
Abstract
Drug repurposing has broad importance in planetary health for therapeutics innovation in infectious diseases as well as common or rare chronic human diseases. Drug repurposing has also proved important to develop interventions against the COVID-19 pandemic. We propose a new approach for drug repurposing involving two-stage prediction and machine learning. First, diseases are clustered by gene expression on the premise that similar patterns of altered gene expression imply critical pathways shared in different disease conditions. Next, drug efficacy is assessed by the reversibility of abnormal gene expression, and results are clustered to identify repurposing targets. To cluster similar diseases, gene expression data from 262 cases of 31 diseases and 268 controls were analyzed by Uniform Manifold Approximation and Projection for Dimension Reduction followed by k-means to optimize the number of clusters. For evaluation, we examined disease-specific gene expression data for inclusion, body myositis, polymyositis, and dermatomyositis (DM), and used LINCS L1000 characteristic direction signatures search engine (L1000CDS2) to obtain lists of small-molecule compounds that reversed the expression patterns of these specifically altered genes as candidates for drug repurposing. Finally, the functions of affected genes were analyzed by Gene Set Enrichment Analysis to examine consistency with expected drug efficacy. Consequently, we found disease-specific gene expression, and importantly, identified 22 drugs such as KM 00927, I-BET, alvocidib, and vorinostat as candidates for repurposing. These were previously noted to be effective against two of the three diseases, and have a high probability of being effective against the other. The two-stage prediction approach to drug repurposing presented here offers innovation to inform future drug discovery and clinical trials in a variety of human diseases.
Introduction
Drug repurposing is a method of developing new targets for existing drugs, that is, discovering new efficacy for a previously approved drug, for which safety and pharmacokinetics have been demonstrated in humans. While de novo drug development typically takes 6–9 years and costs 2–3 billion dollars, drug repurposing will lead directly to preclinical testing and clinical trials (Rapicavoli et al., 2022), thereby significantly reducing the time, cost, and side effects, leading potentially to a higher success rate for introduction in clinical practice because the compounds have already been tested for safety and pharmacokinetics in humans (Jourdan et al., 2020).
Drug repurposing has broad importance in planetary health for therapeutics innovation in infectious diseases as well as common or rare chronic human diseases. Drug repurposing has also proved important to develop interventions against the COVID-19 pandemic. In recent years, drug repurposing has reached 30% of new drugs and vaccines approved by the U.S. Food and Drug Administration (FDA) (Kwon et al., 2019). However, the main questions often arise from how to customize or optimize the repurposing methods into efficient drug repurposing pipelines (Jin and Wong, 2014). Developing promising and affordable approaches for the effective treatment of complex diseases is difficult without prior knowledge of the complete drug-target network. This is now the greatest challenge to advancing drug repurposing technology (Zeng et al., 2020).
The present study presents a unique two-stage approach to drug repurposing that (1) harnessed machine learning (ML) to identify significantly altered gene expression profiles based on comparative data under diseased and normal conditions, and (2) analyzed the data on gene expression changes due to drug treatment, and (3) estimated the expected normalization of expression changes caused by a disease. To fully validate this approach, we analyzed Gene Ontology (GO) for a group of disease-variant genes.
Conceptual background on the study
Screening of drugs for repurposing includes a variety of methods, such as target-based, knowledge-based, signature-based, pathway- or network-based, and mechanism-targeted methods (Rapicavoli et al., 2022). The oldest case of drug repurposing is sildenafil for erectile dysfunction, which is usually considered accidental (Roundtable on Translating Genomic-Based Research for Health et al., 2014). However, serendipity-based discoveries cannot be expanded, and various methods have been used to enable systematic discovery of the off-label effects of drugs.
Phenotypic screening is a relatively more proactive and controlled method (Ciallella and Reaume, 2017), but it is not usually systematic and comprehensive enough, although it can occasionally successfully identify lead compounds. Since a disease usually emerges as a complex interaction between multiple genetic variants (Hirschhorn and Daly, 2005), computer-based approaches must be used to achieve systematic or comprehensive repurposing.
With the advent of high-throughput technologies for exploring biological systems, an impressive amount of data awaits computational analysis and mining tools to be explored and harnessed (Rapicavoli et al., 2022). Systems biology approaches to drug repurposing utilize pathophysiological mapping of diseases to identify targets that modify them, and potential compounds that can hit those targets (Turanli et al., 2018). In addition to computer simulations and target docking using algorithmic solutions other than medicinal chemistry, data mining based on gene expression provides clues to active pharmaceutical ingredients that may be formulated into clinically viable drugs (Chen et al., 2017).
