
Abstract
Abstract
Phoma stem canker (blackleg) is a disease of world-wide importance on oilseed rape (Brassica napus) and can cause serious losses for crops globally. The disease is caused by dothideomycetous fungus, Leptosphaeria maculans, which is highly virulent/aggressive. Cyclophilins (CYPs) and FK506-binding proteins (FKBPs) are ubiquitous proteins belonging to the peptidyl-prolyl cis/trans isomerase (PPIase) family. They are collectively referred to as immunophilins (IMMs). In the present study, IMM genes, CYP and FKBP in haploid strain v23.1.3 of L. maculans genome, were identified and classified. Twelve CYPs and five FKBPs were determined in total. Domain architecture analysis revealed the presence of a conserved cyclophilin-like domain (CLD) in the case of CYPs and FKBP_C in the case of FKBPs. Interestingly, IMMs in L. maculans also subgrouped into single domain (SD) and multidomain (MD) proteins. They were primarily found to be localized in cytoplasm, nuclei, and mitochondria. Homologous and orthologous gene pairs were also determined by comparison with the model organism Saccharomyces cerevisiae. Remarkably, IMMs of L. maculans contain shorter introns in comparison to exons. Moreover, CYPs, in contrast with FKBPs, contain few exons. However, two CYPs were determined as being intronless. The expression profile of IMMs in both mycelium and infected primary leaves of B. napus demonstrated their potential role during infection. Secondary structure analysis revealed the presence of atypical eight β strands and two α helices fold architecture. Gene ontology analysis of IMMs predicted their significant role in protein folding and PPIase activity. Taken together, our findings for the first time present new prospects of this highly conserved gene family in phytopathogenic fungus.
Introduction
P
Phoma stem canker (blackleg) is a disease of world-wide importance on oilseed rape, (Brassica napus, canola, colza, rapeseed, Raps) and can cause serious losses for crops globally (Howlett, 2004; West et al., 2001). The disease is caused by dothideomycetous fungi, Leptosphaeria maculans or L. biglobosa (Mendes-Periera et al., 2003). Leptosphaeria maculans was previously classified as highly virulent/aggressive or as A group type or Tox+ group, in contrast to L. biglobosa, which was classified as weakly virulent/nonaggressive or as B group type orTox0 group (Rouxel et al., 2004; Shoemaker and Brun, 2001; Williams and Fitt, 1999). Leptosphaeria maculans and L. biglobosa were distinguished on the basis of morphological differences of pseudothecia (Shoemaker and Brun, 2001). Both fungi cause phoma leaf spots; however, it was suggested that L. maculans causes more serious phoma stem cankers, while L. biglobosa affects upper stem and causes superficial stem lesions (Fitt et al., 2006; Rouxel et al., 2004; Rouxel and Balesdent, 2005). The diversity between L. maculans and L. biglobosa species complex was also investigated in order to decipher the phylogentic relationship (Mendes-Pereira et al., 2003).
The family of immunosuppressant receptors called Immunophilins (IMMs) includes cyclophilins (CYPs) and FK506-binding proteins (FKBPs) (Fischer et al., 1984; Harding et al., 1989). The family is characterized as having two important properties: peptidyl-prolyl cis/trans isomerase (PPIase) and drug binding. Cyclophilins bind to the immunosuppressive drug cyclosporin A (CsA) (Handschumacher et al., 1984), while FK506 binding proteins (FKBPs) bind to FK-506/rapamycin-binding proteins. Despite their lack of sequence and structural similarity, these two families share a common PPIase, catalyzing the cis/trans isomerization of proline imidic peptide bonds (Fisher et al., 1989).
All CYPs share a conserved domain, the cyclophilin-like domain (CLD), while FKBPs have a conserved FKBP_C domain. Depending upon the functional modules present, IMMs have been broadly classified into two groups: single domain (SD) and multidomain (MD) (Ahn et al., 2010). SD is characterized by the presence of a single catalytic CLD or FKBP domain exhibiting PPIase activity. In contrast, MD possesses other functional domains in conjunction with single or multiple catalytic CLD or FKBP domains. The additional functional domains include tetra-peptide repeat (TPR), WD40, coiled-coil domain (CCD), and internal repeats domain (RPT), Zinc Finger, and RRM, in addition to others (Ahn et al., 2010; Burkhard et al., 2001; Scheufler et al., 2000). The additional functional domains are involved in various processes, such as protein–protein interactions (Ke et al., 1993; Taylor et al., 2001) and RNA binding (Gasser et al., 1990; Luan et al., 1994).
Biochemical and sequence analysis following genome sequencing projects have led to the identification of a large number of IMMs in various organisms. Saccharomyces cerevisiase contains 12 genes (8 CYPs and 4 FKBPs) (Arevalo-Rodriguez et al., 2004), Caenorhabditis elegans: 24 genes (16 CYPs and 8 FKBPs), and Aspergillus nidulans: 16 genes (11 CYPs and 5 FKBPs) (Pemberton, 2006). Human have a total of 24 CYPs and 18 FKBPs (Galat, 2003). Notably, photosynthetic organisms possess a large number of IMMs, with 52 genes (29 CYPs and 23 FKBPs) in Arabidopsis (Romano et al., 2004), 56 in rice (27 CYPs and 29 FKBPs) (Ahn et al., 2010), and 49 in Chlamydomonas (23 CYPs and 26 FKBPs) (Vallon, 2005). The higher number of IMMs in green photosynthetic organisms was predicted because they are more targeted towards the photosynthetic apparatus (He et al., 2004; Peltier et al., 2002). The numbers of IMMs predicted in phytopathogenic fungi are more or less similar to non-photosynthetic organisms except human (Singh, unpublished data).
IMMs are highly conserved ubiquitous proteins (Galat, 1999) localized in all cellular compartments and encode diverse functions (Wang and Heitman, 2005). They are involved in many processes, such as spliceosome assembly (Horowitz et al., 2002), protein trafficking and maturation (Ferreira et al., 1996), receptor complex stabilization (Leverson and Ness, 1998), apoptosis (Lin and Lechleiter, 2002), receptor signaling (Brazin et al., 2002), RNA processing (Krzywicka et al., 2001), development of asexual reproductive structures and cellular turgor generation in appressoria in fungi (Viaud et al., 2002), RISC assembly (Lki et al., 2012), and others. However, the diverse roles and still emerging novel functions suggest that the IMMs encompass far more functions than the already defined function of protein folding in different organisms.
Several reports reveal the involvement of IMMs in response to different types of biotic and abiotic stresses (Ahn et al., 2010; Aviezer-Hagai et al., 2007; Marivet et al., 1992; Meiri et al., 2010; Pogorelko et al., 2014). The expression of IMM genes has also been previously shown to be induced by both biotic and abiotic stresses, including fungal infection (Godoy et al., 2000), viral infection, salicylic acid, ethephon (an ethylene releaser), and HgCl2 (Marivet et al., 1995), cold shock, salt stress, drought, and heat (Ahn et al, 2010; Marivet et al., 1994; Scholze et al., 1999; Sharma and Singh, 2003). Very few studies of IMMs in fungi have been reported. In the phytopathogenic fungus Mangnaporthe grisea, the CYP gene acts as a determinant of virulence during rice blast disease (Viaud et al., 2002). Cyclophilin A of Cryptococcus neoformans exhibited distinct functions that are important for growth and virulence (Wang et al., 2001). Thus, IMMs act as virulence factors for both plant and animal diseases caused by fungi.
