
Abstract
Abstract
Machine learning (ML) is being ubiquitously incorporated into everyday products such as Internet search, email spam filters, product recommendations, image classification, and speech recognition. New approaches for highly integrated manufacturing and automation such as the Industry 4.0 and the Internet of things are also converging with ML methodologies. Many approaches incorporate complex artificial neural network architectures and are collectively referred to as deep learning (DL) applications. These methods have been shown capable of representing and learning predictable relationships in many diverse forms of data and hold promise for transforming the future of omics research and applications in precision medicine. Omics and electronic health record data pose considerable challenges for DL. This is due to many factors such as low signal to noise, analytical variance, and complex data integration requirements. However, DL models have already been shown capable of both improving the ease of data encoding and predictive model performance over alternative approaches. It may not be surprising that concepts encountered in DL share similarities with those observed in biological message relay systems such as gene, protein, and metabolite networks. This expert review examines the challenges and opportunities for DL at a systems and biological scale for a precision medicine readership.
Introduction
I
Continued growth of omics research has required improvements in instrumentation (e.g., longer reads in gene sequencing and increased resolution in mass spectrometry), bioinformatics algorithms, data science methods, and access to computational resources. Increasingly, the scale of omics data generation has been challenging researchers' abilities to integrate and model often noisy, complex, and high-dimensional data. Deep learning (DL) is a subdomain of machine learning (ML), which has emerged as a powerful approach, which can both encode and model many forms of complex data (e.g., numeric, text, audio, and image) both in supervised (e.g., biomarker identification) and unsupervised (e.g., anomaly detection) settings. In particular, precision medicine presents a unique opportunity for omics data to be integrated with many other types, including electronic health records (EHRs), medical imaging, Internet of things (IoT) sensors, and pharmacological entity structures, which if realized, is poised to greatly improve healthcare quality.
This review focuses on opportunities and challenges for DL applied to omics data analysis and integration with specific examples in precision medicine settings. To simplify the analogy between biological and neural network-based message passing systems, this review often limits discussion of omics to information flow between genes, proteins, and metabolites. However, DL modeling has already been successfully applied to a wide variety of omics tasks (Ching et al., 2018). The combination of omics and DL are likely to transform many domains beyond precision medicine; however, further innovation is required to fully enable DL to deal with the unique properties of omics data.
Omics Data Possess Considerable Challenges for DL
DL applications have had greatest successes in areas of biology where data can be represented as images (Poostchi et al., 2018; Poplin et al., 2018) and text (Gómez-Bombarelli et al., 2018), on data with complex nonlinear relationships (Kearnes et al., 2016), with data sets that appropriately sample the experimental complexity space and those large enough to support cross-validation, train/validate/test strategies, and hyperparameter tuning. Omics data do not readily conform to the deterministic assumptions underlying many mainstream DL implementations and require domain-specific approaches for dealing with biologically unrelated modes of variance. For example, let us consider a metabolomic genome-wide association study (mGWAS) focused on relationships between metabolite concentrations and gene polymorphisms to identify differences between sick and healthy individuals.
It may be useful to consider an analogy between common challenges in mGWAS and application of DL for image classification. In this example, missing values might be absent pixels; batch effects, a systematic distortion in one or all of the color channels; and low signal to noise, an increase in transparency, identification of gene and metabolite biomarkers of disease, and a classification of images of related objects, which vary in similarity based on the treatment effect size. Individual measurements, such as single-nucleotide polymorphisms (SNPs) or metabolites, may represent portions of each image or specific objects consistently reproduced among many images. Reconstructing the biochemical domain of origin for plasma or urine metabolites might be akin to assembling an image that has been ripped into pieces and mixed together.
Encouragingly, the flexibility of DL architectures enables the capability to generate meaningful inferences, given enough training data even from images treated in the scenarios described above. The question remains, how can this flexibility in model architectures be applied to omics analyses and their integration?
Many omics analyses are impeded by low signal to noise, high analytical variance, and limitations in experimental design. Even highly controlled quantitative omics applications such as signaling lipidomics (Grapov et al., 2012) can struggle to decouple genetic from environmental indicators of pathophysiological states. Unlike the massively overwhelming scales of free data available to mainstream DL implementations, omics researchers pay a high cost for each sample. Data set sample sizes are often limited by experimental design, implementation (laboratory experiments and human trials), throughput in data acquisition (gene expression and chemical analysis), data processing (quality controls and normalizations), and biological interpretation methods.