The signature-based drug repurposing approach relies on the use of genetic signatures derived from disease-wide data such as microarrays, RNA-seq, which can identify unknown off-targets or unknown disease mechanisms. Since the required information may be difficult to obtain from the existing literature, obtaining genetic signatures for these diseases from publicly available genomic data becomes the best option (Rapicavoli et al., 2022).
In this context, artificial intelligence (AI) tools such as ML are powerful because they can identify patterns at scale. The history of AI can be briefly described in terms of three paradigms: good old-fashioned artificial intelligence (GOFAI) (1950s−1960s), expert systems (late 1970s−1980s), and ML (2010−present). GOFAI focused on the creation of general logic systems and led to the development of fundamental techniques such as heuristic searches. Expert systems narrowed the focus from general intelligence to human experts in specific fields, such as chemistry and medicine, and attempted to replicate their knowledge and decision-making processes. This led to the first major medical AI systems such as MYCIN (Garvey, 2018). While these yielded some practical results, none of these AI paradigms became “thinking machines.”
However, the current ML paradigm has overcome some of the hurdles associated with the real world, thanks to the ever-increasing amount of human-generated data, the massive increase in computing power, and the renaissance of neural networks and other ML algorithms. These “learning” algorithms can be “trained” to infer patterns from human-generated data, and therefore do not require explicit representation of knowledge by the programmer (Garvey, 2018).
Although AI is still in its infancy in drug development, AI and ML algorithms have unprecedented potential to accelerate the discovery of effective new drugs. DSP-1181 is reportedly the first off-targets drug created using AI to enter clinical trials. Exscientia, which developed it, noted that it took <12 months from initial screening to the end of preclinical testing, compared with 4 years using traditional methods (Farghali et al., 2021). To date, many computational methods for drug repurposing using ML techniques are continuously being proposed and improved as new problems arise. Over the past several decades, computer tools, such as quantitative structure-activity relationship modeling, were developed to identify potential bioactive molecules quickly and inexpensively from great numbers of candidate compounds.
As ML approaches evolve into deep learning approaches, they become more powerful and efficient way to deal with the massive amounts of data generated from modern drug discovery approaches (Farghali et al., 2021). For example, to address the challenge of how to derive drug repurposing from drug-disease interactions, a methodological approach that focuses primarily on drug properties has been established (Napolitano et al., 2013). These ML approaches involving data integration are established by integrating information from different layers based on similarities such as chemical structures, molecular targets, and induced gene expression characteristics, and they focus on predicting the therapeutic class of United States Food and Drug Administration (FDA)-approved compounds without considering data about the disease.
The method reclassifies specific drugs and purposefully interprets the parts that do not match the original classification as genuine reclassification opportunities, while showing promising and highly accurate results by integrating different information layers and maximizing their efficacy through the dimensionality reduction-based computational procedures classical multidimensional scaling and principal component analysis (PCA). However, this approach only starts from the direction of drugs, and while it does address problems such as the complexity, variability, and sparsity of data currently available for diseases, the inability to explore specific diseases with strong targeting also largely limits its use.
Another representative modified method, drug repurposing in Alzheimer's disease (DRIAD), combines the use of omics datasets on drug-induced perturbation of neuronal cells with the molecular changes that occur in the brains of individuals suffering from different stages of Alzheimer's disease (AD) to generate a drug-associated gene list (Rodriguez et al., 2021). DRIAD was used to effectively decouple gene set enrichment and predictor performance by filtering the transcriptomic space for genes associated with drugs before model training and predictor evaluation. Simultaneous prefiltering to a limited set of features also addresses issues with overfitting and enables a direct, unbiased quantification of the association between the effects of a drug and AD progression.
However, DRIAD is performed to determine whether drug-induced changes correlate with molecular markers of disease severity by measuring what happens to nerve cells in the human brain when they are treated with drugs, which has some limitations for application in other diseases. Also, the method is based on a specified list of drugs for further analysis of the genes they affect, which means that only the list of drugs specific to the user can be screened, resulting in a local optimum in the list, but still relying to some extent on the user's drug screening strategy.
The k-means method, a commonly used clustering method, has good scalability with sample size increase. However, since it relies on computing the distance between the clustering centers given randomly and each sample, it requires a large feature space in high dimension that can result in expensive computation, large memory requirement, and poor clustering performance (Hozumi et al., 2021). Since the gene expression data are inherently highly dimensional, dimensionality reduction should be effective to avoid this problem, and finding a group of diseases sharing a hidden core of abnormality. PCA is often used for such purposes due to its intuitiveness and mathematical simplicity, but as a linear algorithm, PCA performs poorly on the features with nonlinear relationship.