Fungi, especially S. cerevisiae, have been used extensively as a reduced and easily manageable model system in biological research (Grutzamann et al., 2013). Combination of bioinformatics and omics-based tools has enabled the discovery of new members of several gene families in plants such as ABA repressor 1 (ABR1) (Mishra et al., 2013), homeobox gene family (Jain et al., 2008), glycerol-3-phosphate acyltransferase (GPAT) (Misra and Panda, 2013), protein phosphatase 2C family (Xue et al., 2008), nucleocytoplasmic transport receptors family (Huang et al., 2010), and others.
In spite of the fact that IMMs are highly conserved in all organisms, the genes are not well studied in phytopathogenic fungi due to unsequenced genomes. However, the availability of the high quality genome sequence of an important fungus, L. maculans, and the lack of information about this multigene family, motivated us to conduct this study. Therefore, in the present study, we performed genome-wide analysis of IMM genes using bioinformatics tools, and thus identified putative members of each family, respectively in the haploid strain v23.1.3 of L. maculans genome, and performed a comparative analysis with S. cerevisiae. Furthermore, comprehensive analysis of the putative identified genes, including domain architecture, chromosomal distribution, phylogenetic analysis, sequence alignment and secondary structure features, exon-intron structure, expression pattern in mycelium, as well as in primary leaf of B. napus during disease infection and Gene Ontology (GO) terms, has also been carried out.
Materials and Methods
Searching protein database, nomenclature, and domain analysis of IMMs
In order to identify the members of the IMMs in L. maculans, the Hidden Markov Model (HMM) profiles unique to cyclophilin (accession no. PF00160) and FKBP (accession no. PF00254) from Pfam database (version 26.0) (http://pfam.sanger.ac.uk/) were retrieved and then searched against the genome browser database of L. maculans (http://urgi.versailles.inra.fr/) using HMMER 3.0 software (http://hmmer.janelia.org/). All the significant hits with positive scores were selected for further classification. The genes identified were thus assigned names, such as “Lm” (L. maculans), followed by “CYP” (cyclophilin) and “FKBP” (FK506-binding proteins), according to the catalytic domain present, and then followed by numbers in increasing order based on highest HMM scores. The identified IMM genes were then analyzed for the presence of the respective catalytic domain (i.e., cyclophilin-like-domain (CLD) in the case of CYPs and FKBP_C in FKBPs using Pfam databases). The scaled positions of domains were drawn using the gplots package of the open source package R using R scripts.
Subcellular localization
Subcellular localization of IMM genes were predicted using PSORT (http://wolfpsort.org/). Signal peptide cleavage sites and mitochondrial target peptide (MTP) were predicted using SignalP 1.1 (http://www.cbs.dtu.dk/) and TargetP 1.1 sever (http://www.cbs.dtu.dk/). Nuclear localization signals (NLS) were predicted using NLS mapper http://nls-mapper.iab.keio.ac.jp/) and Nucpred (https://www.sbc.su.se/).
Chromosomal distribution of IMM genes
Genomic distribution of putative IMM genes were determined using the L. maculans genome browser (http://urgi.versailles.inra.fr/) and mapped on their respective chromosomes. The scaled positions of IMM gene family CYPs and FKBPs on respective chromosomes were drawn using the gplots package of the open source package R using R scripts.
Exon–intron structure determination
In order to determine the gene structures for IMM genes, coding sequences corresponding to each gene (CYP/FKBP) were mapped onto genomic sequences including UTRs, exons, and introns. The coding sequences were obtained from the L. maculans genome database, while genomic sequences were identified from contigs and supercontigs (SCs). The contigs and SCs were aligned and further subjected to FGENESH software (http://linux1.softberry.com/) using the L. maculans genome as a query and with default parameters. The genomic sequences and coding sequences from each CYP/FKBP gene were thus mapped using Gene Structure Display Server (GSDS) (http://gsds.cbi.pku.edu.cn/) and cross checked by using Spidey (http://www.ncbi.nlm.nih.gov/spidey/). The final figures were drawn using GSDS.
Phylogenetic analysis and secondary structure features
Multiple sequence alignment of amino acid sequences belonging to CYP and FKBP from L. maculans and S. cerevisiae were performed using ClustalX (version 2.0) (http://www.ebi.ac.uk/) with default parameters and built in Jalview software version 2.7 (http://www.jalview.org/). Secondary structure features were predicted using Jpred3 server (http://www.compbio.dundee.ac.uk/www-jpred/). The phylogenetic trees were generated using ClustalX implementing neighbour-joining (NJ) method. Bootstrap values were calculated at 1, 000 iterations. The final phylogenetic tree was then edited using ITOL (http://itol.embl.de/).
Expression analysis of IMMs
The transcriptomes of L. maculans IMMs were analyzed in mycelium and infected leaves of B. napus at two time points: 7 and 14 disease post infection (dpi). The expression data were obtained from GSE27152 of Gene Expression Omnibus (GEO) (Soyer et al., 2014). The results were log transformed values of the average of three replicates. The hierarchical clustering analysis and the heatmaps were made using the gplots package of R software, as described earlier (Singh et al., 2011).
Conserved motif analysis and sequence logo
To determine the functional motifs within IMM genes, Multiple Em for Motif Elicitation (MEME) version 4.9.0 (http://meme.nbcr.net/meme/) from the Plant Intron Exon Comparison and Evolution database (PIECE) (http://wheat.pw.usda.gov/piece/) was employed with default parameters. Therefore, the conserved motifs in respective sequences were represented as block diagrams. The sequence logos of motifs were also generated using MEME suite.
Gene ontology (GO) term identification
The GO terms, including Biological Process (BP), Molecular Function (MF), and Cellular Process (CC) for IMMs were retrieved from the L. maculans genome database (http://genome.jgi-psf.org/). The datafile was obtained using BLAST with L. maculans predicted proteins (12, 469) against the nonredundant (nr) database to obtain the similarity with known sequences and thus mapped onto the Gene ontology, geneinfo, gene2accession, and PIR databases (Rouxel et al., 2011).
Results
Conservation and characterization of IMM genes in L. maculans
Members of the IMM genes in L. maculans genome were determined using HMM profiles unique to CYPs and FKBPs from Pfam (see Materials and Methods). All sequences returned by such searches were selected for further classification and then examined individually for accessory domains. Following this strategy, a total of 12 distinct CYPs (LmCYP1-LmCYP12) and 5 FKBPs (LmFKBP1-LmFKBP5) genes were identified (Table 1).
Included are nomenclature, molecular weight, isoelectric point, amino acid length (a.a), predicted subcellular localization, nuclear localization signal (NLS), mitochondria targeting peptide (MTP), signal peptide (SP), and transmembrane.
The characterization of 12 CYP and 5 FKBP genes, including nomenclature, accession number, length of amino acids (aa), isoelectric points (pI), molecular weights (Mw), subcellular localization, nuclear localized signal (NLS), signal peptide (SP), mitochondrial targeting peptide (MTP), and transmembrane (TM) profile are listed in Table 1. These reveal that the lengths of CYP proteins varied from 663 aa. (LmCYP6) to 165 aa (LmCYP12; LmCYP2). However, in the case of FKBPs, the longest protein, LmFKBP1, has 549 aa, and the shortest, LmFKBP4, has only 113 aa (Table 1). The isoelectric point ranged from 5.01 (LmCYP12) to 8.98 (LMCYP3) in the case of CYPs, and from 4.58 (LmFKBP1) to 5.81 (LmFKBP4) (Table 1) for FKBPs. The molecular weight of CYP proteins were found to be lowest in LmCYP12 (16.8 kDa) and highest in LmCYP6 (74.1 kDa), in contrast to FKBP proteins, which ranged from 12.2 kDa in LmFKBP4 to 59.7 kDa in the case of LmFKBP3 (Table 1).
