
Abstract
Abstract
The COP9 Signalosome (CSN) is a highly conserved eight subunit protein complex associated with a wide range of essential biological functions in eukaryotic cells, and directly involved in processes including deneddylation, phosphorylation, and ubiquitination. Despite its significant role, very few studies have been undertaken to reveal the interactions between the CSN and its binding partners, and none in human T cells. Here we present a purification method for the CSN and binding proteins via the Streptavidin-Binding Peptide (SBP) fused to CSN Subunit 1 (CSN1). Using this method, coupled with liquid chromatography-mass spectrometry analysis, we identified all eight subunits of the CSN, as well as expected and putative novel binding partners such as a tumor suppressor under the control of Cullin4a-ligase complex; Neurofibromin 2 (Merlin). This work presents a method for fast, reliable, and specific affinity-based purification of a protein complex from a nonadherent cell line. The purification of the CSN and binding partners from T cells can elucidate the roles of CSN in a cell type where it has never been studied before. This proteomic-based approach can broaden our understanding of the functions of the CSN in contexts such as viral–host interactions or immune activation in their natural milieu.
Introduction
Another important function of the CSN complex performed either as a whole or by independent subunits, is the association with kinase activity. Several targets are known to be phosphorylated by the CSN-associated kinases, some of which are yet to be discovered, at least in vitro. Those targets include c-Jun, IκBα, NF-κB p105 precursor, and most importantly p53 (Bech-Otschir et al., 2001; Seeger et al., 1998). Furthermore, several of the CSN subunits, specifically CSN2 and CSN7, are known to be phosphorylated by CKII and PKD kinases with yet-to-be-discovered consequences (Uhle et al., 2003). The best characterized role of individual CSN subunits is that of subunit 5 (CSN5) in Jun signaling and AP-1 activation. CSN5, also known as Jun-activating binding protein 1 (Jab1), directly interacts with c-Jun and facilitates its binding to JNK kinase, resulting in phosphorylation and stabilization of c-Jun upon binding to AP-1 promoter sites (Claret et al., 1996; Kleemann et al., 2000).
The findings to date indicate that CSN is a major player in most cell processes, and yet only one wide-ranging proteomic study analyzing the binding partners of the CSN by liquid chromatography/mass spectrometry has been undertaken (Fang et al., 2008). Furthermore, virtually nothing is known about the interaction of the CSN in T cells with the ubiquitin ligase machinery or other proteins. This knowledge will facilitate the elucidation of how these interactions are affected by processes such as stimulation or viral infection. In order to address this question, we have purified the entire CSN from a human T-cell line through one of its subunits utilizing the Streptavidin-Binding Peptide (SBP) tag. Purification of the CSN and binding partners was further analyzed by liquid chromatography-mass spectrometry.
Materials and Methods
Cells
Human T-cell line SupT1 was obtained from the American Type Culture Collection (ATCC, Manassas, VA). Cells were maintained in complete RPMI 1640 media supplemented with 10% fetal bovine serum (Gemini Bio-Products, West Sacramento, CA), glutamine (2 mM), penicillin G (100 units/mL), and streptomycin (100 μg/mL). Phoenix GP cell-line (Nolan Lab, Stanford University, CA) was maintained in Dulbecco's Modified Eagle's media supplemented with 10% fetal bovine serum (Gemini Bio-Products), glutamine (2 mM), penicillin G (100 units/mL), and streptomycin (100 μg/mL).
Antibodies and reagents
The antibodies to CSN1 (A300-026A), CSN2 (A300-027A), CSN4 (A300-014A), CSN7b (A300-240A), and Cullin-4a (A300-738A) were obtained from Bethyl Laboratories (Montgomery, TX). Antibodies to CSN3 (sc-100693), CSN6 (sc-137122), and Merlin (sc-55575) were obtained from Santa Cruz Biotechnology, Inc (Santa Cruz, CA). The CSN8 antibody (BML-PW8290-0025) was provided by Biomol, Inc. (Plymouth Meeting, PA). The antibody for Nedd8 (2745) was obtained from Cell Signaling (Beverly, MA). The antimouse IgG-HRP (115-035-003) and antirabbit IgG-HRP (115-035-045) antibodies were provided by Jackson Immunoresearch (West Grove, PA). Dynabeads MyOne Streptavidin Beads (656-01) were purchased from Invitrogen (Carlsbad, CA).
