lin et al (2020) proposed a new ML pipeline using the support-vector regression (SVR) algorithm that is a regression version of SVM. Using models trained from AA2AR, DHI1, and AL5AP, respectively, they demonstrated its ability in predicting the binding affinity of drugs to specific proteins and assessed the similarity of drug binding profiles between proteins by the Spearman correlation coefficient (SCC) of coefficients between models, which suggested that the models could be applied for screening of candidate drugs.
The goal of generative adversarial network (GAN) is to train two NNs, where one generative model tries to generate images similar to the real training samples by replicating the distribution of the data, and the other discriminative network predicts the probability that the generated images are from the real training set. These two models compete with each other so as to ultimately produce image results similar to the real training samples (Fanny and Cenggoro, 2018; Goodfellow et al, 2014).
Fanny and Cenggoro (2018) proposed a deep learning approach for imbalance data classification using class expert generative adversarial network (CE-GAN), which attaches a generator of GAN to the classifier instead of sharing a single architecture for differentiation and classification, and effectively increases the size of the data set and improves the classification.
Gene expression clustering
The goal of ML is to extract feature patterns from data that can represent the relationship between input data features and the output targets to be predicted and to use these patterns to make predictions, and the representation of these input features directly affects the nature and quality of the acquired patterns, and to some extent, it can be said that the selection of features determines the upper limit of ML. Therefore, it is particularly important to extract meaningful features from large and noisy data sets, and clustering and dimensionality reduction methods are introduced to assist in feature value selection.
Eisen et al (1998) used hierarchical clustering to classify the generated dendrograms according to predefined criteria and is considered the most commonly used clustering method, which results in side-by-side display of dendrograms and genetic heat maps (D'haeseleer, 2005). However, since it is based on the principle of creating a hierarchical nested tree by calculating the similarity between different classes of data points, in bioinformatics, gene expression information tends to be somewhat correlated, usually leading to the appearance of larger errors.
Another popular unsupervised learning method, k-means, which aims to partition the data set and update the center of mass by minimizing the sum of squares within clusters, is characterized by good scalability as the sample size increases (Chaudhuri and Chaudhuri, 1997; D'haeseleer, 2005; Hozumi et al, 2021). However, since it relies on computing the distance between randomly given cluster centers and each sample, it requires a large high-dimensional feature space that can lead to expensive computations, large memory requirements, and poor clustering performance (Hozumi et al, 2021) and is therefore often used together with dimensionality reduction methods in genomics research, as we will present in the next section.
Considering that the effectiveness of the k-means method is largely influenced by the k-value and the selection of the initial value of cluster centers and is more sensitive to noise and outliers, it extends to variants such as k-means++, intelligent k-means, genetic k-means, k-medians, and so on. It is worth to be mentioned that a completely unsupervised kernel-based clustering algorithm called Intelligent Kernel K-Means (IKKM) has been proposed to cluster the kernel matrix without any information. It has been used in experiments targeting gene expression clustering in human colorectal cancer and positive results have been obtained (Handhayani and Hiryanto, 2015).
The self-organizing map (SOM) method starts from a predetermined number of place names, and each iteration moves a place name closest to the selected gene to that gene, eventually forming a grid of clusters, which neighboring clusters show related expression patterns (D'haeseleer, 2005; Tamayo et al, 1999).
Clustering methods based on multi-omics data
A large number of multi-omics approaches for data integration exist for different objectives. These methods can be classified into three types according to their biological objectives and the way they process the data: regression/association-based methods, clustering-based methods, and network-based methods (Vahabi and Michailidis, 2022). Here, we focus on the clustering-based methods that are of great importance in precision medicine as well as drug research; for specific analytical evaluation of integration methods, please refer to the studies by Chauvel et al (2020) and Vahabi and Michailidis (2022).