These and other considerations such as rare cases often lead to biological experiments with relatively small sample sizes, high dimensionality, and different degrees of noise and error. Mitigation of unwanted variation requires consideration during experimental design such as addition of replicated measurements (Jacob et al., 2016) for precision, reference samples for accuracy, and quality controls for batch and sample normalizations. Without these measures, other downstream analytical approaches may lack reproducibility (Lin et al., 2014). Dealing with sample handling and data acquisition errors may require many forms of normalization.
Common approaches include combinations of sample and variable-wise methods, quality control-based signal correction such as splines and loess (Uusitalo et al., 2016) and domain-specific approaches such as reference-based signal drift correction (De Livera et al., 2015; Sysi-Aho et al., 2007) for drifts in chromatography or normalization to invariant housekeeping (De Kok et al., 2005), and negative control genes (Gagnon-Bartsch and Speed, 2012) in genomics. Biologically linked sources of variation such as differences in concentrations of biofluids, genetic background, or demographic diversity (De Livera et al., 2012) remain other modes of variance requiring consideration.
Sample size limitations and low signal to noise in omics have led to selective application of ML methods, which can give useful inference even with relatively small data and are easy to visualize and interpret. Dimensional reduction such as principal component analysis and variations of partial least squares (O-PLS/-DA) are widely used in omics to explore and explain variation in many measurements through fewer uncorrelated principal components or latent variables. These techniques support complexity reduction and minimization of information loss, and enable visual exploration of linear relationships between sample scores, variable loadings, and similarities within (Yamamoto et al., 2009).
More recently, nonlinear methods such as t-distributed stochastic neighbor embedding for dimensional reduction (Li et al., 2017b) and random forest predictive models (Breiman, 2001), robust to overfitting, have emerged as valuable approaches, which are useful even for relatively small data sets.
DL Promises Unique Opportunities for Multiomics Data Integration
Multiomics data integration is often required to robustly model complex biochemical systems. For example, elevation in a gene's messenger RNA expression might not lead to increased protein expression or elevated protein expression may not translate into increased activity. Furthermore, a single measurement of any one omics domain might not reveal dynamic or time-dependent mechanisms (metabolic flux and circadian fluctuations) (Olafsdottir et al., 2016). These kinds of challenges require thoughtful combinations of omics measurements to sufficiently characterize biochemical signatures reflective of the phenotype at the very moment the sample is taken.
Omics integration is a powerful approach (Fabres et al., 2017) that can link even small data sets across orthogonal biochemical domains, and thereby magnify biologically relevant signals (Fig. 1). This is commonly done using empirical relationships (correlation), functional contexts like pathways (Wanichthanarak et al., 2015), and with data derived from a single experimental design or through meta-analyses combing results from multiple studies (Lin et al., 2014; Song et al., 2012). However, only DL offers both unsupervised and supervised omics data integration possibilities. For example, researchers studying growth dynamics in Escherichia coli used an ML-based data integration strategy, including a recurrent neural network (RNN), to develop a multiomics database and predictive model (Kim et al., 2016).