Plasmids
The retroviral transfer vector pBMN.i.mCherry was constructed by amplifying mCherry from pmCherry-C1 (Clontech, Palo Alto, CA) using the forward primer with extending NcoI site ATCGATGGATCCCCACCATGGTGAGCAAGGGCGAGGAG and reverse primer with extending XhoI site ATGGACGAGCTGTACAAGTAACTCGAGGATCGATC, and inserting it into partially digested pBMN-i-eGFP (Gary Nolan, Stanford University) with NcoI/SalI. The SBP-Citrine construct was a kind gift from Shari Kaiser (Fred Hutchinson Cancer Research Center, Seattle, WA). The construct pBMN.SBP-Citrine.i.mCherry was created by amplifying SBP-Citrine from SBP-Citrine with the forward primer with extending BglII site TATAGCTAGCAGATCTC-CACCATGGACGAGAAGACCACCGGC and reverse primer with extending XhoI site ATGGACGAGCTCTATAAATAACTCGAGTATA, and inserted into pBMN.i.mCherry digested with BamHI/XhoI. The SBP tag was cloned into pcDNA3.1/Zeocin (Invitrogen) by amplifying the SBP sequence with the forward primer TATAGCTAGCAGATCTCCAC-CATGGACGAGAAGACCACCGGC, which contains a Kozak sequence and a BglII site and a reverse primer TATACTCGAGTCTAGAG-GATCCGGGCTCCCTCTGGCCCTGGGGG that contains a linker consisting of an XbaI and an XhoI site. This product was ligated into pcDNA3.1/Zeocin using BamHI/XhoI restriction enzymes. pcDNA3.1/Zeocin SBP-CSN1 construct was created by amplifying CSN1 from HeLa cDNA (courtesy of Christopher Glembotski, SDSU) by using a forward primer containing an XbaI site TATAGGATCCTCTAGACC-GCTGCCGGTTCAGGTGTTT and a reverse primer containing an XhoI site TATACTCGAGTCACATGTTGGTGCTCATCCGGG, digesting it with XbaI/XhoI and ligating it into pcDNA3.1/Zeocin. The construct pBMN.SBP-CSN1.i.mCherry was created by digesting pcDNA3.1/Zeocin SBP-CSN1 with BamHI/XhoI and ligating the extracted fragment into pBMN.i.mCherry digested with BamHI/XhoI.
Virus production and transductions
For the production of MLV based virus, a 10 cm2 plate of Phoenix GP cells at 50% confluence was transfected with 3 μg of the packaging vector (pBMN.SBP-Citrine.i.mCherry or pBMN.SBP-CSN1.i.mCherry) and 3 μg of a vector expressing the Envelope glycoprotein of the Vesicular Stomatitis Virus (pCI-VSVg) by mixing the plasmids in 125μL of FCS-free DMEM and 30μg of Polyethylenimine (linear, MW 24000; Polysciences, Inc., Warrington, PA). Media (DMEM with 10% FCS, Pen-Strep, L-Glutamine) was replaced 24 h posttransfection and viral supernatant was collected 48 h after transfection and filtered with 0.45-micron PTFE filters (Pall Corporation, East Hills, NY). The supernatant was used to spin-infect naive SupT1 cells in a six-well plate format. Briefly, viral supernatant was mixed with polybrene (5 μg/mL final concentrations) and added to the cells, the mixture plated in a six-well plate and spun at 1500×g, 32°C for 80 min in a hanging bucket rotors centrifuge (Becton Dickinson, Fullerton, CA). Twenty-four hours postinfection, cells were resuspended in fresh media.
Flow cytometry and sorting
Flow cyometry and sorting were performed on a BD FACSAria with 405-, 488-, and 633-nm lasers. Data was collected on FACSDiva 6.1.1.
Pull-downs and Western blotting
Cells were spun and resuspended in modified NP-40 buffer (150 mM NaCl, Tris-Cl 50 mM, 10% Glycerol, 0.25% NP-40) supplemented with Complete Protease Inhibitor cocktail (Roche, Indianapolis, IN) at a concentration of 108 cells/mL. After incubation on ice for 30 min, cell lysates were spun at 14,000×g for 15 min at 4°C and supernatants were transferred to new tubes. The lysates were incubated for 1 h with 100 μL of Dynabeads® MyOne Streptavidin T1 beads (Invitrogen) on a rotating rack at 4°C. The beads were separated from the sample with a magnetic rack, and the supernatant was retained for lysate control. The beads were washed at least five times with 1 mL lysis buffer, and the bound proteins were eluted with lysis buffer containing 2 nM D-biotin. Lysates and elutes were boiled in SDS sample buffer, run in a 12% polyacrylamide gel and then transferred to a PVDF membrane. After blocking with 5% milk in PBST (PBS+ 0.05% Tween 20), the membrane was incubated with the indicated primary antibodies followed by HRP-conjugated secondary antibodies. Proteins were subsequently detected using the Amersham ECL kit.
