
Abstract
Abstract
Quantitative proteomics experiments are usually performed using proteolytic peptides as surrogates for their parent proteins, inferring protein amounts from peptide-level quantitation. This process is frequently dependent on complete digestion of the parent protein to its limit peptides so that their signal is truly representative. Unfortunately, proteolysis is often incomplete, and missed cleavage peptides are frequently produced that are unlikely to be optimal surrogates for quantitation, particularly for label-mediated approaches seeking to derive absolute values. We have generated a predictive computational tool that is able to predict which candidate proteolytic peptide bonds are likely to be missed by the standard enzyme trypsin. Our cross-validated prediction tool uses support vector machines and achieves high accuracy in excess of 0.94 precision (PPV), with attendant high sensitivity of 0.79, across multiple proteomes. We believe this is a useful tool for selecting candidate quantotypic peptides, seeking to minimize likely loss owing to missed cleavage, which will be a boon for quantitative proteomic pipelines as well as other areas of proteomics. Our results are discussed in the context of recent results examining the kinetics of missed cleavages in proteomic digestion protocols, and show agreement with observed experimental trends. The software has been made available at http://king.smith.man.ac.uk/mcpred.
Introduction
We believe this problem is particularly acute for targeted proteomics strategies using selected peptides as proteotypic (or quantotypic) surrogate markers of protein abundance, such as that required for the AQUA (Gerber et al., 2003) or QconCAT approach (Beynon et al., 2005; Pratt et al., 2006). It is essential in such cases to select peptides that are genuinely “quantotypic.” This means that that they readily ionize and are observed in the mass spectrometer, and also that no signal is lost owing to missed cleavage or post-translational modification. These are important criteria to consider in targeted absolute quantitation experiments using selected reaction monitoring (SRM) (Lange et al., 2008; Picotti et al., 2009, 2008, 2010). Signal loss from missed cleavages can also potentially affect relative quantitation strategies; although the splitting of the peptide signal can be partly mitigated if sample preparations are identical, this is usually not perfectly achieved, and adds a further source of variance to the experimental pipeline. Indeed, some label-free pipelines prefer to omit missed cleavages from consideration, and we suggest that they are generally best avoided in quantitative proteomic pipelines.
In this study, we have built upon a previous missed cleavage predictor (Siepen et al., 2007) to generate a much expanded dataset of missed and fully cleavage peptides, and derive a superior tool using machine learning techniques. The new tool outperforms the previous one, producing an overall prediction success of 94% positive predicted value (PPV), at a good sensitivity (recall) of 79%, and an overall validated area under the ROC curve (AUROC) of 0.88. Its intended use is as part of a pipeline selecting candidate quantotypic peptides for targeted proteomics experiments exploiting SRM, but we believe it has wider applicability in quantitative proteomics. We demonstrate its superior performance across multiple example proteomes, and have made it available via http://king.smith.manchester.ac.uk/mcpred.