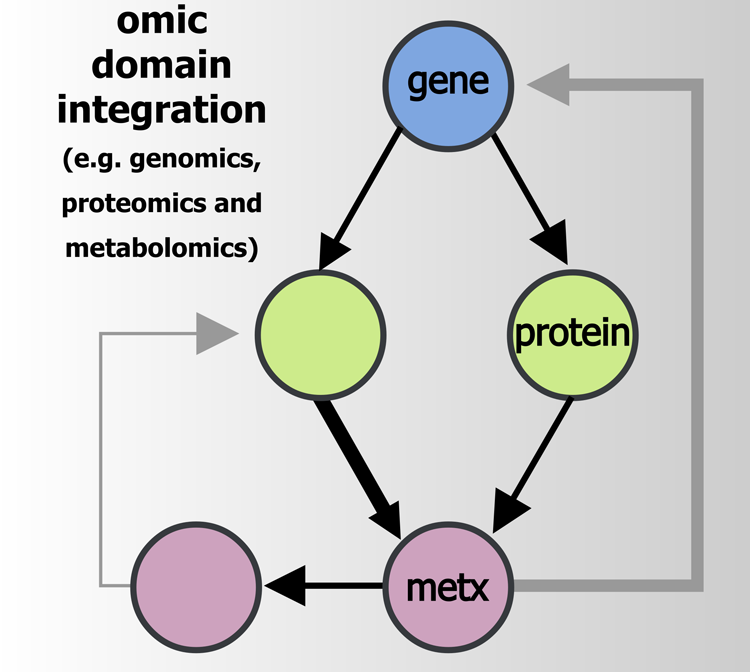
Multiomics data integration utilizes empirical, functional, and other techniques to combine information from multiple omics domains. This systems approach enables robust characterization of biochemical signatures reflective of organismal phenotypes.
This strategy was used to effectively integrate genomic, proteomic, metabolomic, and phenotypic data, and enabled prediction of genome-wide effects on protein and metabolite concentrations, and E. coli growth dynamics. This impressive application of ML was largely made possible by the authors' care in designing their data quality control pipeline and effort to maximize sampling diversity of the biological space. However, large data size requirements often hamper DL implementations in biological settings. The amount of data required depends on many factors such as the ability to benefit from pretrained models through transfer learning (Ching et al., 2018) (Fig. 2), ability to incorporate domain context (leverage biochemical databases, biomarker to disease ontologies, and scientific articles), the ratio of samples to variables, and the experimental effect size.
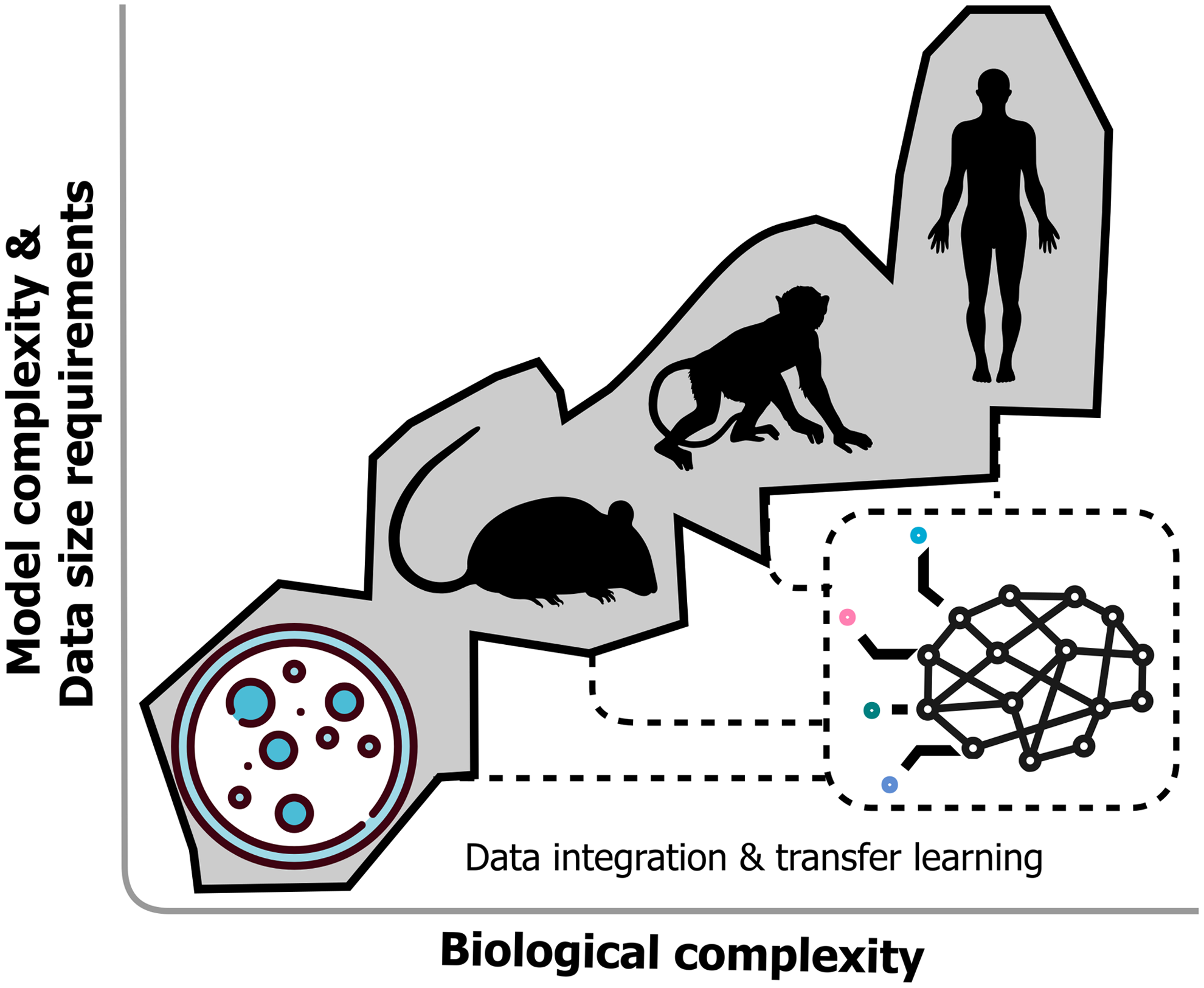
DL architectures may provide unique opportunities to encode locally optimal predictors in a variety of organisms (cellular, mouse, primate, and human) and then integrate their representations of omics layers. Through transfer learning, researchers may leverage larger expert-derived models to improve DL performance for their smaller data sets.
Adoption of academic and industry-wide laboratory data standardization, ontology and sharing methods are key to enabling large-scale experimental data integration opportunities. Initiatives such as www.allotrope.org, which seek to implement analytical platform agnostic formats for experimental data sharing and results encoding (equipment, processes, and materials) through controlled vocabulary and extensible ontologies are needed to increase routine data integration between different laboratories.
Similarly, EHR standards such as Fast Healthcare Interoperability Resources (FHIR), which implement application programming interface ideals such as HTTP-based RESTful protocols, will be needed to effectively generate EHR data sets, which can be integrated with other omics data. Transfer learning or domain adaptation may offer another unique opportunity to utilize extracted features from DL models trained on large data sets or other domains and apply these in custom modeling tasks on far smaller data (Fig. 2).