Preparation of samples for liquid chromatography-mass spectrometry
Samples were first run in a 12% Mini-PROTEAN TGX gel (BioRad, Hercules, CA), and the appropriate gel slices containing the sample were sent to Stanford University Mass Spectrometry Core (SUMS) for LC-MS analysis.
Results
Purification of the CSN holocomplex from SupT1 cells
In order to purify the CSN complex from T cells we decided to engineer a tagged version of one of the CSN subunits, and express the tagged subunit endogenously. For that purpose, we utilized a 38-amino acid peptide (SBP) shown to have high binding affinity to streptavidin (SA) (Keefe et al., 2001). Previous studies have demonstrated that it is feasible to purify the entire complex through CSN1 (Menon et al., 2005). We thus introduced the SBP sequence at the N′ terminus of CSN1, cloned into the pBMN.i.mCherry retroviral vector. In this vector, referred to as pBMN.SBP-CSN1.i.mCherry, the translation of the SBP-CSN1 protein is coupled to the IRES-mCherry cassette (i.mCherry) (Fig. 1A). This vector was used to create MLV particles and infect naive SupT1 cells, as previously described (Wolkowicz et al., 2004), which were then FACS-sorted, clonally selected on the basis of high mCherry expression, and probed with anti-CSN1 antibody to confirm the expression of SBP-CSN1 (Fig .1B–D). As a control, we utilized a similar construct where CSN1 was substituted with Citrine, a modified yellow fluorescent protein chosen to exclude the nonspecific protein–protein interaction of the SBP-tag fusion protein. Citrine, a protein of nonmammalian origin, serves here as a stricter control than the empty vector. Total extracts from SBP-Citrine- and -CSN1-expressing cells were then subjected to streptavidin-coated magnetic beads in order to pull down SBP-tagged proteins (Fig. 2).