Materials and Methods
Tryptic site dataset generation
The process for assembling the tryptic site context data is summarized in Figure 1. It is worth noting that generation of a learning set for this task is non-trivial, particularly for the set of cleaved bonds associated with limit peptides; care must be taken to ensure that no missed cleaved sites contaminate the cleaved dataset. To this end we adopted a fairly conservative strategy that nevertheless generated a substantive dataset of peptides and associated cleaved/uncleaved tryptic sites, which is summarized in Table 1. The datasets were generated from 76 non-ICAT experiments downloaded from PeptideAtlas (Deutsch, 2010), covering three model organism proteomes, comprised of 32 from
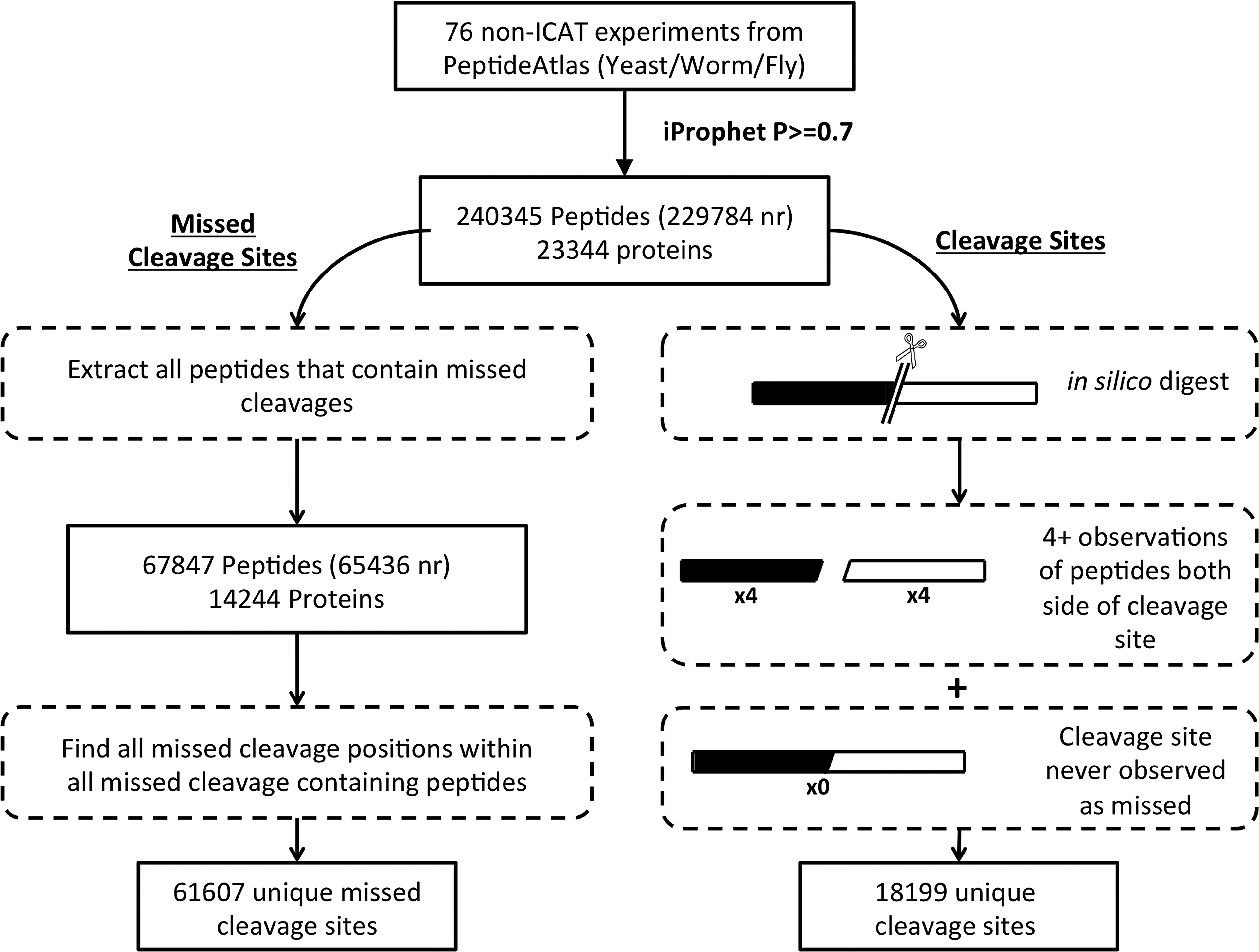
Flowchart describing the generation of the two datasets used to train and test the missed cleavage predictor. Peptides that satisfy an iProphet probability threshold of
The non-redundant counts of the peptides and tryptic sites are displayed in parentheses (mc=missed cleavage).