According to the algorithmic approach, clustering methods for multi-omics are classified into three categories: (1) early integration by joining multi-omics data to form a single matrix with multiple omics features on which a single histological clustering algorithm is applied, (2) intermediate integration by building a model containing all omics data and thus achieving clustering, and (3) late integration by clustering each omics data individually and then integrating the resulting clustering solution. The specific review methods are described in detail in Rappoport and Shamir (2018).
Early integrations, such as the LDACluster model proposed by Wu et al (2015), assume that the features of different omics data are random variables obeying a hidden parameter distribution, and clustering is performed on this benchmark by obtaining the parameter matrix and decomposing it into a lower order representation of the original data (Rappoport and Shamir, 2018).
The Cancer Genome Atlas (TCGA) team proposed Cluster-Of-Cluster-Assignments (C-of-C/COCA) as one of the late integrations to deepen the understanding of cancer at the molecular level, which uses data from five different genomic/proteomics platforms to cluster tumors from different tissues to define breast cancer subtypes (The Cancer Genome Atlas Network, 2012). After that, COCA was used again to perform a comprehensive analysis of samples from 12 cancer types through 5 genome-wide platforms and 1 proteomics platform and successfully revealed a uniform classification of 11 major subtypes (Hoadley et al, 2014).
There are more types of methods included in intermediate integration. Similarity network fusion (SNF), which is recognized as a proven classical algorithm, is a similarity-based method for intermediate integration. SNF considers the complementarity contained in different omics data and first builds a fully connected network on each histology with samples as nodes and similarity as weights, and subsequently updates the network using iterative methods (Rappoport and Shamir, 2018; Wang et al, 2014).
In 2018, a robust and adaptive to noise clustering algorithm, the RSC-OTRI, was established to better identify noisy high-dimensional gene expression clusters with different histological features. It aims to maximize the separation in terms of survival curves by building a gene data matrix and computing eigenvalues and eigenvectors. In the article, it was compared with SNF and t-mixture (TMIX: model-based clustering based on Student's t-distribution), respectively, and the results showed that it was able to combine good and sparse gene correlation estimates and performed robustly to noise and survival analysis (Coretto et al, 2018).
Dimensionality reduction assisted clustering of large-scale data sets
In the previous section, we mentioned that for commonly used clustering methods such as k-means, high-latitude gene expression data can lead to expensive costs with poor performance. In fact, most clustering methods are prone to performance degradation because the feature space becomes sparse in high-dimensional space (Coretto et al, 2018), and dimensionality reduction should effectively avoid this problem.
Principal component analysis (PCA) is one of the oldest and most widely used. It is based on the principle of projecting data matrices onto a low-dimensional space, and being a descriptive tool that does not require assumptions makes it well suited for various types of adaptive exploration methods but has limitations for data sets that are relatively more important to maintain local distances, such as genome sequences (Hozumi et al, 2021; Jolliffe and Cadima, 2016).
UMAP is a relatively new dimensionality reduction method based on multibody theory for topological data analysis techniques (McInnes et al, 2018). The principle lies in optimizing the spectral layout of the data in the lower dimensional space so that the error between two topological spaces will be minimized (McInnes et al, 2018). Since it can capture the global and topological structure while maintaining the local distance and its fast and effective operation, it has advantages for large data sets and is better for capturing recognition interactions and analyzing transcriptome data as well as visualize genetic interactions with biomolecular spatial relationships (Dorrity et al, 2020).
In addition, it is considered to be the most suitable dimensionality reduction method for use with k-means clustering methods that the combination has been used in genome phylogenetic analysis (Hozumi et al, 2021) and drug repositioning (Cong et al, 2022). It has been shown to have significantly improved clustering accuracy and is gradually becoming one of the popular dimensionality reduction methods recently in genomics and other areas.
t-distributed Stochastic Neighbor Embedding (t-SNE) is a nonlinear dimensional reduction algorithm, as an improvement of the SNE, it uses a symmetric probability formula and does not resort to a normal distribution such as the SNE when calculating the probabilities between sample points in low-dimensional space, but instead uses a t-distribution. In measuring the scatter distance between two probability distributions, t-SNE uses the Kullback–Leibler (KL) divergence as SNE does (Hozumi et al, 2021; van der Maaten and Hinton, 2008).