Domain architecture of IMM genes
Leptosphaeria maculans IMMs are categorized into Single Domain (SD) and Multi Domain (MD) depending upon the functional modules present (Ahn et al., 2010). In L. maculans, the number of SD IMMs is remarkably higher compared to MD. In the case of CYP, 8 out of 12 genes viz. LmCYP1, LmCYP2, LmCYP3, LmCYP4, LmCYP5, LmCYP9, LmCYP10, and LmCYP12 were found to be SD (Fig. 1). However, 4 of 12 CYPs were found to be MD: LmCYP6, with a N-terminal WD40 domain; LmCYP7, that possesses a 2 C-terminal TPR; LmCYP8 with a N-terminal Rtf2 domain; and LmCYP11, which has a C-terminal RRM domain (Fig. 1). Notably, there was no MD found with multiple catalytic domains in L. maculans. In the case of FKBPs, 4 out of 5 genes have been classified as SD members (i.e., LmFKBP1, LmFKBP2, LmFKBP4, and LmFKBP5) and possess only single FKBP catalytic domain (Fig. 1). LmFKBP3 is the only MD member, possessing vacuolar membrane protein (VMP) in addition to FKBP (Fig. 1).
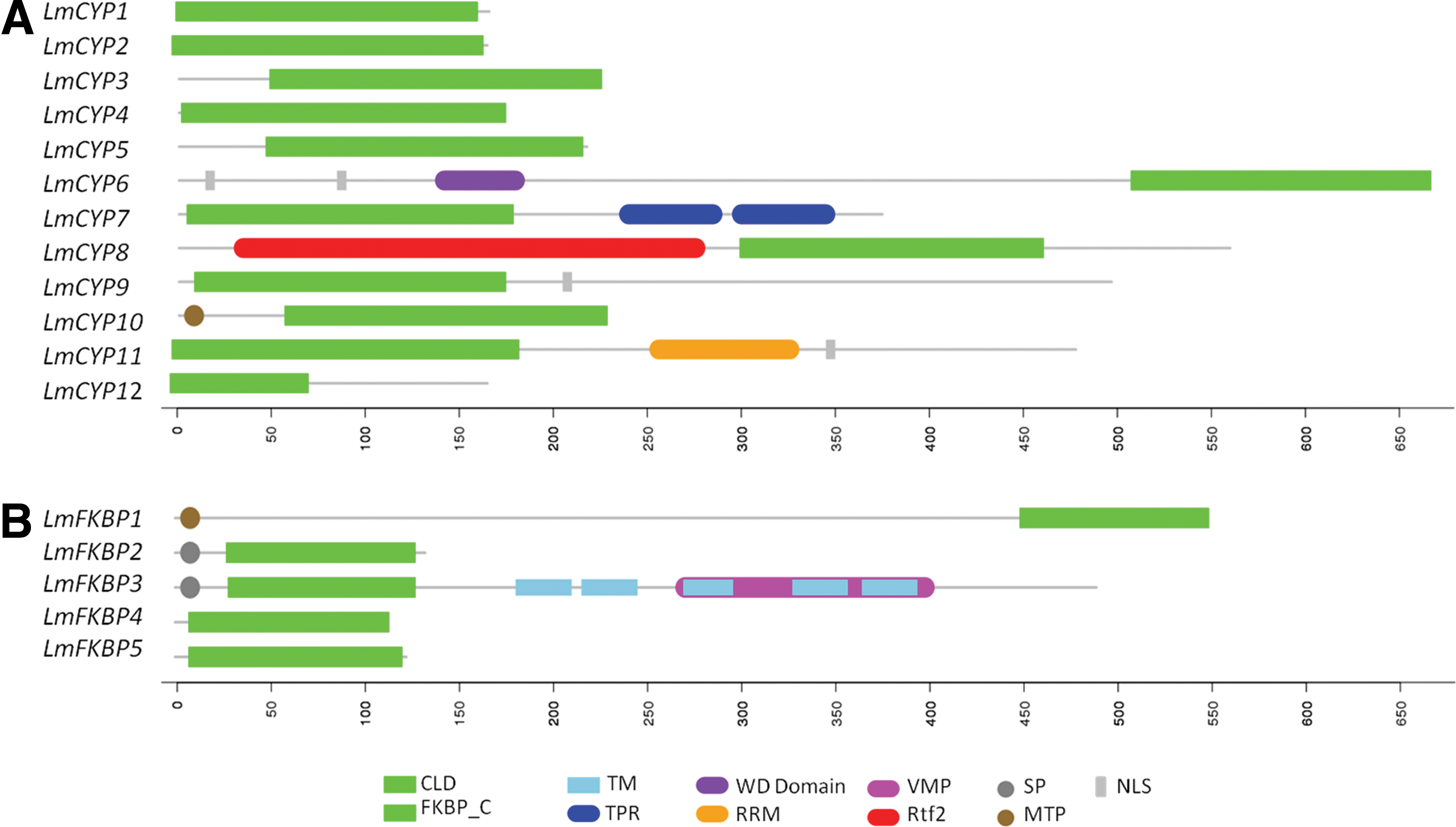
Domain architecture of the L. maculans IMM gene family members:
Subcellular localization of IMM genes in L. maculans
IMMs are present in all major subcellular compartments. In L. maculans, the majority of the proteins were found to be cytosolic, nuclear, and mitochondria localized. Out of 12 CYP, 7 viz. LmCYP1, LmCYP2, LmCYP4, LmCYP5, LmCYP7, LmCYP8, and LmCYP11 were predicted to be localized in cytoplasm (Table 1). 4 CYPs, viz. LmCYP3, LmCYP6, LmCYP9, and LmCYP11 are localized in nucleus (Table 1). LmCYP6 possesses an N-terminal NLS, while both LmCYP9 and LmCYP11 have NLS at the C-terminal (Fig. 1). LmCYP10 is the only CYP localized in mitochondria (Table 1) and also possesses an N-terminal MTP (Fig. 1). There were no CYP found to be present in endoplasmic reticulum (ER) and chloroplast.
In the case of FKBPs, 3 out of 5 are localized in cytosol. Similarly, one is localized in the plasma membrane and one in mitochondria. Cytosolic FKBPs include LmFKBP2, LmFKBP4, and LmFKBP5 (Table 1). LmFKBP2 is a cytosolic CYP possessing an N-terminal signal peptide (SP), compared to LmFKBP4 and LmFKBP5 (Fig. 1). Although LmFKBP3 is located in the plasma membrane, it also contains SP and transmembrane (TM) profiles, which make this protein unique among other FKBP family members. Mitochondrial LmCYP1 has MTP at the N-terminal (Fig. 1).
Genomic distribution of IMMs
The L. maculans genome has been assembled into 17–18 chromosomes. There are 10 chromosomes that are completely annotated while others are incomplete. IMMs were found to be unevenly distributed on different chromosomes. In the case of complete chromosomes, Chromosome 0 (SC0) is the longest among all (4.25 Mb) and possesses LmCYP7 and LmFKBP3. Chromosome 1 (SC1; 3.37 Mb) has LmCYP9, and chromosome 4 (SC4; 1.91 Mb) contains LmCYP1. LmCYP2 was located on chromosome 5 (SC5; 1.86 Mb), and chromosome 7 (SC7; 1.76 Mb) possesses LmCYP10. LmCYP6 was located on chromosome 9 (SC9; 1.77 Mb). Chromosome 21 (SC21; 3.30 Mb), which comprises two other supercontigs (i.e., SC20 and SC23), is 3.30 Mb long and possesses only single FKBP gene, LmFKBP2 (Fig. 2) (Rouxel et al., 2011).