This conservative definition produced 18,199 unique cleaved sites. For both the missed and cleaved datasets a 9-
Sites to features
In preparation for support vector machine (SVM) training, the tryptic site datasets were converted into numerical feature vectors that retained both the position and residue-specific information. The 20 amino acids (plus Z) were indexed (
Support vector machines (SVM)
SVMlight was chosen as the implementation of SVM for training and testing (Joachims, 1999). The missed cleaved dataset was assigned as the positive target, and the cleaved dataset as the negative target for the training step. The radial basis function was used for learning, for which the gamma parameter was optimized to maximize sensitivity×PPV, and training errors were weighted to account for the unbalanced dataset.
Prediction performance and validation
The dataset was split into two groups, 10% held back for validation, and 90% used for 10-fold cross-validation training and testing. The performance of the predictor was assessed on both the cross-validation dataset and further validated on the unseen 10%. Prediction performance was measured using the usual statistics of sensitivity, PPV, and AUROC. In this case, a high sensitivity means a high proportion of the missed cleavages are correctly predicted (few Type II errors), and high PPV reflects that a high proportion of predicted sites are truly missed (few Type I errors), both of which are desirable in this case.
The final SVM predictor was compared with our previous published missed cleavage predictor, which used an information theoretic approach and a smaller learning set (Siepen et al., 2007).
Results
Amino acid propensities
As a first look at the missed and cleaved datasets, the amino acid propensities at each position in the 9
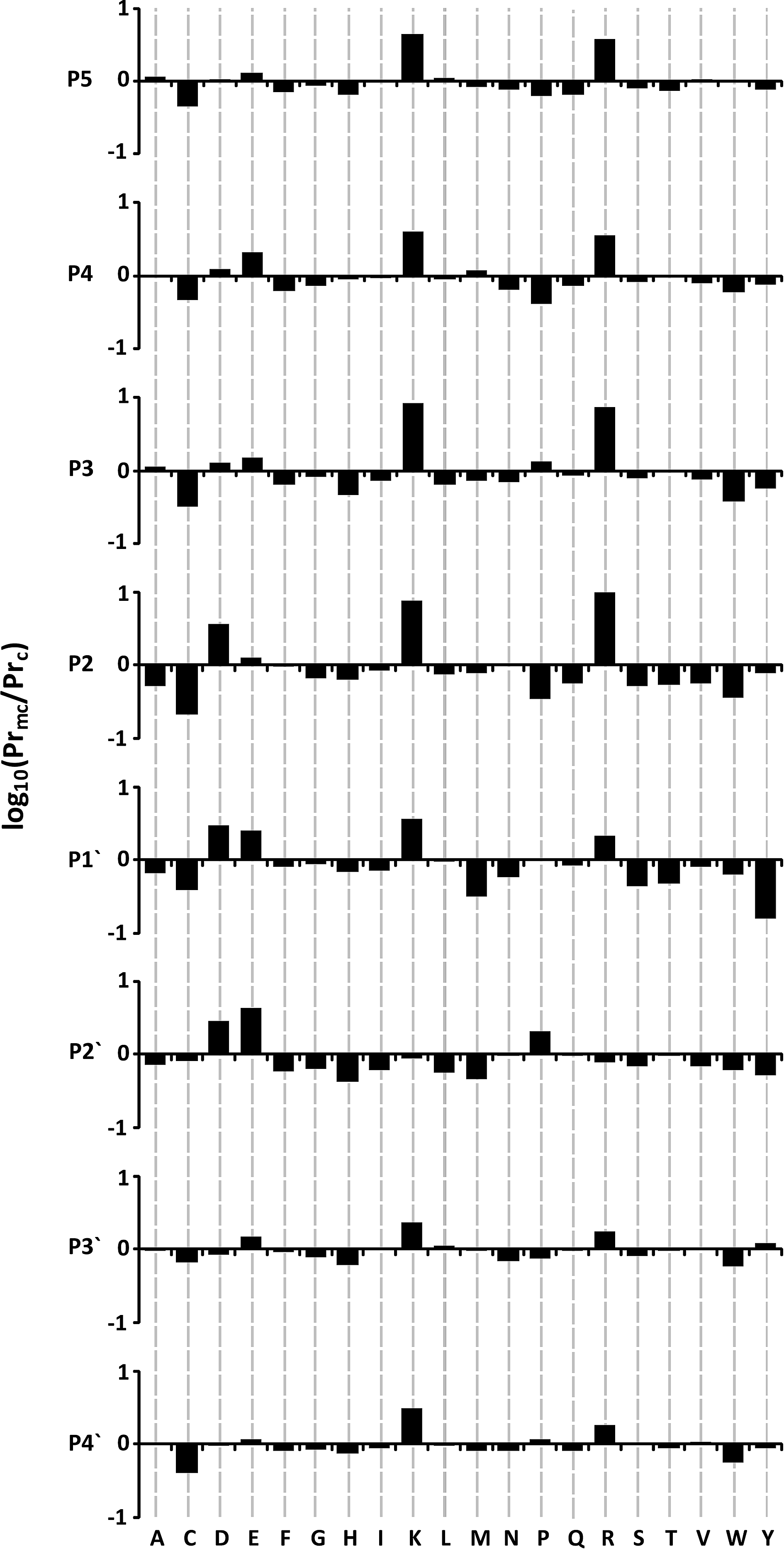
Histograms are shown plotting log10 ratio propensities for given amino acids to lead to missed cleavage around tryptic sites. Graphs are plotted for the four residues immediately N and C terminal of a generic tryptic site following the pattern P5-P4-P3-P2-P1-P1′-P2′-P3′-P4′, where the tryptic site is cut between P1 and P1′. Data for P1 are not shown due to the lack of new information content (since they are all lysine or arginine).
