
Abstract
Abstract
Enabled by diverse high-throughput technologies, the rapidly evolving field of “-omics sciences” offers the potential to study health and disease in breadth and depth at the human population level. We have recently linked genomics and metabolomics to present the first genome-wide association study of metabolic traits in human urine providing new insights into the functional background of chronic kidney disease. We propose systems epidemiology as a novel approach to study the complexities of human pathophysiology by integrating various population-level omic-metrics and to identify new trans-omic biomarkers.
Introduction
Systems Epidemiology
Recent advances in high-throughput -omic platforms such as expression arrays and mass spectrometry, with their exquisite sensitivity, and specificity, have led to the possibility of accumulating a wealth of genetic, transcriptomic, proteomic, and metabolomic data to study health and disease in breadth and depth at the human population level. Based on the increased amount of detail available to describe an individual phenotype, we propose systems epidemiology as a new research field that integrates -omics together with physiological, epidemiological, and environmental data to create a systems network that can be used to predictively model multilevel causes of health and disease (Fig. 1). Further, the combination of complementary -omic levels could be implemented for the identification of novel trans-omic prognostic and diagnostic biomarkers. Therefore, we do think that deep phenotyping (Tracy, 2008)—the comprehensive and thorough description of the physical state of an individual—will be the corresponding principle of systems epidemiology on a population level, to lay the foundation for the analyses of dynamic feedback and interaction patterns among its multiple levels (Fig. 1).
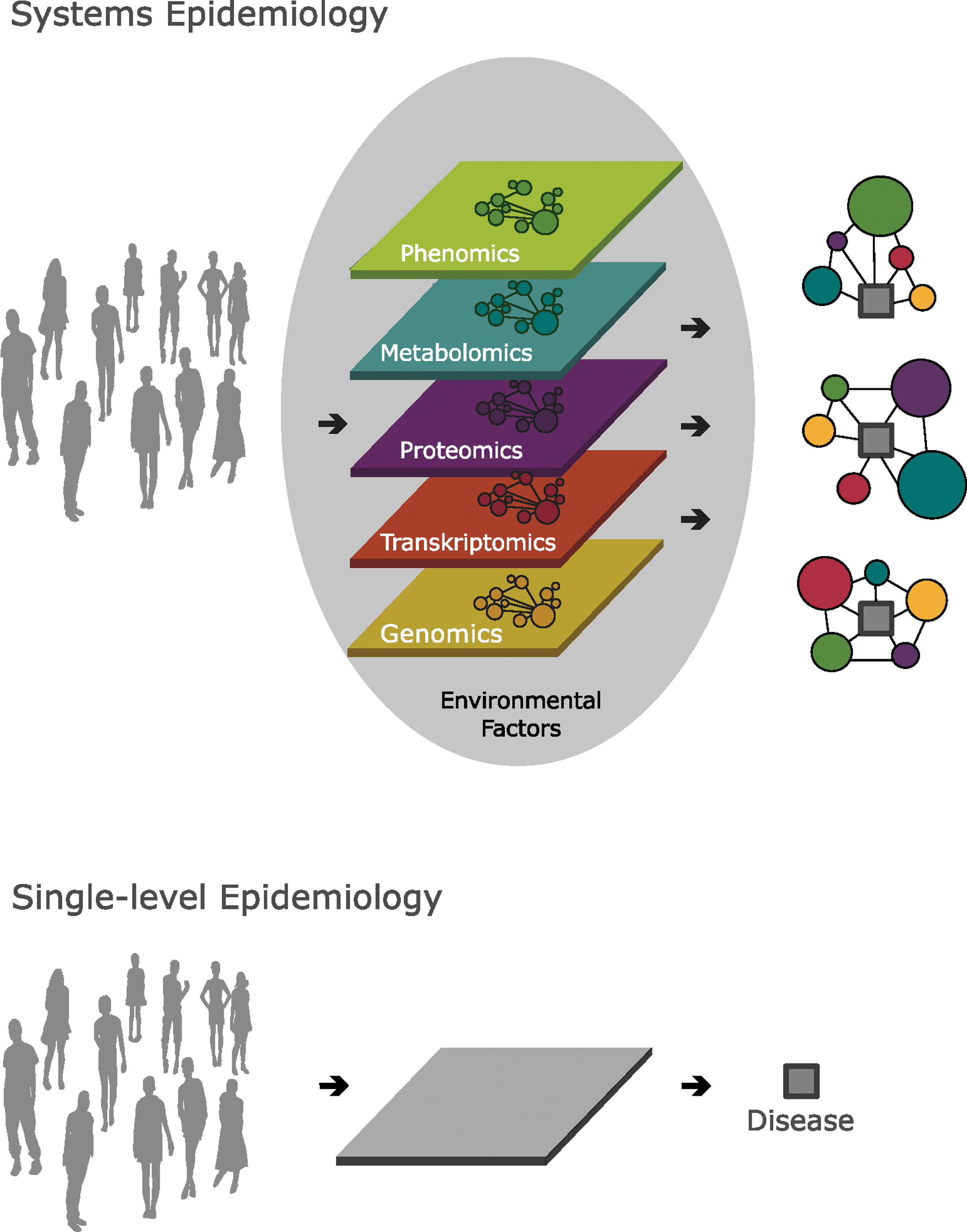
Systems epidemiology versus the classic single-level paradigm to study health and disease at the human population level. Integrating various population-level omic-metrics including the
Moreover, the here proposed systems epidemiology approach does not only concern the measurement of the molecular underpinnings of human disease, but also multiple environmental interaction components including behavioural, sociodemographic, and group levels that may influence health and disease. When considering human health in a wider perspective, it is clear that most major diseases are subject to environmental influences. For example, obesity has been recently reported to cluster in communities such that friends have an even more important effect on an individual's risk of obesity than genes do (Christakis and Fowler, 2007). Although epidemiology may be described as an effort in measurement, systems epidemiology could turn out as synthesis after measurement; an advanced pattern recognition approach integrating various -omics level (Fig. 1). This is not to say that epidemiology would relax its historic focus on populations, but need to absorb and apply the advancing scientific understanding at molecular and cellular levels to the study of health and disease in human populations.