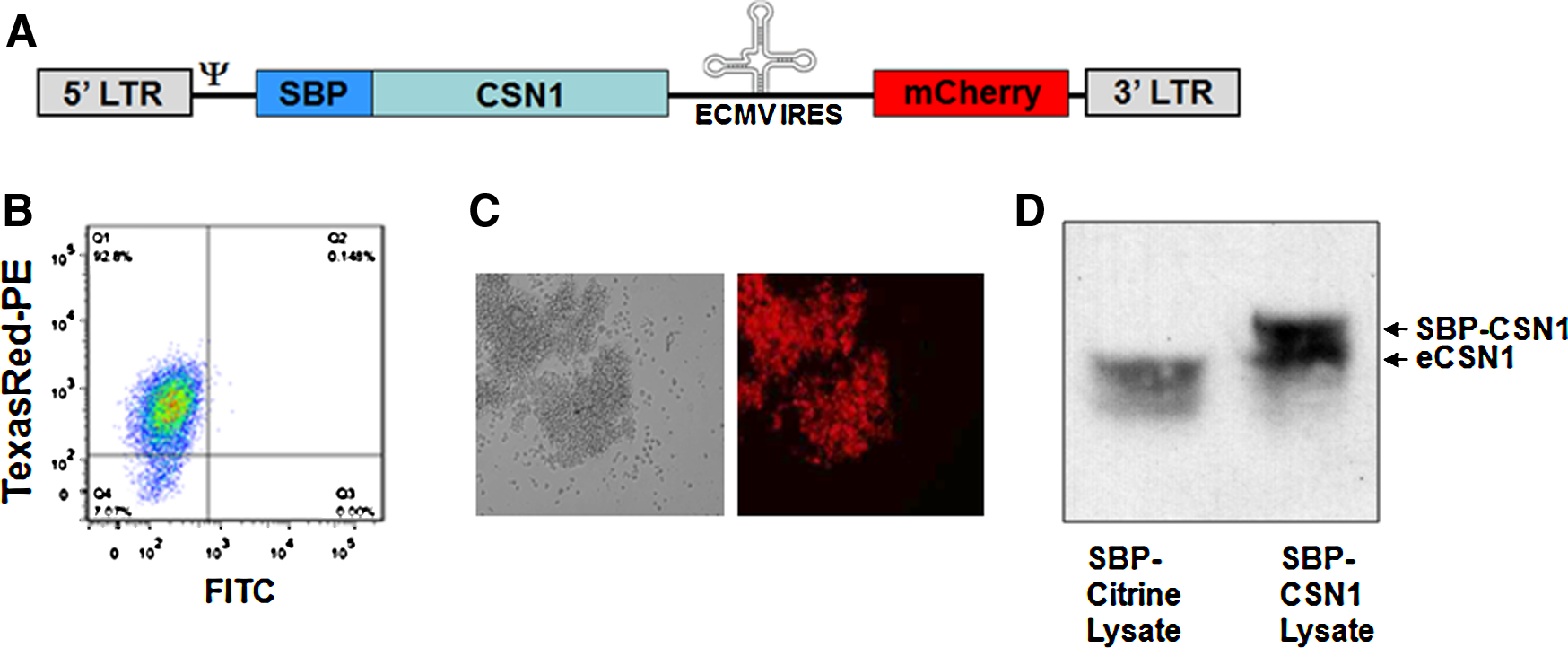
Establishment of an SBP-CSN1 expressing cell line. (
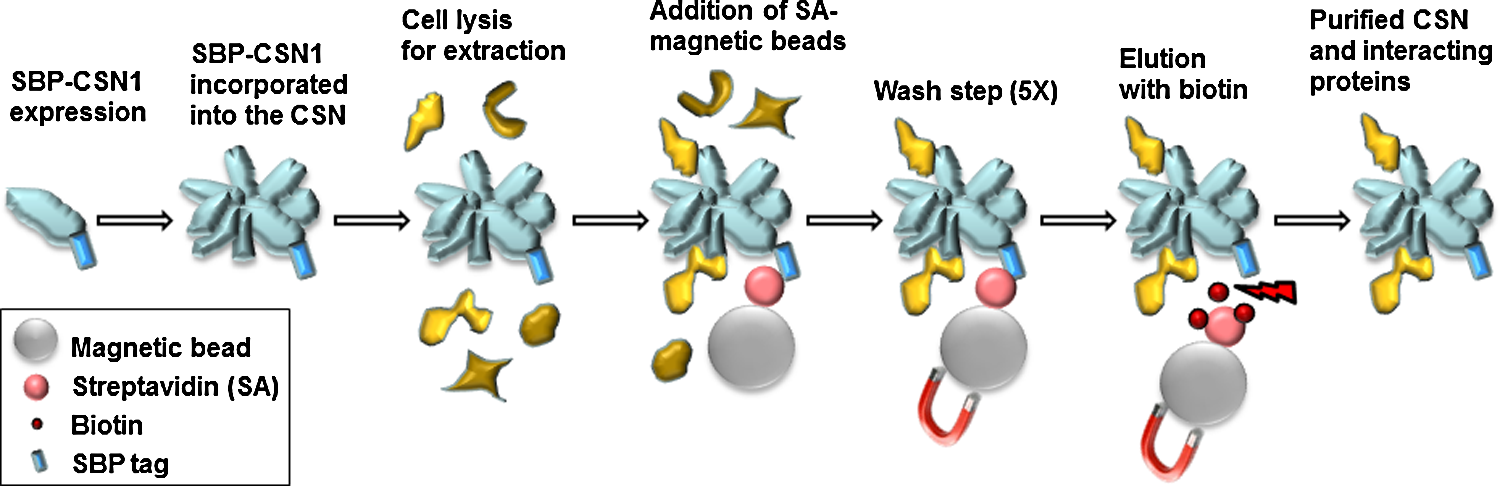
Schematic representation of the SBP-based pull-down technique for the purification of the CSN and binding partners.
The eluted fractions were then analyzed for the presence of CSN subunits. Preliminary results (data not shown) indicated that the lysis buffer utilized was too stringent to pull down the entire CSN holocomplex, probably due to the high concentration of the nonionic Nonodet P-40 detergent and the presence of the ionic sodium deoxycholate detergent, both of which can disrupt weak protein–protein interactions. Interestingly, at these high-stringency conditions (150 mM NaCl, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris, pH 8.0), CSN subunits 1, 2, 3, and 8 could be detected. This finding is consistent with a recent study demonstrating that the CSN subunits form two distinct minicomplexes, one of which is composed of CSN1, 2, 3, and 8, that together, assemble into the holocomplex (Sharon et al., 2009). Decreasing the stringency of the lysis buffer by reducing the amount of NP-40 detergent and gentler mechanical lysis resulted in the purification of all eight of the CSN subunits from SupT1 cells, including both known isoforms of CSN7, as confirmed by Western blots and LC-MS (Fig. 3 and Table 1).