The goal of linear discriminant analysis, which also uses projection, aims to minimize intra-class variation and maximize directional projection of inter-class variation (Fisher, 1936).
AI approaches in COVID-19 drug discovery
AI methods are widely used in public health, disease prediction, and drug development. With the advent of DL, automatic feature extraction from raw data has led to improved performance compared with other computer-aided models. Different DL algorithms are used to fight the COVID-19 pandemic, including artificial neural network, CNN, and long short-term memory (Prasad and Kumar, 2021). Next, we discuss some representative examples of AI-driven drug development that used in COVID-19.
Generative models were proposed for antiviral drug discovery in the early days of the pandemic. Nguyen et al (2020b) proposed a mathematical deep learning (MathDL) model for generating a low-dimensional representation of high-dimensional chemical/physical interactions. They integrated this representation into different DL models such as CNN and GAN for predicting the pose and energy of the interaction. They applied this model for finding inhibitors for 3CLpro of SARS-CoV-2 (Nguyen et al, 2020a).
3CLpro is an important potential drug target that is critical for the inhibition of viral replication. On this basis, Gao et al (2020) evaluated the binding affinity of drugs to SARS-CoV-2 3CLpro using a structure-based drug repositioning (SBDR) ML model, and 314 SARS-CoV-2/SARS-CoV-3CL inhibitors were trained on a two-dimensional fingerprint-based DL gradient-enhanced decision tree model from 8565 drugs were evaluated and scored, and finally, the top 20 drugs approved by FDA and the top 20 non-marketed drugs in the study were selected as effective inhibitors of SARS-CoV-2 3CL protease.
Hozumi et al (2021) successfully performed a phylogenetic analysis of large-scale SARS-CoV-2 genomic sequence data sets by UMAP-assisted k-means method and also compared the performance of various dimensionality reduction methods used for assisted clustering to further analyze the effectiveness of different methods in terms of speed and scalability, which has positive implications in analyzing mutation patterns of viruses and predicting transmission routes for effective drug discovery and vaccine production. Meanwhile, UMAP, being validated as the most suitable dimensionality reduction method for auxiliary clustering, could be used in the future for more drug repurposing as well as other fields.
Richardson et al (2020) integrated biomedical data from structured and unstructured sources through the BenevolentAI knowledge graph to propose a list of potentially effective drugs including the anti-HIV lopinavir plus ritonavir combination. Although some of the compounds obtained in this study result in serious side effects and cannot be useful as effective treatments in this infection today, it is undeniable that the BenevolentAI knowledge graph, which can respond quickly at an early stage, has potential in areas such as future prevention of infectious diseases.
Kowalewski and Ray (2020) presented a ML drug discovery pipeline that identifies at least 6 potential lead drugs that may be effective against COVID-19 from FDA-registered chemicals and approved drugs as well as ∼14 million purchasable chemicals.
Ke et al (2020) developed an AI-based model using DNN to identify 80 promising marketed drugs and tested the activity of all AI-predicted drugs against feline coronavirus (FCoV) in a cell-based in vitro assay, 8 of which inhibited proliferation of feline infectious peritonitis virus (Mottaqi et al, 2021).
Beck et al (2020) used a DL-based DTI prediction model called Molecule Transformer-Drug–Target Interaction (MT-DTI), which they proposed in 2019, to identify a list of commercially available antiviral drugs that could disrupt the components of the SARS-CoV-2 virus. Prediction using established drug repurposing methods is a fast and efficient approach, which can help to provide a timely response in the face of an unexpected and complex situation.
By constructing a compound database with virtual drug screening, molecular docking, and supervised ML algorithm identification, Kadioglu et al (2021) obtained a list of the best compounds targeting spike protein, nucleocapsid protein, and 2′-o-ribose-methyltransferase and identified the top nine compounds with the highest protein–drug interactions.