Uniform Manifold Approximation and Projection for Dimension Reduction (UMAP), an ML algorithm, is a topological data analysis technique based on many-body theory, which provides significant improvements in data localization and preservation of local structure compared with PCA. Also, in capturing similar word vector groups, UMAP has advantages over T-distributed stochastic neighbor embedding for large datasets, because it captures global and topological structures, and the error between two topological spaces will be minimized by optimizing the spectral layout of data in the low-dimensional space (Hozumi et al., 2021; McInnes et al., 2018). However, the method does not provide the boundary of clusters that can often be unapparent. In practice, logical clustering is mandatory for consistency and validity.
In the present study, we propose a five-step method for clustering. First, UMAP is applied to downscale gene expression data from different diseases. Second, data are classified using the k-means method with Silhouette analysis to determine k, the appropriate number of groupings. Third, those genes with significantly deviated expression for each disease were identified. Forth, L1000CDS2 knowledge base is searched to identify chemical compounds revert impaired gene expression. Finally, the obtained lists are compared among the disease cluster to highlight possible drug repurposing. The result was examined with gene set enrichment analysis (GSEA) to address possible mechanisms shared across the group of diseases.
We use the data from microarrays to identify genes that may be up-/downregulated in disease microarrays, then searches the literature on drugs known to have opposite effects. By combining the gene expression responses of cell lines caused by diseases with data on drug-induced changes in gene expression, which not only solves the problem of scarce genomic data for diseases and quantifies the association between drug effects and diseases, but also generates a list of candidate drugs for specific diseases and performs analytical calculations by clustering. Therefore, compared with other existing methods, this method is considered to have wider applicability in practical applications and largely alleviates the bias caused by subjective screening by users.
Materials and Methods
Transformation of gene expression data
From the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/), gene expression datasets were obtained from 262 patients, and 268 healthy human samples associated with 31 diseases that were analyzed on the Affymetrix Human Genome U133A array platform and submitted by Affymetrix. The associated information is listed in Table 1. The logarithm of the ratio of expression intensities from patients' samples to the mean of the corresponding healthy controls was calculated for each disease in turn to obtain log fold-change (logFC) values for disease-specific gene sets (Robinson and Oshlack, 2010). The obtained gene expression data were treated as a matrix of 262 × 22,283 dimensions in subsequent analyses (Supplementary Data S1).
Datasets for Cases
BOEC, blood outgrowth endonuclear cells; GEO, Gene Expression Omnibus; IgA, immunoglobulin A; PBMC, peripheral blood mononuclear cells.
Two different procedures were applied to normalize the experimental data (Park et al., 2003). For data with binary logarithm values, such as robust multiarray average (RMA) and GC-RMA, mean values of healthy samples were simply subtracted from those of diseased samples to yield the binary logarithm of the ratio. For antilogarithms, such as normalization by Affymetrix MicroArray Suite 5.0 (MAS5) algorithm, binary logarithms of values were calculated before subtraction.
Clustering of disease data and selection of target diseases using UMAP and k-means methods
UMAP was used to reduce the hyper dimensionality to two dimensions. UMAP analyzes topological data based on manifold theory, which is characterized by its ability to clearly separate clusters through dimensionality reduction (McInnes et al., 2018). In this study, the final parameters used were n_neighbors = 8, min_dist = 1, n_epochs = 500, metric = correlation, and set_op_mix_radio = 0.3, to allow a clearer classification of the different disorders to adjust the parameter values.
The resulting two-dimensional data were clustered using the k-means method that based on Euclidean distance to visualize target diseases among clearly classified groups. Since the basic idea of the k-means method is to select the centers of k clusters based on a given k-value, and then assign the sample points to be classified to the clusters according to the nearest neighbor principle for calculation, selecting a k-value will have a decisive impact on data clustering. We chose the Silhouette method for the determination of k-value. The Silhouette value measures how similar a point is to its own cluster compared to others, so in which a high Silhouette value is desirable and indicates that the point is placed in the correct cluster (Khyati, 2019).
Disease groups in the same cluster obtained by the k-means method are expected to have similarities in cellular signaling and gene expression and are candidates for off-target drug effects.
Classification of disease-specific gene expression
GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/) in NCBI GEO is a web-based interactive tool that can be used to compare multiple groups in the GEO series. | logFC | > 1 and p-value <0.05 are usually considered statistically significant (Zhang et al., 2020), so genes exhibiting significantly altered expression in a specific disease were identified on it based on the latter specifications.