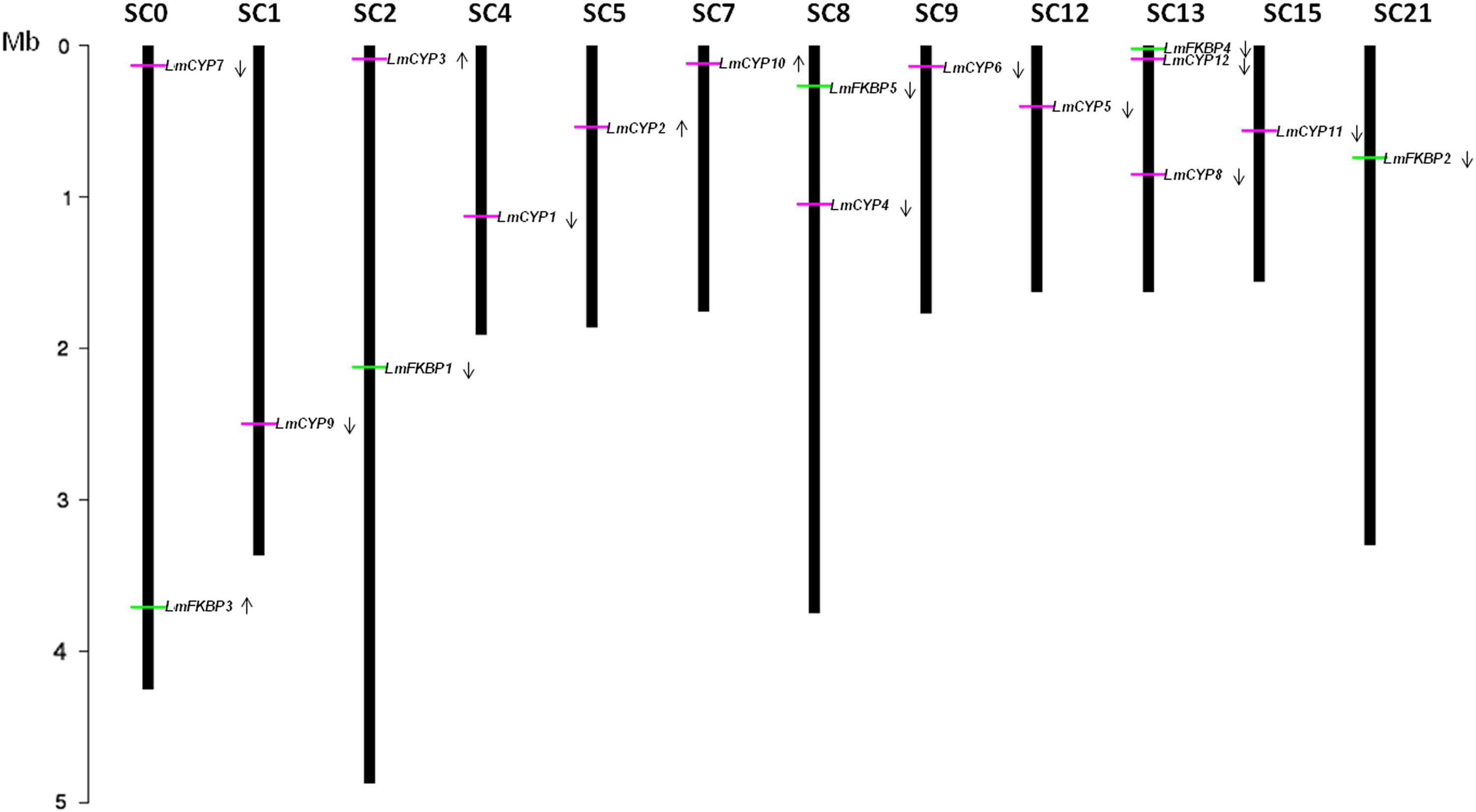
Genomic distribution of L. maculans IMMs on various chromosomes. CYPs and FKBPs represented with horizontal green and pink bars. Arrows show the direction of the ORF specific to the gene encoding CYP and FKBPs proteins (i.e., lower for sense strand upper for antisense strand). Only the chromosome having IMMs are shown; their number is indicated at the top. The chromosome length has been shown in Mb.
Partial chromosomes, including SC2, SC8, SC12, SC13, and SC15, are also included among IMM genes. Chromosome 2 (SC2) and chromosome 19 (SC19) may either form a single chromosome of size 4.87 Mb or two chromosomes sized greater than 3.43 Mb. The exact size is difficult to establish due to lack of one telomere and presence of rDNA repeats. Therefore, we combined both chromosomes (SC2 and SC19 together) and formed one single chromosome named chromosome 2. The chromosome becomes the longest amongst all. Chromosome 2 (SC2) has LmCYP3 and LmFKBP1. The other two chromosomes, chromosome 8 (SC8) and chromosome 10 (SC10), were also assembled together to form a single chromosome of size 3.75 Mb, and named chromosome 8. Notably, one CYP (LmCYP4) and one FKBP (LmFKBP5) were found to be localized on chromosome 8. Chromosomes 12 (SC12), chromosome 15 (SC15), and chromosome 13 (SC13) are dubiously assembled, and thus uncertain. Therefore, we kept the aforementioned chromosomes separate. Chromosome 12 (SC12) has size 1.63 Mb and possesses LmCYP5, and chromosome 15 (SC15) comprises the LmCYP11 gene. Chromosome 13 is still not assembled and had no genetic map evidence (Rouxel et al., 2011). Interestingly, it was found to have the maximum number of IMM genes [i.e., 3, irrespective of its size (1.63 Mb)]. It consists of two CYPs viz. LmCYP8 and LmCYP12, and one FKBP gene, LmFKBP4, as shown in Figure 2 (Rouxel et al., 2011).
Phylogenetic analysis and secondary features of IMMs
Comparative evolutionary relationships between IMM gene family CYPs and FKBPs, from L. maculans and S. cerevisiae, were analyzed by constructing phylogenetic trees supported by bootstrap values with 1000 replications using the clustaX software program. Homologous gene pairs with more than 90% bootstrap values were grouped together (Fig. 3A and 3B). In the case of CYPs, Group1 includes CPR2, CPR5, Group 2 includes LmCYP2 and LmCYP8, Group3 includes CPR4 and CPR8, and Group4 includes LmCYP9, LmCYP11, and LmCYP12 (Fig. 3A). FKBPs formed three major groups: Group5 with FPR3 and FPR4, Group 6 with LmFKBP4 and LmFKBP5, and finally Group7 with LmFKBP2 and LmFKBP3 as shown in Figure 3B. Different orthologous gene pairs between two genomes are shown in Table 2. The secondary structure features were also compared from the deduced aa. sequences of CYPs and FKBPs from both genomes. Remarkably, typical 8 anti-parallel β strands and 2 α helices were observed in the case of CYPs (Fig. 4). In contrast, there were 8 beta sheets and only 1 helix observed in FKBPs (Fig. 5).