The most striking feature of Figure 2 is the large lysine/arginine propensity in missed cleaved sites, particularly N-terminal to the tryptic site (P2–P5), where there is approximately a 10-fold difference in propensity at P2 and P3 compared to cleaved sites. Looking C-terminal to the tryptic site, the propensities for arginine/lysine are reduced, though still present at P1′, P3′, and P4′. There is an increase in glutamic acid propensity at all positions in missed cleaved sites compared to cleaved sites, which is more pronounced at P1′ and P2′. Similarly, the presence of aspartic acid at P1′ and P2′ appears to discourage efficient trypsin cleavage, and also has an influence N-terminal to the tryptic site at P2. These observations are consistent with previously reported studies (Monigatti and Berndt, 2005; Thiede et al., 2000; Yen et al., 2006), which note the difficulty in observing efficient cleavage at dibasic sites (KK, KR, RR, and RK), and sites where there is potential to form a salt bridge between acidic side chains and the basic site at P1.
Interestingly, from the cleaved point of view, the most influential amino acid appears to be tyrosine at position P1′. An unexpected result is the positive influence cysteine seems to have at all 8 positions, with the greater impact being at the N-terminal positions. It is not straightforward to rationalize this result, which we believe may be partly associated with the difficulties encountered in dealing with reduced/oxidized cysteines in proteomics experiments, and their relative low abundance in proteins in general. Of the remaining residues, seven show a consistently higher propensity at all sites for cleavage (phenylalanine, glycine, histidine, asparagine, glutamine, serine, and tryptophan), albeit these residues do not show as high a propensity as those mentioned above, and generally are not as striking as those with strong propensities for missed cleavage.