For example, large biological databases such as the UK Biobank (Sudlow et al., 2015), which contains publicly available data for 500,000 participant's genomic and phenotypic data, including multimodal imaging, genome-wide genotyping, and clinical outcomes, might one day seed other DL models making this approach accessible to far smaller data. Transfer learning has already been successfully used in image classification within a variety of unique omics-related tasks such as cell sorting for malaria detection (Poostchi et al., 2018) and SNP identification in genomics (Poplin et al., 2018).
Common DL Architectures Useful for Omics Data
A prominent difference between DL and traditional ML such as artificial neural networks (ANNs) includes DL models' capacity to learn and fit raw data through representation at multiple levels of abstraction or hidden layers. This essentially produces more refinement of the representation of observed patterns in upper layers, in contrast to the ANNs, which only contain three layers: input, hidden, and output (Ching et al., 2018).
Novel DL architectures are continuously developed (Angermueller et al., 2016; Ching et al., 2018; Lecun et al., 2015; Min et al., 2017a; Miotto et al., 2017; Tran et al., 2018) and include deep neural networks, convolutional neural networks (CNNs), RNNs, and autoencoders, to name a few (see http://www.asimovinstitute.org/neural-network-zoo for an excellent graphical overview of common DL architectures). One of the most promising features of DL approaches is this class of models' consistent interface to data encoding and integration. However, these methods are often criticized as “black-box” and do not in themselves improve “bad” data quality. Simpler approaches have often been shown to be on par or superior to DL for many tasks (Cheng et al., 2018). Table 1 summarizes common DL architectures and their omics applications.
AE, autoencoder; CNN, convolutional neural network; DNN, deep neural network; LSTM, long short-term memory; MDA, mean decrease accuracy; NMR, nuclear magnetic resonance; RNN, recurrent neural network.