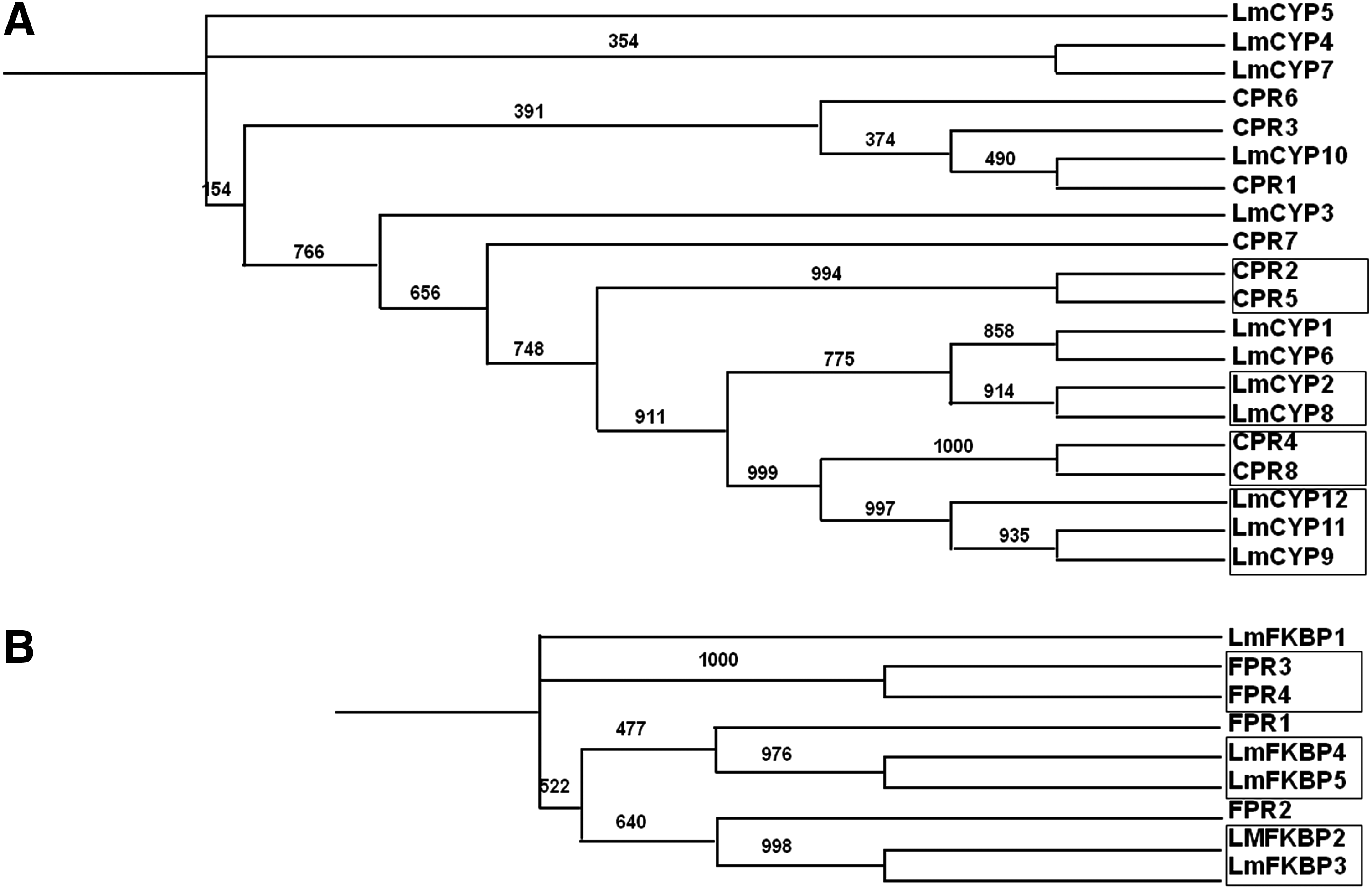
Phylogenetic tree depicting the evolutionary relationship between L. maculans and S. cerevisiae IMMs. CYPs

Multiple sequence alignment of CYPs from L. maculans and S. cerevisiae. The secondary structures are also represented (i.e., α helix (red bar) and β sheets (green bar)).
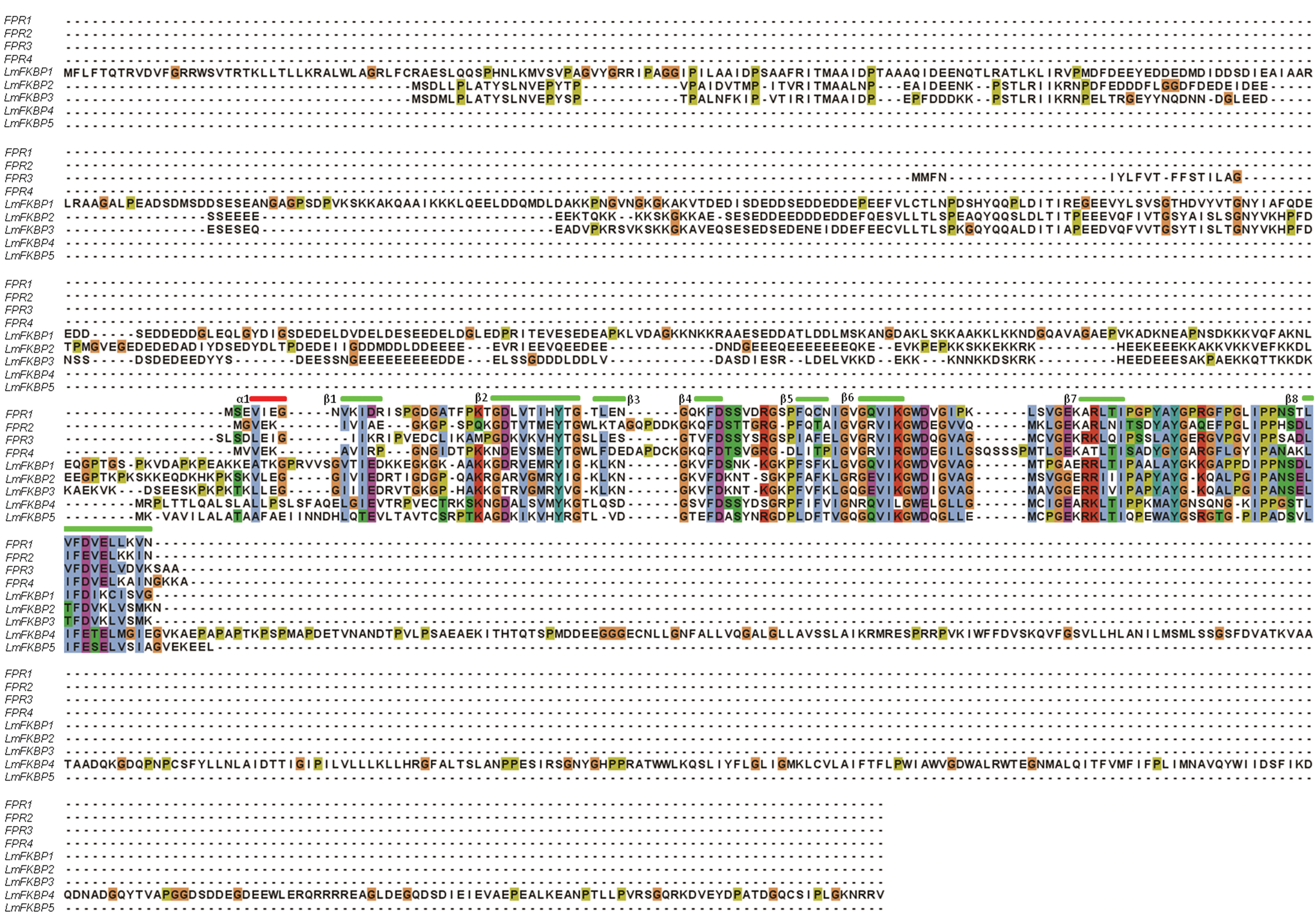
Multiple sequence alignment of FKBPs from L. maculans and S. cerevisiae FKBP genes. The secondary structures are also represented (i.e., α helix (red bar) and β sheets (green bar)).