Taking the amino acid propensities for both missed and cleaved across all sites, the information content was calculated as:
where the information content
The amino acid propensities were also calculated for the three individual proteomes that contribute to the combined missed and cleaved datasets (see Supplementary Figs. 1–3 at www.liebertpub.com/omi). <http://www.liebertpub.com/omi.)>). Comparing the positional propensities via a linear regression (Fig. 3), shows that propensities for 6 out of the 8 positions are highly correlated (R2>0.7,
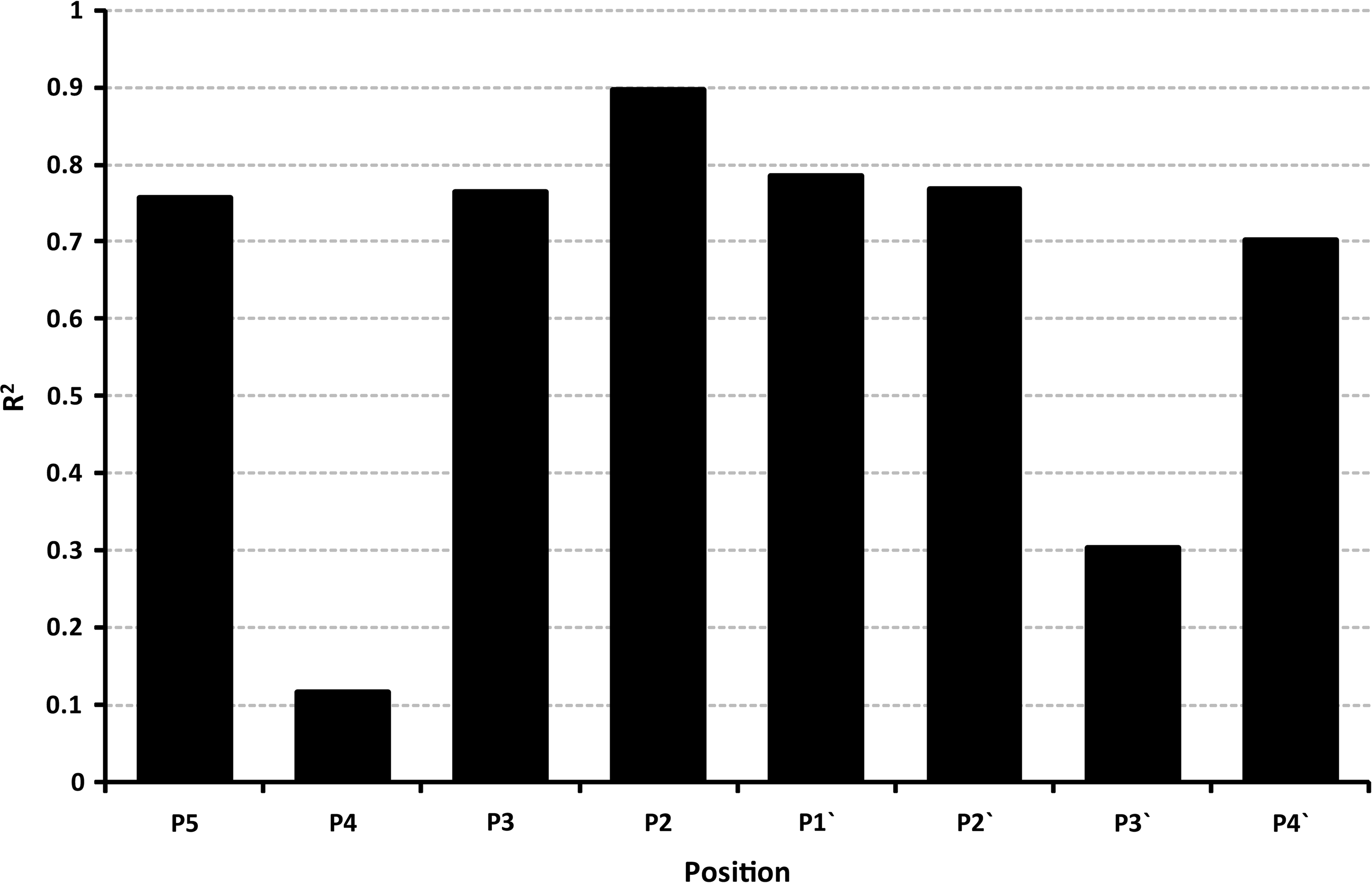
This histogram shows the linear regression (R2) of missed to cleaved propensity ratios at each position across the three proteomes used to create the dataset.