Searching for small-molecule compounds
Using L1000CDS2 (https://maayanlab.cloud/L1000CDS2/#/index), we identified compounds that reversed the changes in gene expression patterns for each disease while searching for drugs (Duan et al., 2016). Using the scatter plots of L1000 perturbation gene signatures and the results of chemical perturbations, genes directly affected by each identified drug were matched and organized.
GSEA and function prediction for gene sets
In preparation for GSEA, we collated raw genetic data for target diseases and inserted a “Description” (value = na) column for each disease to obtain three collated data tables. GSEA was performed using the “Human_AFFY_HG_U133_MSigDB.v7.4.chip” chip platform, and GO information was extracted for normalized p-value below 0.05 and false discovery rate (FDR) below 0.25 (Thomas et al., 2011). Finally, disease-specific expressed genes were classified using GO terms and their functions were compared.
Results
Dimensionality reduction with UMAP and clustering with k-means
For the datasets for cases and controls obtained from the NCBI GEO, by transforming gene expression data to analyze similarity between different diseases using clustering, and by comparing diseases classified in the same group to obtain a list of alternative drugs, we further processed the combined data and used UMAP and k-means methods for analysis.
UMAP was applied to the transformed expression data to reduce the hyper dimensionality of gene expression levels into two-dimension (Fig. 1a). Then, they were grouped by applying the k-means method to obtain similar changes in expression of gene sets, expecting they reflect similar changes in cytological conditions. The Silhouette Score reaches its global maximum at the optimal value (k-value = 19, Fig. 1b) as represented by the peak in the figure. The visualized groupings resulting from UMAP are shown in Figure 1c.
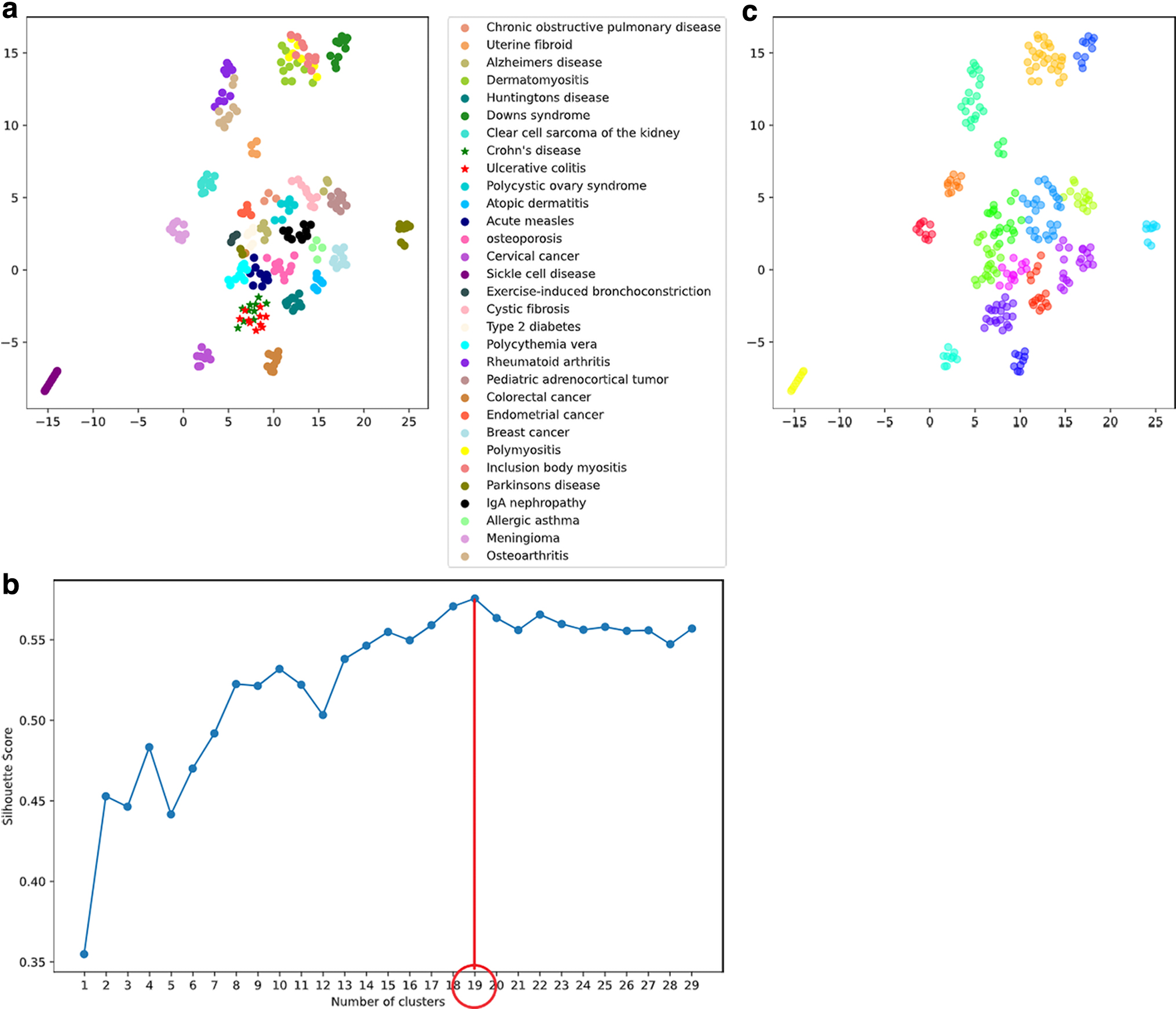
Clustering of disease data.