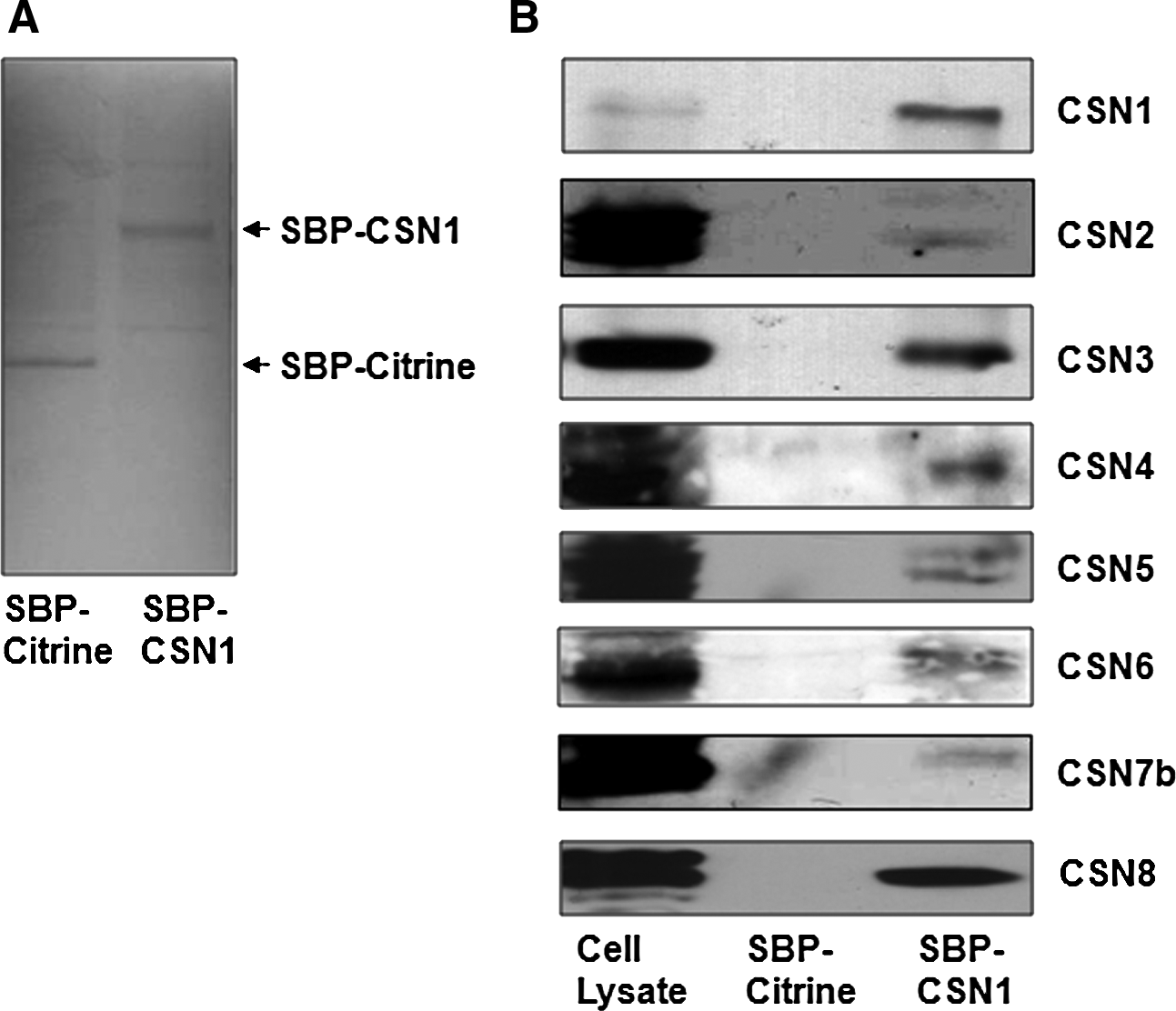
Purification of the CSN from T cells. (
Known and predicted binding partners of the CSN isolated from SupT1 cells
One of the major functions of the CSN is the cleavage of Nedd8 from Cullin proteins (Cope et al., 2002). Prior to LC-MS analysis, we probed our eluted fraction with a Nedd8 antibody in order to ascertain if any binding proteins were pulled down along with the CSN holocomplex. Several protein bands were detected, indicating the presence of neddylated proteins in the eluate from the lysate of the SBP-CSN1 cell line (Fig. 4A). We next addressed whether this technique enabled us to isolate the binding partners of the CSN, either as previously described in literature or predicted by bioinformatics tools in silico (Table 2). The eluted fractions from SBP-CSN1- and SBP-Citrine whole-cell lysates were analyzed by LC-MS at the Stanford University Mass Spectrometry facility. Hits from the SBP-Citrine pull-down were used to eliminate nonspecific binding proteins. A total of 355 proteins were identified as putative CSN-binding partners. Out of 355, 78 showed an 80% or greater protein identification probability, as determined by the probability of peptide identification and the number of hits identified per protein (Supplementary Tables 1 and 2). The specific hits listed in Table 2 (16 in total), represent hits that have been characterized in other studies, predicted to interact with the CSN in