Although some of these candidates, such as the antiviral Remdesivir, have been shown to be effective in adaptive platform trials (Beigel et al, 2020), most have not been proven in clinical trials or have been found to be ineffective suggesting that efficient and accurate identification of drug candidates is still a problem that needs to be addressed.
Conclusions and Future Perspectives
Through this sudden explosion of COVID-19, drug repositioning is back in the spotlight with its advantages in terms of time and cost in the drug development process. For ease of viewing, the methods we discuss this time are briefly summarized in Table 1. Although the most of these studies cannot yet be considered clinically successful, empirical facilitation of standardization through computational methods and predictive capabilities can lead to faster and more accurate responses in the face of possible future outbreaks of epidemics.
Summary of Important Artificial Intelligence-Based Methods and Studies
AI, artificial intelligence; BLM, bipartite local model; CE-GAN, class expert generative adversarial network; CNN, convolutional neural network; CNNC, convolutional neural network for coexpression; COCA, Cluster-Of-Cluster-Assignments; DCNN, deep convolutional neural network; DNN, deep neural network; DTI, drug–target interaction; GAN, generative adversarial network; IKKM, Intelligent Kernel K-Means; LDA, linear discriminant analysis; MathDL, mathematical deep learning; mRNA, messenger RNA; MT-DTI, Molecule Transformer-Drug–Target Interaction; n.a., not available; NLP, natural language process; NN, neural network; PCA, principal component analysis; PREDICT, PREdicting Drug IndiCaTion; RNN, recurrent neural network; SBDR, structure-based drug repositioning; SNF, similarity network fusion; SOM, self-organizing map; SVM, support-vector network; SVR, support vector regression; t-SNE, t-distributed Stochastic Neighbor Embedding; UMAP, Uniform Manifold Approximation and Projection for Dimension Reduction.