Included are subcellular localization, amino acid length (a.a), molecular weight (Mw), and isoelectric point (pI).
Exon–intron architecture analysis
To understand the structural components of L. maculans IMMs, their exon–intron organization was analyzed. Interestingly, we found that IMM genes in L. maculans contain short introns compared to exons (Fig. 6), as is the case in other fungi (Grutzmann et al., 2014). Based on their intron length, in the case of CYPs, LmCYP1 has the smallest intron of 48 bp, while the longest one is in LmCYP5, with 188 bp. However, the length of exons in CYPs varies drastically, from 1854 bp of the longest one in LmCYP6 to 33 bp of the smallest one in LmCYP1. In the case of the FKBP family, LmFKBP3 contains the shortest intron (49 bp), and LmFKBP5 has the longest one (303 bp). Similarly, LmFKBP1 has the longest exon with 715 bp, compared to the shortest one in LmFKBP5 with 21 bp. Interestingly, we found that 2 genes are intronless, namely LmCYP8 and LmCYP9.
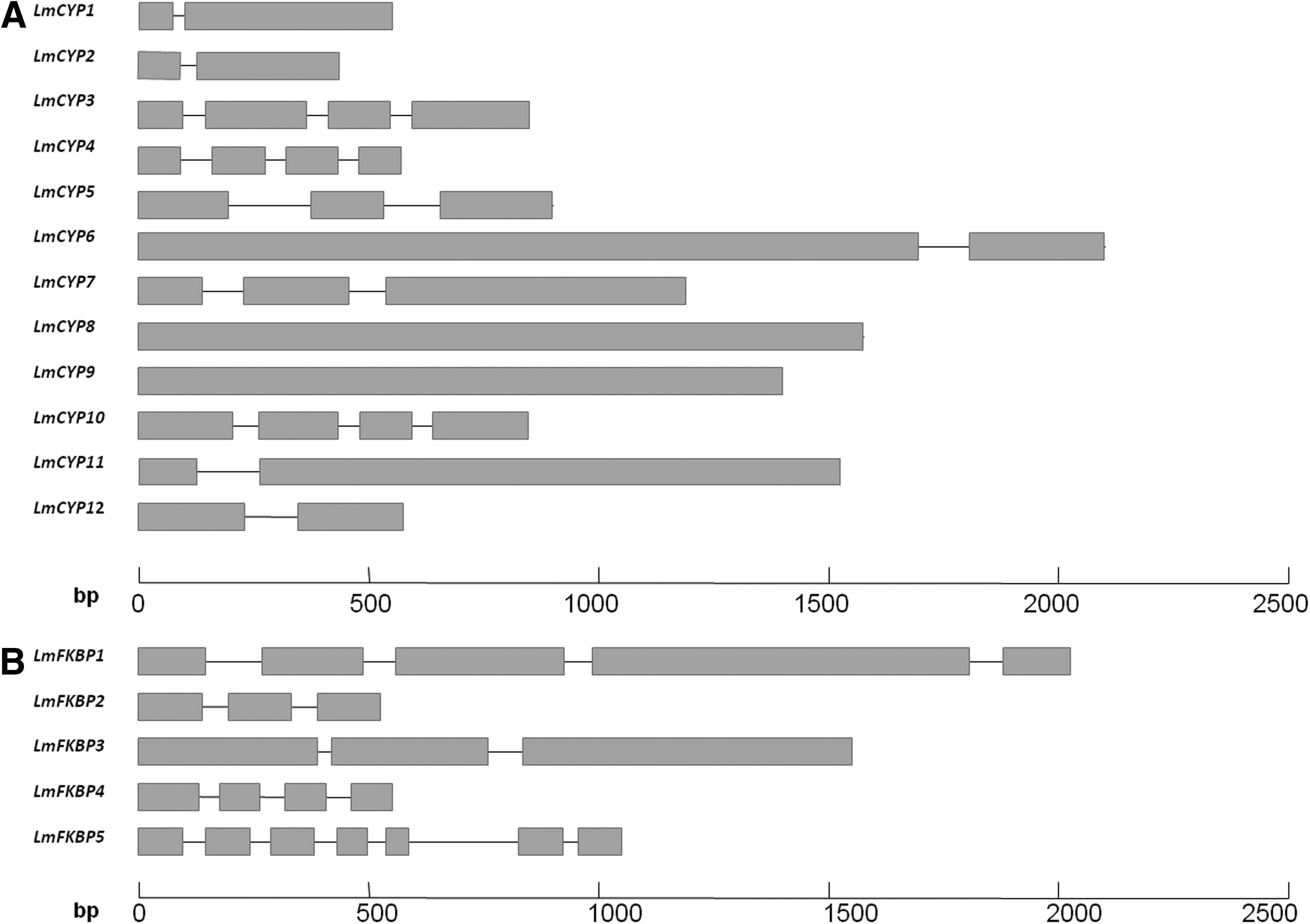
Exon–intron architecture of L. maculans IMMs. CYPs
In terms of the number of exons and introns, LmCYP4, LmCYP3 and LmCYP10 possess 4 exons and 3 introns each (Fig. 6). LmCYP5 has 3 exons and 2 introns. Likewise, LmCYP1, LmCYP2, LmCYP6, LmCYP7, LmCYP11, and LmCYP12 all equally possess 2 exons and 1 intron. Despite IMM gene family CYPs and FKBPs sharing a common peptidyl-prolyl isomerase (PPIase), catalyZing the cis/trans isomerization of proline imidic peptide bonds (Fisher et al., 1989), the exon–intron architecture varies drastically. It was observed that FKBPs in L. maculans contain remarkably higher number of exons and introns compared to CYPs, irrespective of size (Table 2). LmFKBP5 consists of 7 exons and 6 introns, LmFKBP1 possesses 5 exons and 4 introns, both LmFKBP2 and LmFKBP3 have 3 exons and 2 introns, and LmFKBP4 has 4 exons and 3 introns.
Transcriptomic expression analysis of IMMs
We have used publicly available transcriptome data for carrying out expression analysis of IMM genes identified in this study (“Materials and Methods”). Heat maps showing information about mycelium and primary leaf infection specificity of the expression of IMM genes of L. maculans, have been generated using gplots from the R-statistical software package (Fig. 7). Based on the expression patterns of CYPs, they have been subgrouped into three classes, viz. Highly expressed, Moderately expressed, and Low expressed (Fig. 7A). IMM genes, such as LmCYP1, LmCYP2, LmCYP5, and LmCYP10, were constitutively highly expressed in mycelium and infected primary B. napus leaves, thereby indicating their enormous significance during infection. In contrast, IMM genes, such as LmCYP3, LmCYP7, LmCYP8, and LmCYP12, were found to be moderately expressed during in vitro growth (mycelium), as well as during primary B. napus leaf infection. Interestingly, LmCYP3 and LmCYP7 were found to be more expressed during mycelium growth compared to LmCYP12 and LmCYP8, showing their limited effect during plant infection (Fig. 7A). The expression of LmCYP4, LmCYP6, LmCYP9 and LmCYP11 were found to be low in mycelium as well as in infected primary B. napus leaves, suggesting them less significant during infection (Fig. 7A).