Predictor performance
Moving on from propensities, we evaluated the performance of the SVM trained on this data. The performance statistics for the fully-trained SVM and the information theory predictor are shown in Table 2, covering sensitivity, specificity, PPV, sensitivity×PPV, and AUROC. As a binary classifier, the SVM predictor achieves a respectable sensitivity of 0.79, coupled with a high PPV of 0.94, on the cross-validated test data. Compared to the recommended 0.5 score cut-off for the information theory approach, the SVM is clearly superior, although this earlier classifier was trained on a considerably reduced dataset compared to the current version. Indeed, the sensitivity of the information theory predictor only achieves 0.02 on the current dataset, which suggests that the 0.5 cut-off needs revising. It is also worth noting that the original predictor was designed for a different task: namely, predicting clearly missed cleavage cases with high PPV (rather than good sensitivity). Indeed, optimizing the SM − SC threshold score used for the information theoretic method on the current test dataset to maximize sensitivity×PPV results in a lower threshold of −0.12, which generates a considerably improved sensitivity of 0.70 with a PPV of 0.88. This is still inferior to the SVM predictor, however. The precision-recall plot in Figure 4A shows that the SVM predictor achieves a higher PPV at any given sensitivity than the information theory approach for the test data, demonstrating its superiority.
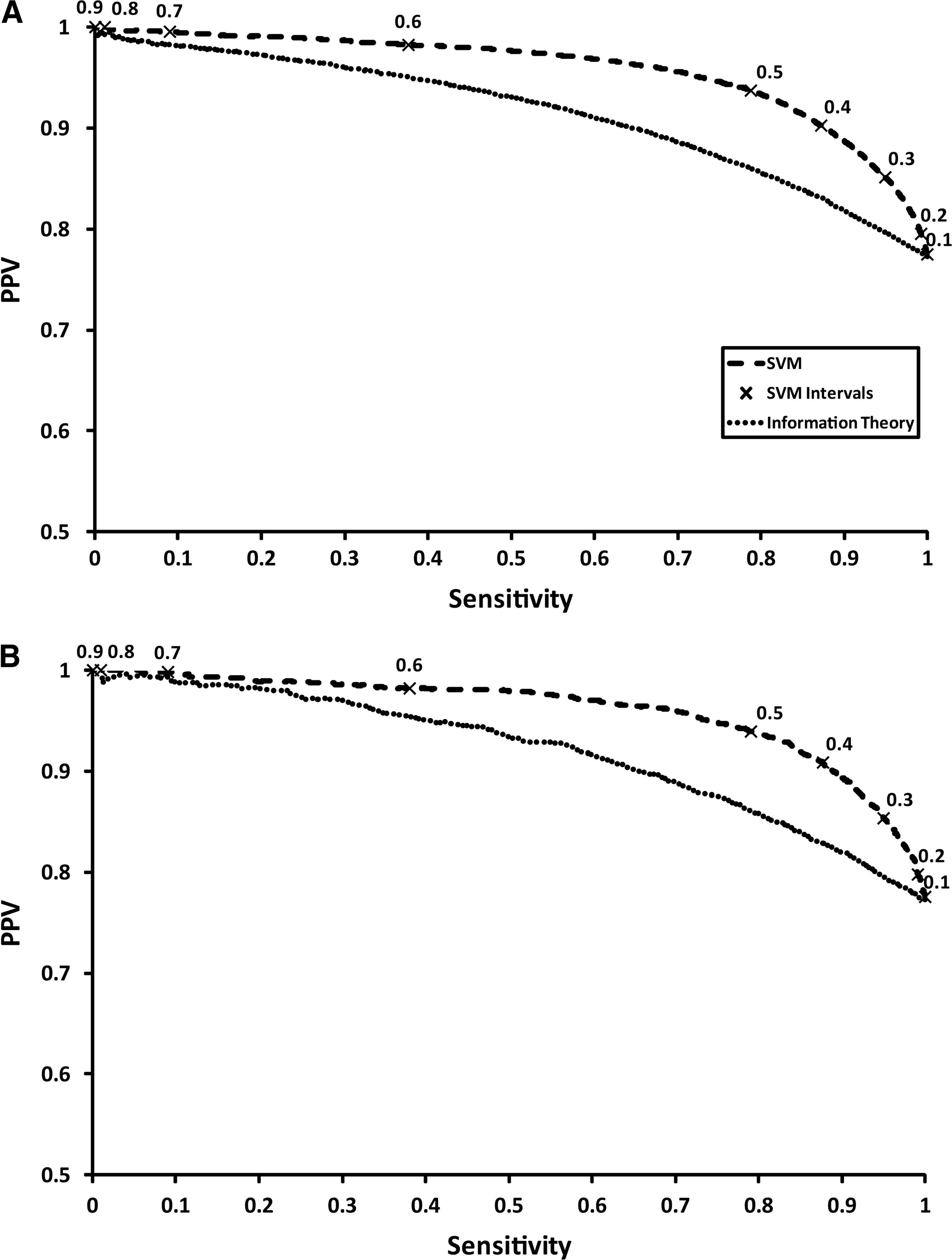
Precision-recall plots showing the performance of both the support vector machine (SVM) predictor and the information theory approach for the test data (
SN, sensitivity; AUROC, area under the ROC curve; PPV, positive predicted value.
The SVM predictor performance on the validation dataset is marginally better than on the test data, suggesting that the SVM has not been over-trained and has produced a good generalized model. The marginal improvement of the validation data over the test data is noticeable for the specificity, increasing to 0.83 from 0.82. Again, the information theory approach appears to be a poorer performer using the recommended threshold (see Table 2, validation data). Using the SM – SC threshold optimized on the test data (−0.12), a sensitivity of 0.70 and PPV of 0.89 are reached (data not shown). Despite this, the SVM can still outperform the information theory approach (Fig. 4B).
The trained SVM provides the distance from the hyperplane for each prediction as part of its output, and we assume that the distance from the hyperplane is indicative of a more confident class prediction. In order to provide a more meaningful and intuitive score for end users, the hyperplane distances are scaled from 0 to 1. For both the test data and validation data, performance statistics were plotted on Figure 4A and B for scaled hyperplane