If one accepts the case for deep phenotyping, including the limitation that increased costs will necessarily yield smaller sample sizes, there are at least three study design strategies that might be employed in systems epidemiology. The first involves a longitudinal study design making multiple measurements over time with fairly short time periods between measurements. Data, for example obtained yearly over 10 years, would enable the investigator to closely monitor subclinical disease progression and to detect dynamic changes in the nature of the phenotype over time. A second operational approach accounts for the fact that for many biomarkers the within-subject variability is larger compared to the change in the biomarker over time. Therefore, a mean biomarker value calculated based on two or three blood draws spread over the day are likely to eliminate this within-subject variability. Finally, the case of deep phenotyping is likely to reduce the inaccuracy and misclassification of disease outcomes present in most epidemiological and clinical studies, by increasing an individual's phenotypic information and refining risk classification.
Network analytic methods provide the computational framework for data integration and biomarker selection in systems epidemiology (Adourian et al., 2008). For example, network-based computational approaches and longitudinal data from the Framingham Heart Study revealed a number of surprising insights into the dynamics of smoking (Christakis and Fowler, 2008), development of obesity (Christakis and Fowler, 2007), and metabolic determinants of diabetes risk (Wang et al., 2011). Similarly, network-based analyses of known disease-gene associations revealed a number of surprising connections between diseases, forcing us to rethink apparently distinct pathophenotypes and their nomenclature (Goh et al., 2007). Taken together, the proposed integration of population-level omic-metrics promises to advance our understanding of human disease (Barabasi, 2007; Barabasi, et al., 2011; Loscalzo et al., 2007) and to enable the identification of new trans-omic biomarkers (Rantalainen et al., 2006).
We recently provided a proof of concept for the integration of population-level omic-metrics employed in systems epidemiology using genomic and metabolomic data for the first genome-wide association study of metabolic traits in human urine (Suhre et al., 2011). Through the identification of genetic variants related to metabolism, specific “genetically determined metabotypes” have the potential to uncover additional risk factors for common diseases and may provide new insights into the pathophysiology of these diseases. Using nuclear magnetic resonance spectroscopy to quantify 59 metabolites in urine from 862 male participants of the population-based epidemiological Study of Health in Pomerania (SHIP), we identified genetic variants that have been previously linked to important clinical outcomes including chronic kidney disease and coronary artery disease. The revealed plausible relationships between the associating metabolic traits and the genetic variants' encoded protein functions provided new insights into the metabolic basis and functional background of related pathophysiological processes. Thus, the study of genotype-dependent metabolic phenotypes may provide new functional insights for many disease-related associations and may constitute potential trans-omic biomarkers for diagnosis and monitoring (Suhre et al., 2011).
Challenges
Although disease manifestations have been shown to be derived via different pathways in different individuals, epidemiological research using multiple phenotypic levels to study human disease is scarce. Especially results from genome-wide association studies, perhaps the most vibrant research field of the last 5 years, have thus far proven to be surprisingly disappointing, partly because of the unexpected complexity of the human genome and the difficulties in accurately and unequivocally describing human phenotypes (Maher, 2008). Furthermore, biological systems exhibit robustness and dynamic stability where phenotypic changes are fairly resistant to scattered omic-level fluctuations (Hillenmeyer et al., 2008). Thus, competing risks are buffered by regulatory networks that use alternative mechanisms to ensure phenotypic stability (Nobrega et al., 2004). For example, competing risk patterns emerging from the nearly 600 genome-wide association studies reported nearly 800 significant single nucleotide polymorphisms (SNP)-trait associations with few variants having large effects, but most having small effects (Manolio, 2010).
Another important challenge may be the lack of biomarker standardization and harmonization. As advanced computational modeling techniques are used, well-integrated platforms and data sets are required to conquer the intersection of highly dynamic parameters and to assess the relationships between omic-level metrics and their phenotypic manifestations (Connor et al., 2010). Furthermore, the proposed study design enables a dynamic exposure assessment requiring a close iteration between experimental data input and theoretical modelling (Kohl et al., 2010). For instance, it is increasingly recognized that the understanding of complex metabolic and cardiovascular diseases or cancer requires an integrated analyses of its molecular and cellular components, as well as their relationships, pathways, and interconnectivity. To address the risk of increased data noise and false positive findings in this extremely data-rich research environment resulting from high-throughput multiomics technology, the suggested application of high-performance computing technologies may facilitate high-volume data analysis and close the scientific gap between increasing amount of data, correlation identification, and plausible causal pathways (An, 2010).
Conclusions
The network-based integration of deep phenotypes in systems epidemiology may provide a novel approach to study the complexities of human pathophysiology, and to account for the paradox of ever-increasing measurement capabilities followed by decreasing abilities to translate basic mechanistic knowledge into clinically effective therapeutics (An, 2010; Lenfant, 2003). Thus, systems epidemiology aims to account for the large variability of interindividual disease onset, manifestation, and progression and may thereby support an individualized medicine grounded on a multiscale and nonreductionist analytical approach.
Footnotes
Acknowledgments
I would like to express my sincere gratitude to Prof. M.D. Ramachandran S. Vasan for his generous support and guidance in the process of this manuscript. The authors did not receive any specific funding to write this article.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.