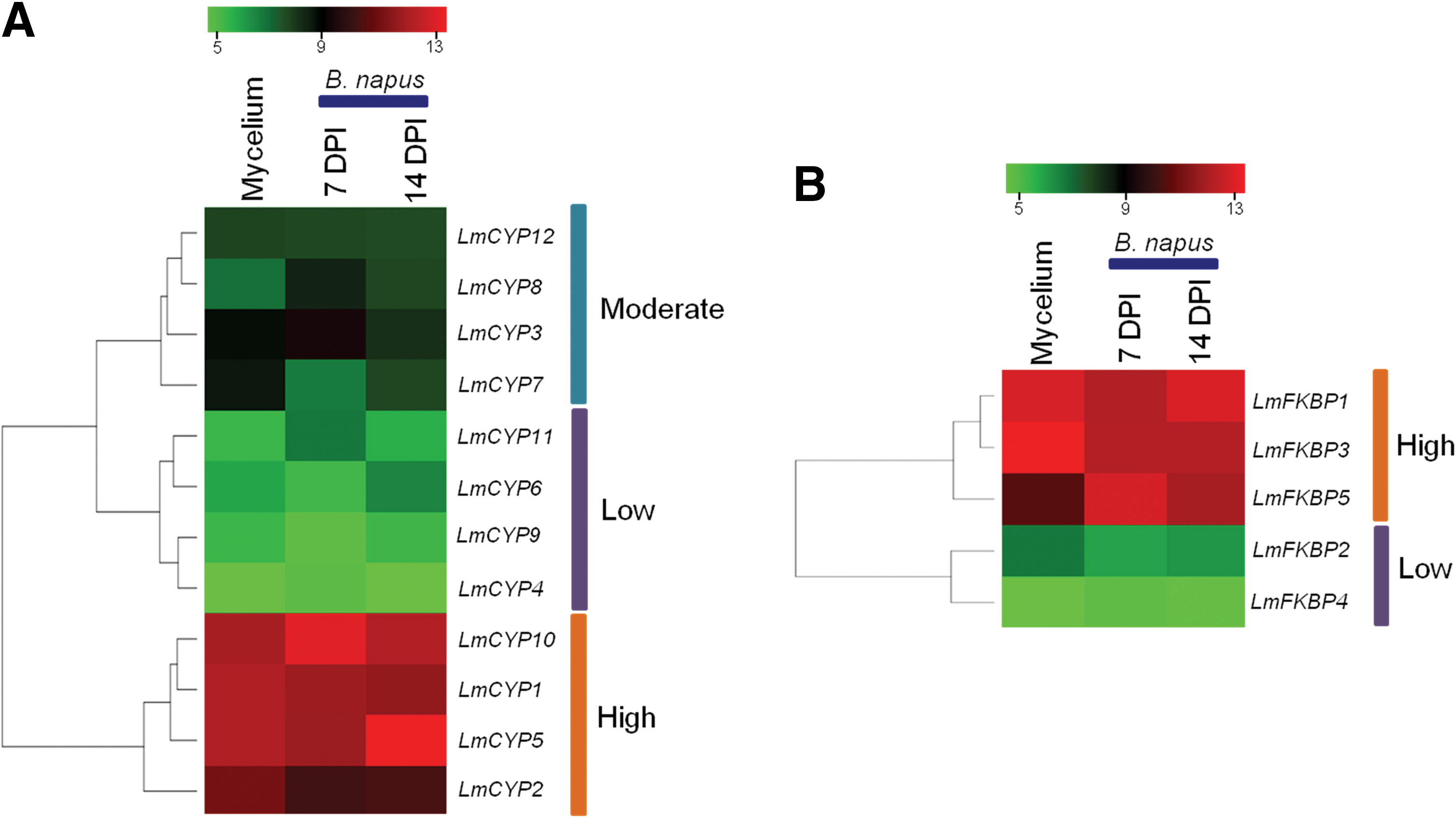
Expression analysis of L. maculans IMMs in mycelium and during B. napus leaf infection at 7 and 14 days post-infection (DPI). Heat maps showing expression of CYPs
FKBPs were classified into two major classes (expressed at high and low levels) (Fig. 7B). IMMs, such as LmFKBP1, LmFKBP3, and LmFKBP5, were found to be highly expressed during both mycelium growth, as well as during primary B. napus leaf infection. This clearly demonstrates their functional significance during infection. Strikingly, LmFKBP5 expressed strongly in leaves compared to mycelium. In contrast, the expression of LmFKBP2 and LmFKBP4 were found to be constitutively lower during both mycelium growth as well as during primary B. napus leaf infection (Fig. 7B).
Conserved motifs in IMMs
In order to determine the conserved motifs present in L. maculans IMMs, aa sequences were subjected to MEME suit (“Materials and Methods”). It was observed that IMM genes possess highly conserved motifs with significant p value (<0.0001). The conserved motifs and sequence logo for CYPs and FKBPs are thus represented in Supplementary Figures S1 and S2, respectively.
Gene Ontology (GO) term identification
GO terms, including Biological Process (BP), Molecular Function (MF), and Cellular Component (CC) were retrieved for all IMMs: BP, including protein folding (GO:0006457) (Supplementary Table S1), MF, peptidyl-prolyl cis trans isomerase activity (PPIase) (GO:0003755), cyclophilin-type peptidyl-prolyl cis trans isomerase activity (GO:0042027), FK506-sensitive peptidyl-prolyl cis trans isomerase activity (GO:0030051), ubiquitin-protein ligase activity (GO:0004842), and nucleic acid binding (GO:0003676) (Supplementary Table S1). Cellular components (CC), including ubiquitin-ligase complex (GO: 0000151) were thus represented in Supplementary Table S1.
Discussion
IMM gene families code for ubiquitous proteins with conserved peptidyl-prolyl cis/trans isomerase (PPIase) activity, and thus are conserved in all organisms. We have systematically retrieved and analyzed the putative IMMs from phytopathogenic ascomycetous fungus L. maculans, the causel agent of phoma stem canker (blackleg) which is a disease of world-wide importance on agronomically important crop (i.e., oilseed rape) (Fitt et al., 2006; Howlett, 2004; West et al., 2001). IMMs have been implicated in a wide variety of processes. The conservation of IMMs in phytopathogenic fungi shows their crucial role in a vast majority of processes. IMMs were demonstrated to play a potent role as virulence determinant in both plant and animal diseases.