Improvements in Computational Frameworks and Model Interpretation Methods
Continued advancements in ease of use of DL model implementation, calculation, and interpretation are helping democratize biological researchers' access to these powerful tools. Readily available and highly scalable cloud computing resources (Amazon web services, Google compute engine, and Microsoft Azure) coupled with big data wrangling software (SPARK and pachyderm) and specialized model calculation-supporting hardware (graphical and tensor processing units) have made it easier for omics researchers to train large-scale DL models.
Challenges for reproduction of DL architectures from published research are being addressed by tools such as DLPaper2Code (Sethi et al., 2018), which aim to automate extraction of computational graphs from articles and convert this into execution ready source code in popular DL frame frameworks such as Keras. Despite this, further improvements in DL model interpretation are required for broader adoption. Efforts to develop local interpretable model-agnostic explanations (Ribeiro et al., 2016) for model predictions have improved model interpretation, but further advancements are needed to enable incorporation of biological domain knowledge.
Predictive modeling in omics research settings is often aimed at multivariate feature ranking or selection, which is difficult to determine when using complex DL architectures. DL models pass input signal through a series of computational layers, creating a directional dependence of the signal encoding between previous and downstream layers, which makes it difficult to interpret the original inputs' contributions to the overall model fit and echoes the act of identifying biomarkers within a linked gene, protein, and metabolite interaction network (Fig. 3).
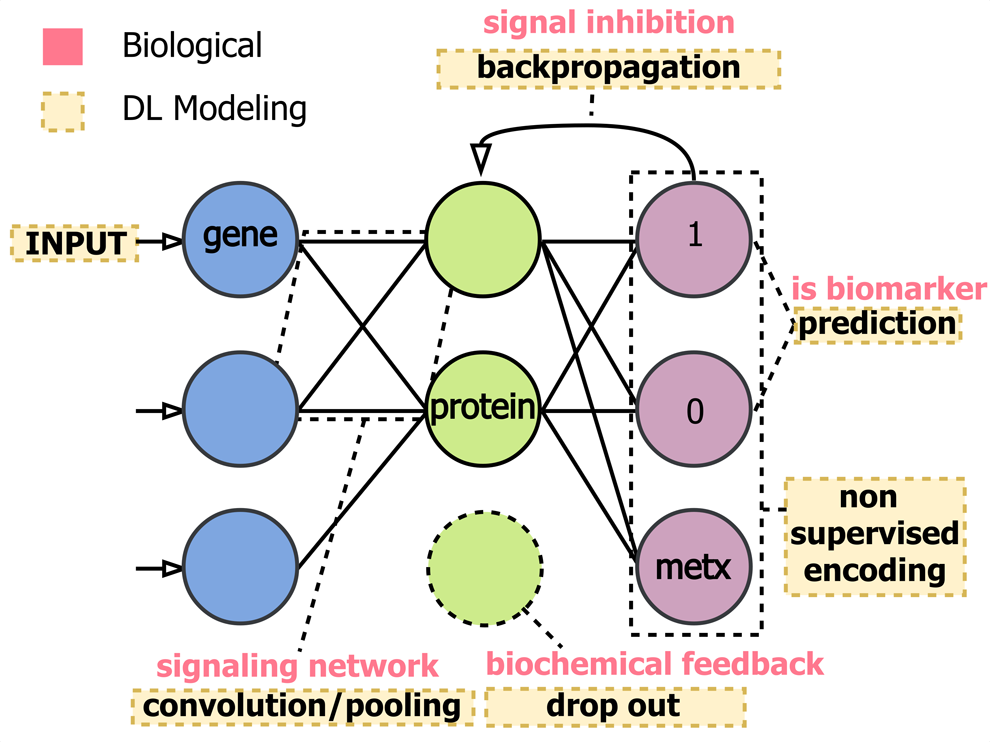
DL model architectures and training techniques share many similarities with biological message passing systems. DL models contain a minimum of three layers: input, hidden, and output. This could mimic representation of relationships between gene transcription, protein expression, and metabolite concentrations, but can also extend other omics layers. Interesting parallels between computational and biological optimizations such as backward propagation in DL and signal inhibition in omics have also emerged.