Target diseases were selected from the results of clustering by the k-means method, through which 31 diseases were clustered into several clearly classified groups. Figure 1c shows that inclusion body myositis (IBM), polymyositis (PM), and dermatomyositis (DM), which were classified into the same yellow cluster located on the upper right according to the results of the k-means method, shared similarity with each other; hence, they were chosen for the following analyses:
Target diseases and samples:
IBM: GSM3676259-GSM3676284 (26 sets) PM: GSM3676317-GSM3676323 (7 sets) DM: GSM3676247-GSM3676258 (13 sets) Healthy: GSM3676285-GSM3676296 (12 sets)
Identification of disease-specific significant changes in gene expression by GEO2R
GEO2R was used to identify genes whose expression levels were specifically increased or decreased in the three target diseases. Figure 2 shows the significance levels and p-values on the ordinate versus the fold change on the abscissa. Genes satisfying the conditions | logFC | > 1 and p-value <0.05 (Supplementary Tables S1 and S2) were used for further analysis to identify common gene expression patterns (Supplementary Tables S3 and S4).
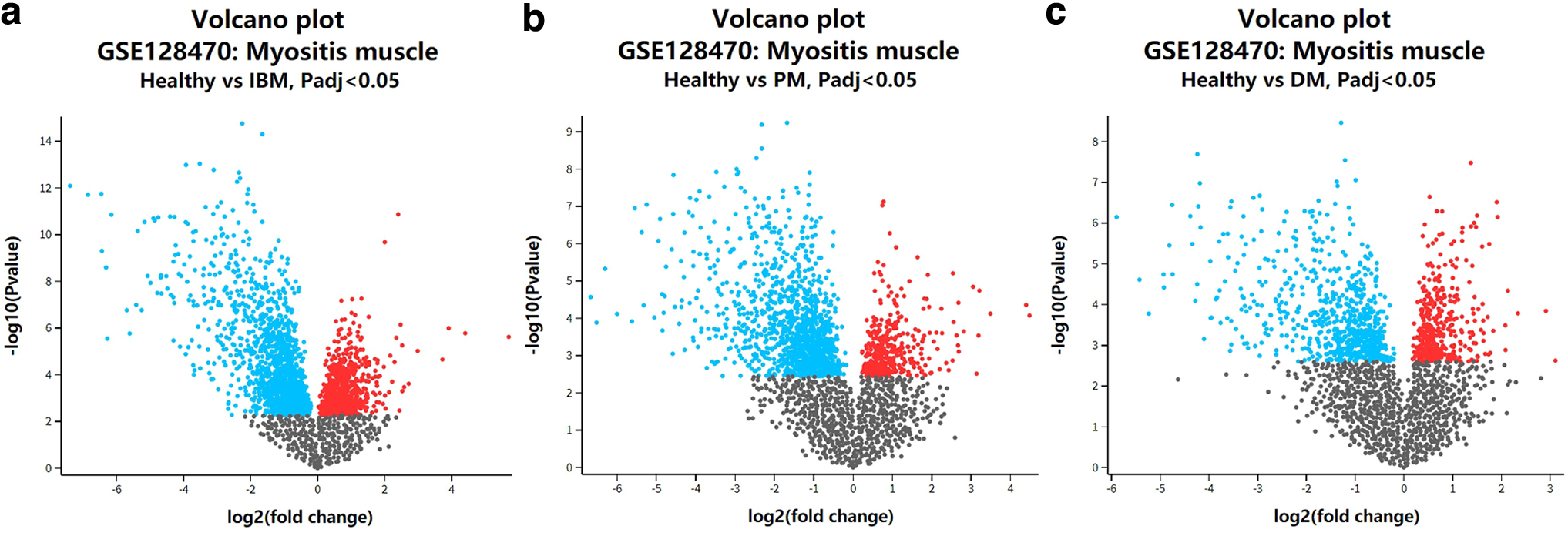
Discovery of differentially expressed genes.