Figure 3 briefly summarizes the current applications of AI and ML in response to COVID-19; a more comprehensive summary of 146 articles is presented by Comito and Pizzuti (2022), and we can find that the majority of AI methods are used for detection/monitoring, prevention/treatment, and pathogenesis analysis, but only few are used for drug repositioning (Mottaqi et al, 2021). In other words, while AI approaches have been more fully researched in areas such as medical image processing diagnosis and its prediction of propagation patterns through ML calculations, there is still much room for progress in areas such as patient treatment and drug development.
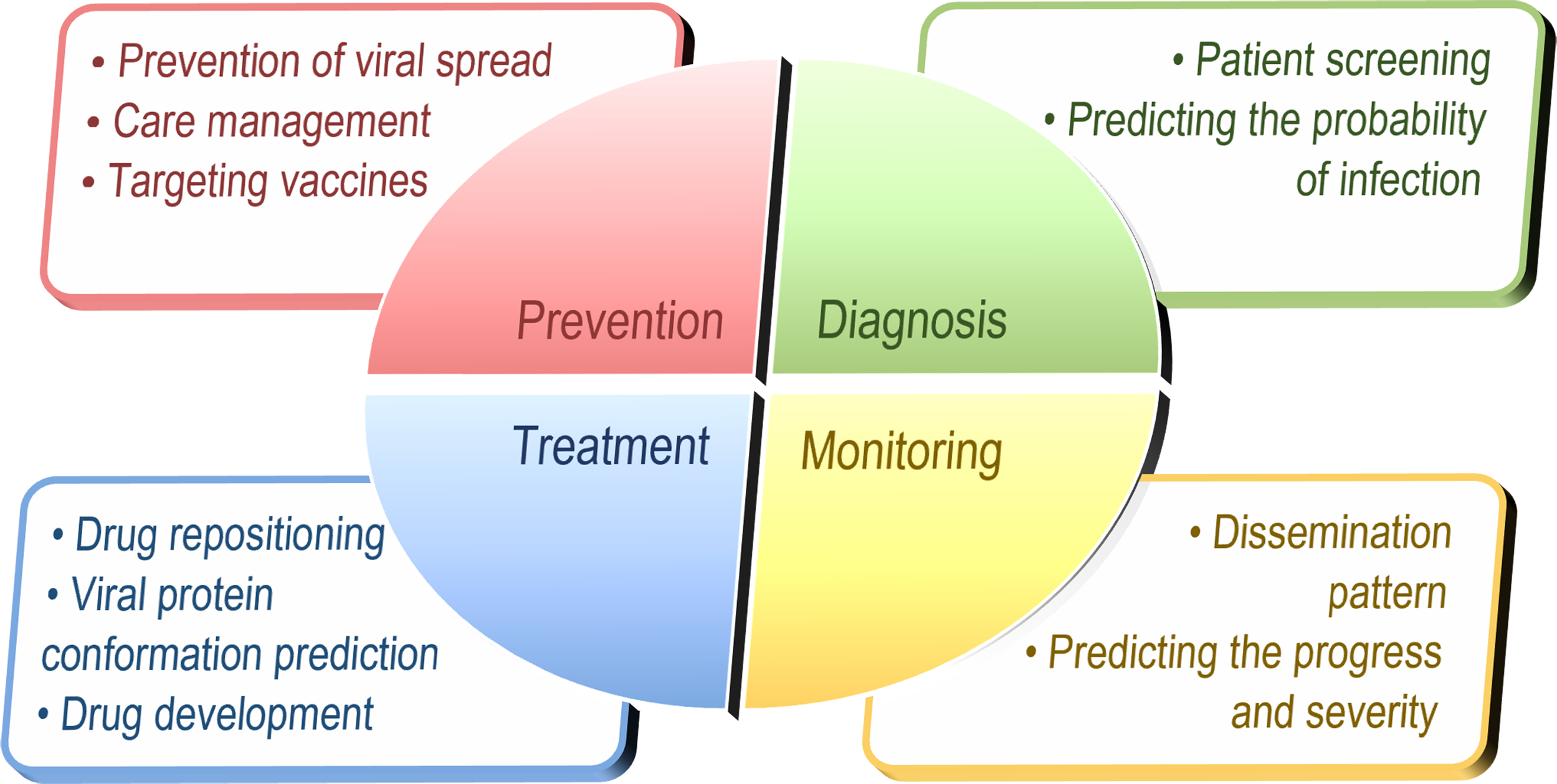
Current applications of AI and ML in response to COVID-19. AI, artificial intelligence; COVID-19, coronavirus disease 2019.
In these areas, the potential of AI is undoubtedly huge, and if more advanced and targeted AI-based drug repositioning methods can be developed, it will undoubtedly be a great help for future efforts to fight infections similar to COVID-19.
AI/ML methods open the door not only drug repurposing with combining existed multi-omics data but also development of new drugs under complex conditions as well. For this sake, establishing relevant databases should be helpful. For emerging viruses, data collection activities are of great importance, as almost all computational methods need to be based on large high-quality data sets.
During this COVID-19 pandemic, the data sharing mechanism of the Global Initiative on Sharing All Influenza Data (GISAID), that the world's largest repository for SARS-CoV-2 sequences, the Centers for Disease Control and Prevention (CDC), WHO, and other databases greatly facilitated the progress of related research, while most of the data resources that have been established before the pandemic and expanded with COVID-19, such as molecular data resources such as GenBank, UniProt, PPI networks resources, or various drug databases, networks resources, and databases for various drugs or compounds have been utilized (Galindez et al, 2021).
It would be prospected that expansion of multi-omics data will be concerted for the drug repurposing as well as drug design by the conductor of AI/ML on the harmony of appropriately organized database in the future.
Footnotes
Acknowledgments
The authors wish to thank the anonymous reviewers as well as the editor for their valuable and constructive suggestions to improve the article.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
No funding was received for this article.