For example, metabolic feedback mechanisms and adaptation to cellular stimuli through upregulation or downregulation are strikingly similar to DL training techniques such as backpropagation and dropout. Mapping DL model variable weights into a context understandable by domain experts remains difficult. More visual interpretation approaches such as network mapping (Grapov et al., 2015), wherein statistical, functional, and ML-based outputs are mapped onto network manifolds (similarity, biochemical and empirical), need to be adapted for the layered DL feature space. Interpretation of large networks of interdependent entities is often limited by resolution in network layout (hairball networks). Classically, this is dealt with methods for calculation of conditionally independent (Zhao et al., 2012) or causal networks, which poorly scale to larger data.
Conversely, graph convolutional networks, DL methods which operate on graph structures, leverage network complexity and have been shown to project complex networks into lower dimensional and more interpretable similarity-based representations (Kearnes et al., 2016). For example, semisupervised classification using a graph convolutional network on molecular structures represented as networks or text strings (Gómez-Bombarelli et al., 2018) has been used to effectively model molecular structure to activity relationships, and may one day enable automated methods for pharmacological activity optimization.
DL Model-Based Data Integration is Poised to Revolutionize Omics Applications in Precision Medicine
Precision medicine is a burgeoning field where omics analyses are enabling systems-biology based methods for preventive medicine, health promotion, and treatment monitoring (Chen and Snyder, 2013). Approaches in cancer diagnostics and treatment efficacy exemplify an area of research requiring complex data integration (Fig. 4, EHR, imaging, clinical, and omics data). Common omics technology applications in precision medicine include disease biomarker panel assessment to quantify risk, diagnose, measure progression, and optimize treatment strategies.
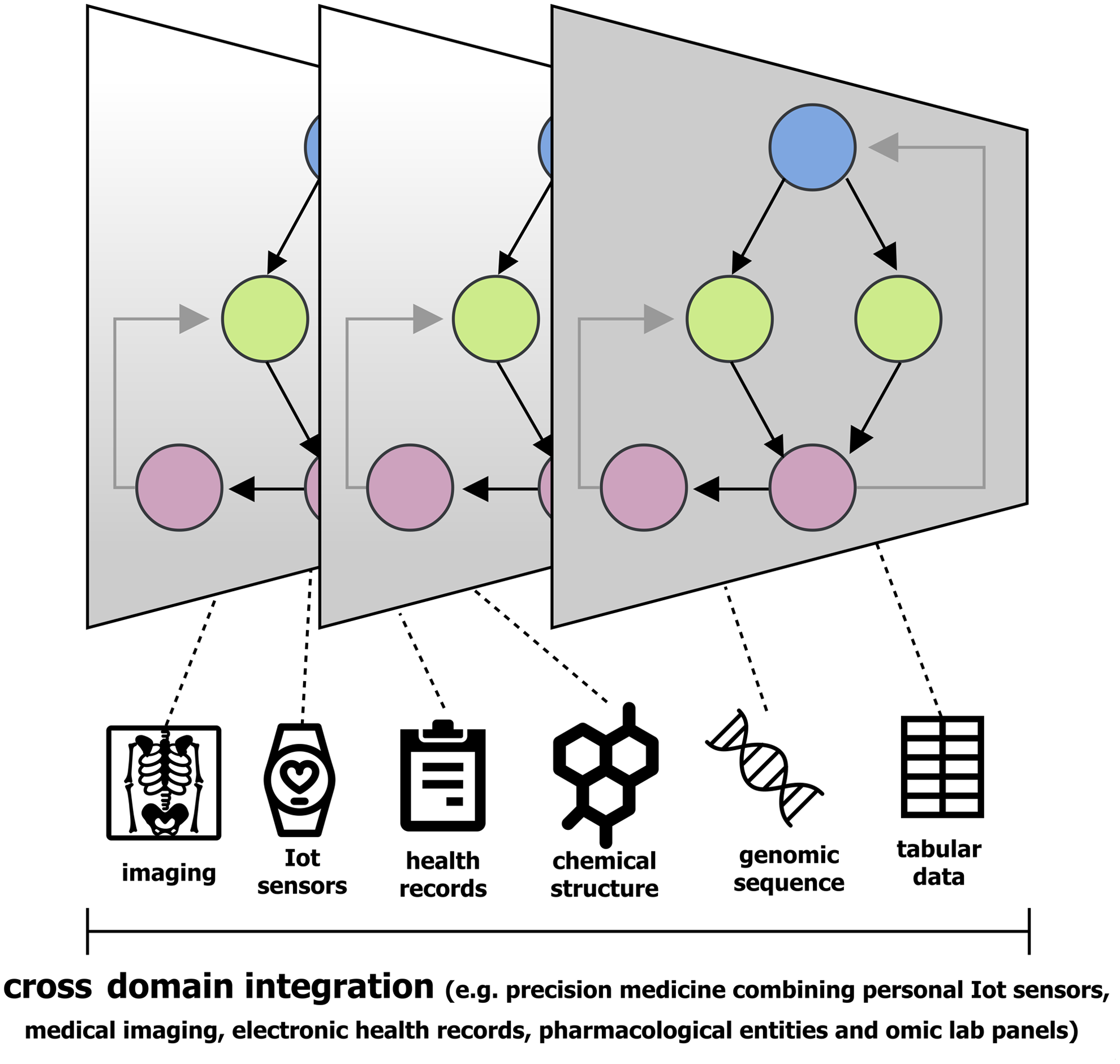
Personalized medicine is a quickly growing area of research that requires complex data encoding and integration tasks, which are well suited for DL.
For example, metabolomics has been used to identify serum diacetylspermine (DAS), a novel prediagnostic marker for non-small cell lung cancer (NSCLC) (Wikoff et al., 2015). Furthermore, the combination of DAS and the pro-surfactant protein B, an independently identified protein marker for NSCLC (Taguchi et al., 2011), improved classification performance relative to either marker alone (Wikoff et al., 2015). Genomic analyses of abnormalities in lung adenocarcinomas (KRAS and EGFR mutations, EML4-ALK fusions) have given rise to specialized treatments such as Erlotinib or Gefitinib for EGFR-mutant NSCLCs (Maemondo et al., 2010; Rosell et al., 2012).