Exploration of small-molecule compounds that recover gene expression patterns altered by disease
Using data for genes with variable expression, we searched for small-molecule compounds that reversed the expression pattern changes for each disease using L1000CDS2. The obtained compounds corresponding to each disease are listed in Supplementary Table S5.
When these compounds were compared according to the L1000CDS2 results, common drugs were identified (Fig. 3, Table 2 and Supplementary Table S6). We found nine drugs that corresponded to IBM and PM, four drugs that corresponded to IBM and DM, nine drugs that corresponded to PM and DM, and ten drugs that corresponded to all three diseases.
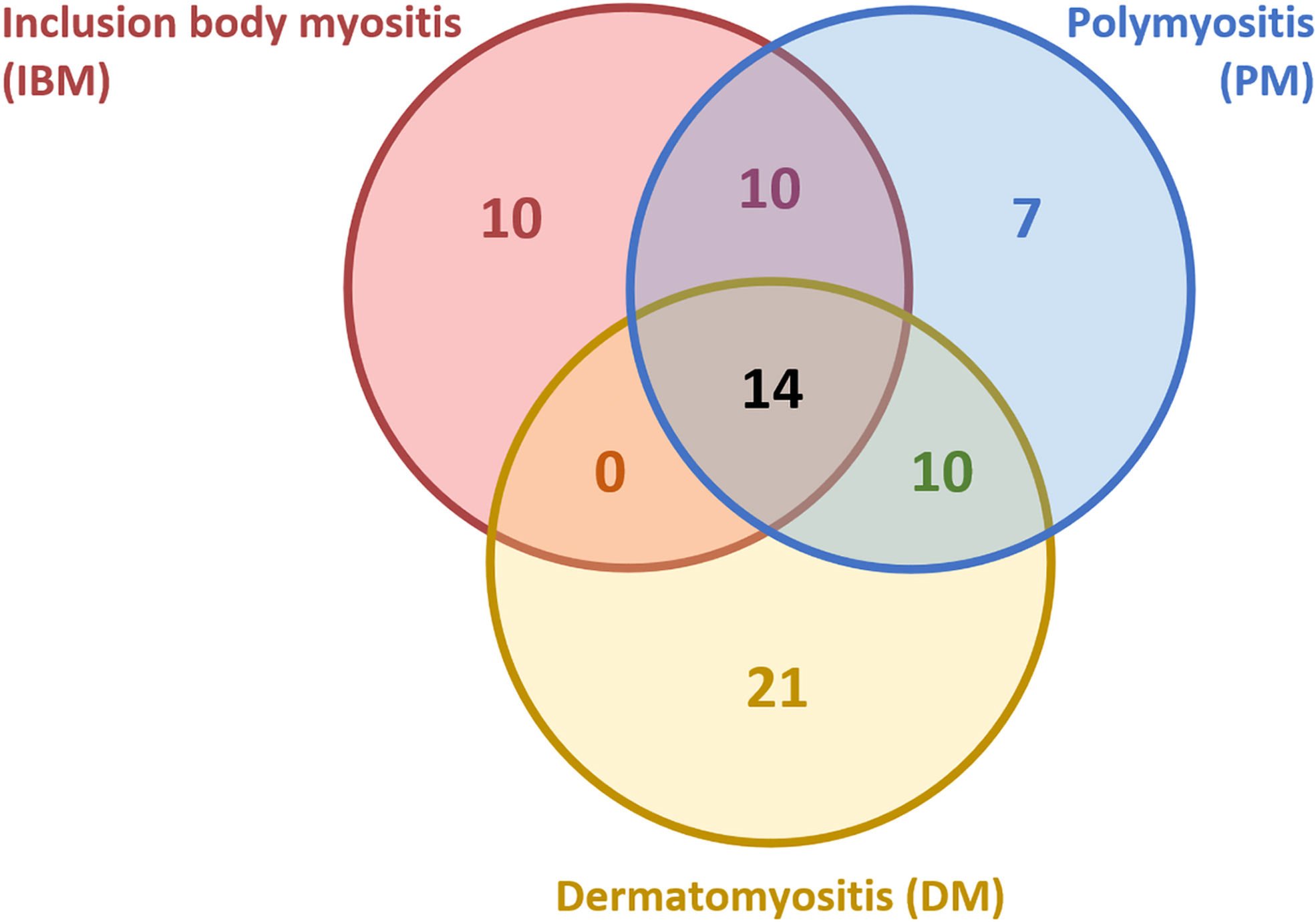
Number of small-molecule compounds for each disease. Numbers in overlapping regions are compounds shared between the disorders.