silico or confirmed in our manuscript. From the putative CSN-binding proteins, we identified two Cullin proteins, Cullin 3 and Cullin 4B, in addition to Cullin 4A, which we detected by Western blot (Olma et al., 2009) (Fig. 4B). We have also identified CAND1 and DDB1, a subunit of the damaged-DNA binding protein DDB. These are two of the proteins known to interact with Cullin 4 during the process of ubiquitination of target proteins by the CSN (Bondar et al., 2006). Another probable hit was Ubiquitin ligase E2N (UBE2N), a human analog of Saccharomyces cerevisiae ubiquitin ligase Ubc13. The interaction between E2N and the CSN has previously been predicted by genome-scale genetic interaction maps, but never confirmed in either yeast or human cells (Huttenhower et al., 2009). Other members of the ubiquitination machinery included Ubiquitin protein ligase E3A (UBE3A) and Isoform 4 of Ubiquitin protein ligase (UBR4).
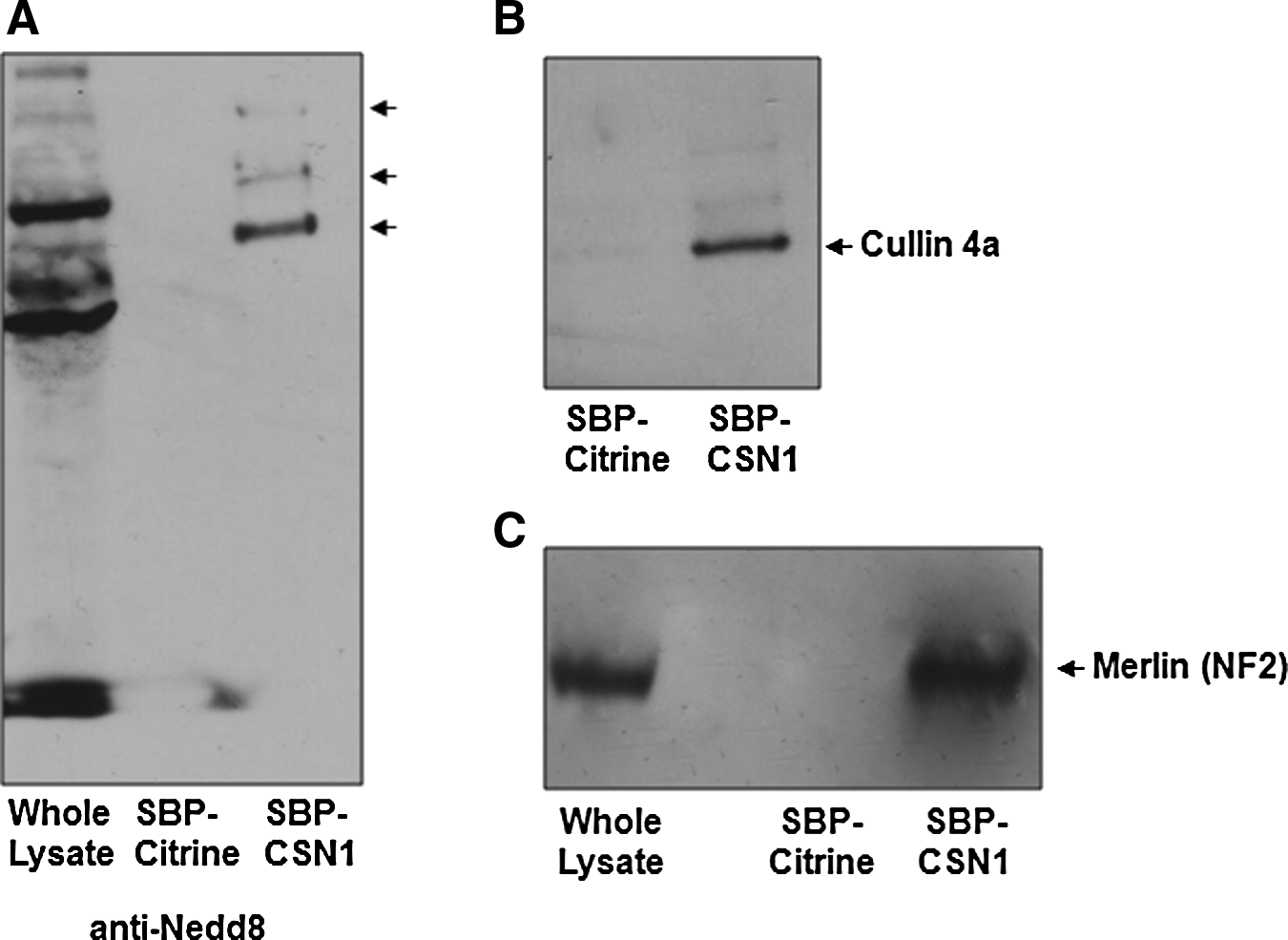
Western blot analysis of SBP-Citrine and SBP-CSN1 cell lysates to confirm CSN binding partners. (
Proteins previously described to interact with the CSN.
Proteins predicted to interact with the CSN by PIPs (Human Protein-Protein Interaction Prediction).
Proteins predicted to interact with the CSN by BioGRID3.1.