intervals from 0.1 to 0.9. As the score increases the PPV increases, but sensitivity is sacrificed. For the purposes of peptide selection for quantitation experiments, a slight over-prediction of missed sites is preferable, permitting some fully cleaved sites to be mispredicted (false-positives); the opposite is certainly more unfavorable. With this in mind a reduction in PPV can be tolerated in favor of an increase in sensitivity. A hyperplane threshold of 0.4 provides high sensitivity >0.85, while still retaining a PPV>0.9. If one would prefer to be more accurate on the missed cleavage prediction the threshold need only be raised, bearing in mind that a portion of real missed cleavages will be wrongly predicted to be cleaved.
Discussion
We report here a novel prediction algorithm that is able to identify candidate tryptic sites likely to lead to incomplete proteolysis with high accuracy, achieving PPVs over 94% at high sensitivity. This tool has been made available to the proteomics community, and we believe it has utility for groups aiming to select candidate surrogate peptides for quantitative proteomics experiments.
The success of this predictor runs parallel to ongoing experimental studies that are attempting to examine the kinetics of incomplete proteolysis for a number of relevant exemplar cases (Brownridge and Beynon, 2011; Brownridge et al., 2011). These studies highlight the effect that dibasic sites can have on the kinetics of cleavage, and importantly rationalize the results for interspersed dibasic sites, where there is a gap between the lysine/arginine and the P1 site. Importantly, the inability of trypsin to act as a dipeptidyl peptidase, cleaving peptides at the N-terminus, are reported. These trends are clearly represented in our data, as shown in Figure 2, where the marked arginine/lysine propensities for missed cleavage are present on the non-prime, N-terminal side, in complete accordance with Brownridge and Beynon (Brownridge and Beynon, 2011). Indeed, both of these studies were motivated by a need to improve our understanding of (in)complete proteolysis in the context of selecting surrogate peptides for absolute quantitation of an entire proteome (Brownridge et al., 2011). For this ambitious and challenging project to quantify the yeast proteome, we have observed peptides that are apparently susceptible to missed cleavage, and see direct evidence that incomplete cleavage can lead to inaccurate quantitation estimates. This was particularly notable in our early designs, and our current approach has been modified to consider these effects directly when selecting Q-peptides for QconCATs. We note that approximately 25% of the candidate peptides available in the yeast proteome between 6 and 30 amino acids in length have a dibasic context at one or both of the N- and C-terminal scissile bonds, highlighting the widespread significance of this phenomenon.