Breakdown of Common Drugs
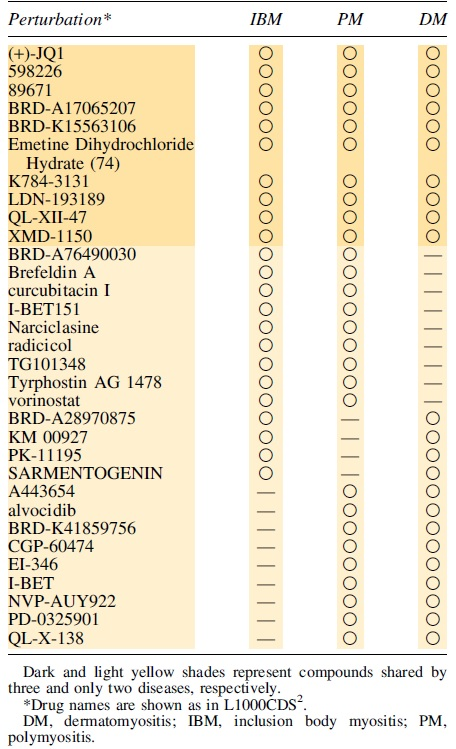
These drugs are small-molecule inhibitors and other interfering agents that reverse the changes in gene-specific expression patterns. Genes regulated by each drug were collated, the number of times the drug appeared was calculated in a sum set of results for drugs that appeared more than once, and areas where two diseases had something in common were sorted (Supplementary Tables S7–S9). The following L1000 perturbation gene signatures scatter plots were generated:
IBM: https://maayanlab.cloud/clustergrammer/l1000cds2/63c5271f7903f4005f0b012a
PM: https://maayanlab.cloud/clustergrammer/l1000cds2/63c5285e7903f4005f0b012c
DM: https://maayanlab.cloud/clustergrammer/l1000cds2/63c52f867903f4005f0b0134
Biological processes significantly affected by the diseases
The three target disease datasets were formatted and GSEA was performed to compare the functions of the expressed genes. The GO results for the top 20 functions with Nominal p-value <0.05 and FDR <0.25 are shown in Supplementary Table S10. Genes obtained from GSEA were classified by function using GO terms (Supplementary Table S11).
Discussion
In this study, we took advantage of the large-scale identification and integration of different levels of information and biological insights that ML offers. The efficiency and accuracy of drug candidate calculations were superior to those of previous studies, effectively improving the likelihood of successful drug repurposing, since all drugs were derived from agents effective against other diseases that clustered together in the same group.
To examine the efficacy of dimensionality reduction by UMAP, we directly clustered the 262 × 22,283 dimensions gene expression data using the k-means method as shown in Supplementary Table S12. The result failed to show effective clustering as suggested by Hozumi et al. (2021), although some of the groups appeared to be clustered appropriately as Osteoarthritis were clustered into the same group (no. 7).
In addition, for the k-value calculation in the k-means method, we first considered using the elbow method, but due to the complexity of the data, a clear inflection point could not be determined. Therefore, we instead used the Silhouette method, which has a wider range of applicability. It combines both cohesion and separation to evaluate the impact on the clustering results produced by different algorithms, or different ways of running the algorithm, based on the same original data. This yielded 22 small-molecule compounds for only one disease for which applicability to other diseases was unconfirmed, nine with unconfirmed efficacy only for DM, four with unconfirmed efficacy only for PM, and nine with unconfirmed efficacy only for IBM.
Of the 266 genes directly affected by the nine compounds that were validated for IBM and PM using L1000CDS2 but not validated for DM, 112 were common to the disease-specific gene expression data for DM obtained using GEO2R. Of the 63 genes directly affected by the four compounds whose efficacy was unconfirmed only for PM, 53 were common to the disease-specific genes for PM obtained using GEO2R. Of the 171 genes directly affected by the nine compounds whose efficacy was unconfirmed only for IBM, 123 were common to the disease-specific genes for IBM obtained using GEO2R.