Several protein hits from our pulldown corresponded with predictions made by the Human Protein–Protein Prediction Interaction database (PIP), a bioinformatic tool that predicts the likelihood of interaction between two proteins (McDowall et al., 2009). These included Long Isoform of 14-3-3 protein beta/alpha (YWHAB, interaction score of 5.28) and Eukaryotic translation initiation factor 5A-2 (EIF5A2, interaction score of 2.68). The CSN has previously been shown to associate with the 26S Proteasome, most likely in order to direct very specific proteolysis (Huang et al., 2005). We indeed were able to identify several of the proteosomal subunits, including Proteasome subunit alpha type-2 (PSMA2) and 4 (PSMA4), beta type-3 (PSMB3), and Proteasome activator complex subunit-1 (PSME1). Interestingly, the BioGrid 3.1 protein interaction database (Stark et al., 2011) and the Drosophila protein interaction map based on a yeast two-hybrid screen (Giot et al., 2003) hinted at the interaction between the Ribosomal protein L23A and CSN3, a high-probability hit obtained in our pulldown (Table 2).
Novel CSN binding partner Neurofibromin 2
One of the strong hits identified in our pulldown was Neurofibromin 2 (Merlin). Merlin is a 70-kDa protein related to proteins that anchor the actin cytoskeleton to specific membrane proteins, and appears to function as a tumor suppressor. A recent study determined that Merlin is targeted to the Roc1-CUL4A-DDB1 E3 ligase complex for degradation by Vpr-binding protein (VprBP) (Huang and Chen, 2008). We further confirmed the presence of Merlin in the SBP-CSN1 eluate by Western blotting (Fig. 4C).