In C. neoformans, the CPA1 and CPA2 cyclophilins proteins play a shared role in cell growth, mating, and virulence (Wang et al., 2001). CYP1, a cyclophilin-encoding gene in the phytopathogenic ascomycetes M. grisea, seems to regulate virulence-related functions including apresorium turgor generation and lipid generation (Viaud et al., 2002). Similarly, IMM gene family cyclophilin (CYPA) in the phytopathogenic fungus Botrytis cinerea were shown to be involved in different aspects of morphogenesis and virulence (Viaud et al., 2003). Similar to other non-photosynthetic organisms, such as S. cerevisiase, C. elegans, and A. nidulans (Arevalo-Rodriguez et al., 2004; Pemberton, 2006), in which putative IMM genes have been identified, L. maculans has also been shown to possess low number of IMMs [i.e., 17 altogether (12 CYPs and 5 FKBPs)]. The number of IMM genes in photosynthetic organisms, such as Arabidopsis, rice, and Chlamydomonas, are remarkably higher, which shows that they are more targeted toward the photosynthetic apparatus (Ahn et al., 2010; He et al., 2004; Peltier et al., 2002; Romano et al., 2004; Vallon, 2005). L. maculans IMM proteins exhibited wide variation on the basis of their amino acid (aa) length, molecular weight (Mw), isoelectric point (pI), and presence of signal peptides at N and C-terminal region; however, their catalytic domains were found to be highly conserved, supporting the previously made observations (Wang and Heitman, 2005). IMMs in L. maculans are also categorized into single domain and multi domain (MD), such as in the other organisms that have been studied to date (Ahn et al., 2010). In the case of MD, the presence of other functional domains, such as tetratricopeptide repeat (TPR), WD40 repeats, RNA-binding domain (RNP) or RNA recognition motif (RRM) and vacuolar membrane protein (VMP), in addition to CLD/FKBPs, may also reveal their role in diverse functions (Bannikova et al., 2013; Li et al., 2007; Scheufler et al., 2000; Taylor et al., 2001).
IMM gene family CYPs and FKBPs are localized throughout the cell and thus perform various functions (Wang and Heitman, 2005). In the present study, in L. maculans, they were predominantly localized in three main compartments (i.e., cytoplasm, nucleus, and mitochondria). However, only one gene was found to be localized in plasma membrane. Cytoplasmic cyclophilin A is a well-known target for the binding of immunosuppressive drug CsA in human (Handschumacher et al., 1984). Cyclophilin gene CYP1, from M. grisea, encodes a protein localized in mitochondria and cytosol, thus playing a crucial role during plant infection causing rice blast disease (Viaud et al., 2002). A similar, bifunctional CYPA from B. cinerea is localized in the mitochondria (Viaud et al., 2003). It is possible that the IMMs from L. maculans can also act as virulence determinants against blackleg disease in B. napus. However, further experimental validation is needed to check the exact role of IMMs in L. maculans.
Nuclear IMMs are required during embryogenesis (Carol et al., 2001). Mitochondrial and nuclear encoding IMMs in L. maculans also possess mitochondrial targeting peptide (MTP) and nuclear localized signal (NLS). Additionally, mitochondrially localized IMM genes are significantly involved in mitochondrial permeability, which leads to apoptosis (Zoratti and Szabo, 1995). In fungi, mitochondrial cyclophilin also accelerates folding of newly imported proteins within the matrix as part of a complex that includes the chaperones Hsp60 and Hsp70 (Matouschek et al., 1995; Rassow et al., 1995). There is no evidence available regarding endoplasmic reticulum (ER) and chloroplast localized genes. Perhaps, L. maculans lacks IMM genes that are localized in chloroplast, a plant-specific organelle, thereby explaining the fact that the plant IMM gene has a conserved function in photosynthesis (He et al., 2004). L. maculans genome was found to encode only one IMM, LmFKBP3, with predicted targeting to the signal peptides (SP) possibly suggesting their roles in secretory pathways as well (He et al., 2004).
Comparative phylogenetic analysis between L. maculans and S. cerevisiae IMMs was performed to understand the relationship between the two genomes, using clustalX and a bootstrap value of 1000 iterations. Notably, the bootstrap value of the parent node, as well as the node of individual clades, was found to be low, indicating high sequence variations in proteins in both genomes. The variation in IMM gene family CYPs and FKBPs from L. maculans and S. cerevisiae possibly indicates their divergent evolution. However, only a few homologous and orthologous gene pairs could be identified. Similarly, homologous genes were clustered in different groups based on their high bootstrap value and thus marked on the phylogenetic tree. The orthologous gene pairs between the two genomes are shown in Table 2. Secondary structure prediction revealed that the CYPs and FKBPs in L. maculans have conserved secondary structure features, such as other organisms studied to date (Ahn et al., 2010; Kay, 1996). In the case of CYPs, typical eight β strands and two α helices were observed, while FKBPs has eight β strands and one α helix. GO analysis of IMMs in the phytopathogenic fungus L. maculans also suggests their role in diverse processes, such as protein folding, PPIase activity, ubiquitin-protein ligase activity, and nucleic acid binding (Ahn et al., 2010; He et al., 2004).
Despite the observation that IMM gene family CYPs and FKBPs share a common PPIase, catalyzing the cis/trans isomerization of proline imidic peptide bonds, there are variations in exon–intron architecture. Gene structure analysis of IMMs in L. maculans showed the presence of smaller introns compared to exons. The small size of introns in genes is due to a compact genome (45.12 Mb), suggesting a correlation between intron and genome size. Intron and exon lengths within a genome can reflect the constraints imposed by splicing. In S. cerevisiae, due to a compact genome, genes usually have small introns (Grutzmann et al., 2014). They also show extended consensus sequences for the 5′ canonical splice site (SS) and the branchpoint region (Kupfer et al., 2004). These features facilitate a structural interpretation of intron sequences, and they suggest low-complex alternate splicing patterns, both of which make fungi attractive models for mechanistic studies of alternative splicing (Grutzmann et al., 2014). Notably, in L. maculans, two CYPs were found to be intronless. However, more strikingly, the FKBPs genes in L. maculans contain remarkably higher number of exons and introns compared to CYPs, irrespective of size.
The expression of IMM genes has previously been shown to be induced by both biotic and abiotic stresses (Chou and Gasser, 1997; Godoy et al., 2000; Marivet et al., 1992). Although there are several reports about the potential role of IMMs in various biotic stresses, only few studies are available about phytopathogenic fungi IMMs and their expression during plant infection (Viaud et al., 2002). Therefore, present studies also demonstrated the expression profiles of IMM genes of L. maculans, depicting their potential role in infection in vitro (mycelium) as well as in infected plant leaves at two time points viz. 7 and 14 dpi. L. maculans IMMs were shown to be differentially expressed in mycelium and B. napus leaves. In total, 7 IMMs out of 17 IMMs were shown to be highly expressed during mycelium and leaves infection. This demonstrates their significant role in disease development and regulation during infection. However, 10 IMMs were found to be either moderately or weakly expressed, thereby suggesting downregulation of these genes in mycelium as well as in infected in B. napus leaves.
Conclusions
Immunophilins have not been well studied in phytopathogenic fungi and their precise number, architecture, role in biotic stress, and transactional regulation is poorly understood. However, their role in biotic stress derived from previous studies have provided us knowledge to identify this family in phytopathogenic fungi L. maculans causing economical important disease ‘Blackleg’ in oilseed rape. Thus the fungus is pivotal in agrigenomics-related research. Our study, for the first time, has identified and classified this important gene family in L. maculans and compared them with model organism Saccharomyces cerevisiae. The expression profile in both mycelium and infected primary of B. napus leaves demonstrated their potential role during disease infection. Taken together, our findings present new prospects of this important gene family in phytopathogenic fungi. Further studies may aim to functionally characterize the differentially regulated immunophilins, furthering our understanding of the underlying regulatory network of gene expression and delineating the precise role of L. maculans immunophilins during disease infection in B. napus.
Footnotes
Acknowledgments
Award of research fellowship from Ministry of Education, Youth and Sports of Czech Republic to KS is thankfully acknowledged. Work carried out in this study has been supported by the Czech Ministry of Agriculture, project no. QJ1310227, QH81127 and Internal Grant Agency of the Czech University of Life Sciences, Prague (CIGA, project no. 20142031).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.