Next, we obtained BRD-IDs and PubChem Names for 22 small-molecule compounds with unconfirmed efficacy for only one of the three diseases using side effect prediction based on the L1000 data (http://maayanlab.net/SEP-L1000/index.html). PubChem names were obtained and their efficacy as drugs was investigated in several databases (Supplementary Table S13). Of the nine compounds that were only validated for IBM and PM, BRD-A76490030 is functionally related to certain α-amino acids, and is predicted to be an EGFR inhibitor, PARP inhibitor and calcium channel blocker at L1000 fireworks display (L1000FWD), although no specific functions have been reported. Brefeldin A is a fungal metabolite with broad antibiotic activity. Curcubitacin I is a plant metabolite and a cucurbitacin that acts as an antitumor agent. I-BET151 induces apoptosis via down-regulation of BCL2 or induces G0/G1 cell cycle arrest. Narciclasine has been reported to exhibit significant anticancer activity in experimental models of brain tumors by targeting GTPases and damaging actin cytoskeletal organization (Van Goietsenoven et al., 2013). Radicicol is a macrocyclic antifungal antibiotic that inhibits signal-dependent transcriptional activation and induces differentiation in leukemia. TG101348 is an ATP-competitive inhibitor of Janus-associated kinase 2 (JAK2) and FMS-like tyrosine kinase 3 (FLT3; CD135; STK1; FLK2) used in the treatment of primary and secondary myelofibrosis. Tyrphostin AG 1478 may have a role as an epidermal growth factor receptor antagonist, antineoplastic agent, geroprotector, and antiviral agent. Vorinostat is a histone deacetylase (HDAC) inhibitor used to treat cutaneous symptoms in patients with cutaneous T-cell lymphoma (CTCL) after systemic therapy.
Of the four compounds that were only validated for IBM and DM, only PK-11195 has been found to act in the assessment of neuronal damage (Weissman and Raveh, 2003). Although the pharmacological actions of the other three have not been reported, there is a clear trend in MOA prediction in the L1000FWD database: BRD-A28970875 has a probability of 0.9477, and SARMENTOGENIN has a probability of 0.4499, that is a protein synthesis inhibitor. The last one, KM 00927, has a 0.6189 chance of being an NFkB pathway inhibitor.
Of the nine compounds that were only validated for PM and DM, A-443654 is a pan-Akt inhibitor with equivalent activity against intracellular Akt1, Akt2 or Akt3, inhibiting tumor cell growth and proliferation. Alvocidib as an inhibitor of cyclin-dependent kinase (CDK), inhibits CDK phosphorylation, down-regulates cyclin D1 and D3 expression, and induces G1 cell cycle arrest and apoptosis. BRD-K41859756 aka NVP-AUY922 is a potent HSP90 inhibitor that effectively destabilizes IGF-1Rβ protein and is involved in cell growth inhibition, autophagy and apoptosis. CGP-60474 is one potent anti-endotoxin agent, effective in inhibition against CDK. EI-346 has a predicted probability of 0.2968 as a cyclooxygenase inhibitor at L1000FWD. I-BET is considered a BET bromodomain inhibitor and is described in the related link for I-BET 151 above. NVP-AUY922 can effectively downregulate and destabilize IGF-1Rβ protein, leading to growth inhibition, autophagy and apoptosis. PD-0325901 targets mitogen-activated protein kinase (MAPK/ERK kinase or MEK) and is expected to have antitumor activity. QL-X-138 is a potent and selective BTK/MNK dual kinase inhibitor that may be useful in the study of B-cell malignancies.
When we contrasted the functions of genes that were specifically altered in one of the target diseases identified by GSEA with the effects of drugs that were found to be effective only in the other two diseases according to L1000CDS2, many overlaps were found, and most were related to immune responses. Since these three diseases are both autoimmune inflammatory muscle diseases (Nishino, 2020), it is likely that the reactions inhibited need to be suppressed for treatment.
The two-stage prediction approach to drug repurposing presented here offers innovation to inform future drug discovery and clinical trials in a variety of human diseases. We predict that drugs shown to be effective for only two diseases may also be effective for other diseases, for which no efficacy was reported previously.
It should be noted, however, that the clustering of gene expression might reflect shared tissue of origin instead of disease mechanism in common. The fact that body myositis (IBM), PM, and DM clustered together could be due to tissue origin (muscle) rather than common disease etiology or mechanism, or both. Nonetheless, we believe that our proposed method would be useful for drug repurposing because the method focuses of the genes with altered expression under the condition of disorders, which is canceled by the treatment. Further study shall give more concrete perspectives.
Footnotes
Authors' Contributions
Y.C. designed the study, performed data analysis, and wrote the article. M.S. contributed the conceptual design. F.I. contributed writing the article. T.E. and N.O. contributed to the research question and edited the article. All authors have made a significant intellectual contribution, read, and approved the article.
Acknowledgments
The authors thank the anonymous reviewers as well as the editor for their valuable and constructive suggestions to improve the article.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
No funding was received for this article.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.