Discussion
Although the COP9 Signalosome plays a major role in many vital cellular functions, few proteomic studies analyzing its binding partners in human cells have been performed, and none in T cells. In this work, we used retroviral technology to efficiently deliver the SBP-tagged CSN1 subunit into a T-cell line. The SBP-tag provides a fast, efficient, and relatively specific one-step method for the isolation of protein complexes, as demonstrated here with the CSN. Fang et al. (2008) successfully purified the CSN holocomplex from fibroblasts utilizing a long tag of around 110 amino acids in length. This tag included two six-Histidine residue sequences, a 75 amino acid-long sequence as in vivo substrate for biotinylation, and a 29 amino acid-long Tobacco Etching Virus protease cleavage site with linkers. The SBP-tag is only 38 amino acids-long and does not rely on the efficiency with which the biotinylation process occurs in the cell. Furthermore, in the SBP-tag system, biotin is used as competitor at the time of purification and purification does not rely on the activity of a protease. Using the SBP-tagged CSN1 subunit expressed endogenously, we were able to recover all eight subunits of the CSN through the pull down, demonstrating that tagging the CSN subunit did not disrupt its ability to interact with the rest of the subunits and form the CSN holocomplex. Previous studies for the purification of the CSN holocomplex in human cells were performed in fibroblasts. Here we have successfully purified the complex from T cells, a nonadherent cell type with very different biological characteristics and functionalities, such as those related to immune activation. Although SupT1 cells are not primary cells, they are easily activated by many physiological and nonphysiological factors, and infectable by viruses such as HIV-1. This is not to say that parallel experiments should not be performed in primary T cells, where similar results should nonetheless be expected. Furthermore, combining this approach with LC-MS yielded a large number of potential CSN binding partners. These included previously documented members of the ubiquitin-ligase machinery, such as CAND1, DDB1, and various Cullins, as well as novel ubiquitin ligases UBR4, UBE3A, and UBE2N. In addition, we were also able to detect several protein components of the 26S Proteasome, a finding that is consistent with the model of the CSN-Proteasome supercomplex, where the CSN directly interacts with the barrel of the 26S Proteasome to specifically degrade protein targets (Huang et al., 2005). Importantly, we were able to detect several proteins predicted by in silico tools such as BioGrid 3.1 and PIP, such as YWHAB, EIF5A2, and L23A. The nature of this interaction is unknown. Nevertheless, the fact that one of the main roles of the CSN in involves phosphorylation via CSN-associated kinases and the fact that both YWHAB and L23A are known to be phosphorylated during cellular processes reassures us that purification of these targets may have a significant biological role (Dai et al., 2004; Liu et al., 2007).
Interestingly, another putative novel binding partner of the CSN with no known relation to the proteasomal and/or ubiquitination machineries was Neurofibromin 2 (Merlin). In a recent study, it was found that the cellular levels of Merlin are controlled by the HIV-1 Vpr-Binding Protein, a protein known to act as a substrate-recognition subunit of the ROC1-CUL4A-DDB1 ubiquitin ligase complex and which appears to direct Merlin for degradation via ubiquitination (Huang and Chen, 2008; Wen et al., 2007). The finding that Merlin is associated with the COP9 Signalosome is consistent with the model describing the control of Merlin expression, as one of the functions of the CSN is to deneddylate Cullin proteins, including CUL4A, in order to negatively regulate the ubiquitination of their targets (Deshaies, 1999).
The identification of all eight subunits of the CSN (including both isoforms of CSN7, as demonstrated by LC-MS and Western blotting) and a number of its binding partners from T cells, including Merlin, demonstrates that the purification method through intracellularly expressed SBP tag is a robust method for protein-binding discovery. A large number of hits were found to be nonspecific and bind to the SBP tag, streptavidin, and/or beads, particularly mitochondrial proteins. The method described here for the purification of the CSN may thus not be appropriate for targeting protein complexes such as those associated with the mitochondria, emphasizing the importance of a control, SBP-Citrine in our experiments, for determining specific binding. The advantages of using the SBP tag include the relatively rapid pulldown procedure (20 min incubation for most protein isolations combined with rapid magnetic rack-based washes) and high specificity of streptavidin to SBP compared to immunoprecipitation methods involving antibodies. This makes the method an attractive alternative for sample preparation for LC-MS analysis.
Purification of the CSN holocomplex together with its binding partners from a natural environment such as a T cell can greatly facilitate the study of the CSN and its interactions as well as the regulation of its binding partners under different conditions, such as T cell activation and HIV-1 infection.
Footnotes
Acknowledgments
R.W. was supported by the California HIV/AIDS Research Grant Program (CHRP) ID09-SDSUF-023 #163058, and the California State University Program for Education and Research in Biotechnology (CSUPERB). The authors thank Dr. Shari Kaiser (Institute for Systems Biology, Seattle, WA) for providing the SBP-Citrine construct, Brett Hilton from the SDSU Fluorescence Activated Cell Sorting facility, the Stanford University Mass Spectrometry Facility, and the San Diego Chapter of the ARCS (Achievement Rewards for College Scientists) Foundation, who supported A.S.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.