Although the prediction tool performs well, it is worth remembering that missed cleavage is not a discrete concept, and that in reality an equilibrium will exist between cleaved/uncleaved states for most susceptible bonds. We were mindful of this when designing our datasets, considering any evidence that a site could be missed as evidence for inclusion in the “missed” dataset. Given the wide variety of experimental conditions used to derive the raw data, there will naturally be some variance beyond our ability to control, although these experiments all sought to digest proteins to completion. The scaled hyperplane distance is therefore a useful metric to rank predicted missed tryptic sites, with larger scores reflecting sites that are more difficult to digest, and the converse for those that are easily cleaved. The full extent of this relationship, however, requires further experimental validation.
Prediction of missed cleavage can have other important applications in experimental proteomics. It has already been used by other groups for improvement of peptide mass fingerprinting scoring (Li et al., 2011; Siepen et al., 2007), and indeed we used it ourselves for the prediction of quantotypic peptides (Eyers et al., 2011). Since this latter method essentially attempts to predict the “detectability” of peptide ions in the mass spectrometer, it could also have applications in label-free quantitation techniques, which normalize spectral counts based on the likelihood of observing a given peptide, such as APEX (Lu et al., 2007).
We suggest some guidelines for the use of our predictor, based on our experience of selecting and designing peptides for incorporation into QconCATs for absolute quantification (Pratt et al., 2006), but will be equally applicable for AQUA approaches (Gerber et al., 2003). Clearly, an absence of missed cleavages is desirable in both the surrogate peptides and the endogenous protein. In the case of the latter, missed cleavages can be generated from two candidate bonds in the endogenous protein surrounding the peptide used for quantitation. In our protocol for QconCATs, peptides are typically selected based on a set of composition filters, avoiding unwanted modifications and fragmentation pathways. The remaining peptides are then ranked by whether they have been previously observed (Deutsch, 2010), and on their predicted detectability (Eyers et al., 2011), in order to select the best possible candidate for the proteins of interest. The ability to predict a peptide's “cleavability” provides an additional filter to rank candidate peptides. The ideal peptide would therefore have the maximal detectability and cleavability. The top-ranked peptide by detectability (i.e., proteotypic) may have poor tryptic contexts, and thus despite being readily detected via mass spectrometry, will lead to lower abundance estimates, as the signal is attenuated through inefficient cleavage of the scissile bonds. This is an important distinction between proteotypic and quantotypic. The former is well served by several good proteotypic predictors (Fusaro et al., 2009; Mallick et al., 2007; Tang et al., 2006; Webb-Robertson et al., 2010), but these tools make no direct inclusion of missed cleavage.
In the case of QconCATs, the predictor has a further application, in the physical Q-peptide order within the construct. A missed cleavage occurring within the synthetic QconCAT protein would essentially result in an overestimate of the analyte peptide, as the reference peptide would be at a lower abundance than the level of QconCAT spiked in. In an attempt to avoid this, the predictor can be used to find the order of peptides providing the optimum cleavage efficiency across the entire QconCAT.
Footnotes
Acknowledgments
This work was supported by the Biotechnology and Biological Sciences Research Council, via several grants to S.J.H. (BB/G009058/1 and BB/I000631/1). We would also like to thank Phillip Brownridge and Rob Beynon at the University of Liverpool for useful discussions and support.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.