However, despite these successes, there remains considerable heterogeneity in patient responses to these specialized treatments. Mechanisms leading to drug resistance and patient-specific characteristics such as sex and smoking status can have a profound impact on drug efficacy (Wang et al., 2012; Westover et al., 2018).
These complexities highlight the need for continued development of omics data integration methods to facilitate optimal treatment strategies. Predictive modeling in medical applications often involves extraction of curated predictor variables from normalized EHR. This is a labor-intensive process, which may discard useful information in each patient's record. More recently, researchers have developed near automated methods to harmonize raw EHR data to FHIR formats (Rajkomar et al., 2018). This work showed that EHR data sets from different hospitals (>100,000 patients) could be integrated with low manual intervention and used to generate DL-based predictions of patient mortality before and after hospitalization events, which were significantly better than the Early Warning Score.
Advantages of DL-based approaches also include the capacity to consider the entire medical record, including physicians' free-text notes. Other applications of DL, for example, CNN, have been widely applied in medical image analysis such as image segmentation (Lee et al., 2017) and RNN for modeling complicated temporal (Lee et al., 2017) and time series-based effects (Prasad and Prasad, 2014). Furthermore, information-rich environment data derived from personal sensors and IoT devices (smart watches, thermostats, fridges, and phones) offer yet another opportunity for DL-based integration within precision medicine settings.
Taken together, DL approaches promise the computational flexibility to effectively model and integrate almost any type of omics and other data given enough context and scale. Currently, identification of causality in complex phenotypes requires custom analyses and domain expert interpretation; however, one might envision a future of medical data accessibility, quality, and scale, which could enable near automated DL-based detection of many clinically relevant events. Needless to say, new technologies and methodologies such as DL also demand technology assessment grounded in responsible innovation, a topic reviewed in detail elsewhere for the readers (Fisher, 2017).
Footnotes
Acknowledgments
J.F.F. is supported by the University of Texas MD Anderson Cancer Center Duncan Family Institute for Cancer Prevention and Risk Assessment. K.W. and S.K. are supported by Chalermphrakiat Grant, Faculty of Medicine Siriraj Hospital, Mahidol University.