
Abstract
With the advancement of high-throughput biotechnologies, we increasingly accumulate biomedical data about diseases, especially cancer. There is a need for computational models and methods to sift through, integrate, and extract new knowledge from the diverse available data, to improve the mechanistic understanding of diseases and patient care. To uncover molecular mechanisms and drug indications for specific cancer types, we develop an integrative framework able to harness a wide range of diverse molecular and pan-cancer data. We show that our approach outperforms the competing methods and can identify new associations. Furthermore, it captures the underlying biology predictive of drug response. Through the joint integration of data sources, our framework can also uncover links between cancer types and molecular entities for which no prior knowledge is available. Our new framework is flexible and can be easily reformulated to study any biomedical problem.
Introduction
Over 18 million new cases of cancer and 9 million deaths were recorded worldwide in 2018. 1 This makes cancer one of the leading causes of death. Cancer is a multi-faceted, complex disease arising from an accumulation of somatic mutations within the genome of normal cells that eventually leads to loss of normal cellular functioning and appearance of tumors that can spread across the body. Technological advances have enabled measurements from patients' tumor biopsies, including gene expression levels, DNA methylations, and somatic mutations. The research into cancer causes, and treatments, has greatly benefited from this wealth of patient data.2,3
Cancer projects, including The Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium (ICGC), have made publicly available wide-ranging, multi-modal, multi-omics cancer data, such as patient whole slide images, genome alterations, transcriptome, and epigenome.4,5 Free access to these large-scale, diverse databases has dramatically facilitated studies of the biological mechanisms of specific cancer types.4,6,7 The available data have also enabled pan-cancer analyses that study cancer in general to identify common mechanisms and differences across cancer types.7,8 Recently, the Pan-Cancer Analysis of Whole Genome (PCAWG) project 7 has informed that our knowledge about cancer is far from complete, as 5% of their cohort was without any known cancer driver mutations. Importantly, these large databases have paved the way for the field of Precision Medicine, whose overarching aim is to improve medical care for patients by tailoring treatment to their individual molecular profiles. 9 Precision medicine has diverse intermediary objectives, for instance, uncovering diagnostic and prognostic biomarkers. This is especially relevant to a heterogeneous disease, such as cancer, which manifests uniquely in every patient.
Complex diseases, such as cancer, can be caused by combinations of genetic, molecular, environmental, and lifestyle factors. Any single type of biological data cannot fully capture such diseases. As such, collective mining of different data has been gaining momentum as a means to extract integrated system knowledge that goes beyond what any single data source can offer. 10 This principle applied to the study of cancer has enabled the discovery of cancer-related genes, or group of genes,11–13 and the identification of cancer subtypes significantly correlated with patient prognoses.12,14
Biological data often have a small number of samples relative to the number of available features. For instance, a typical dataset in TCGA contains a few 100 patients who are each characterized by tens of thousands of features (e.g., expression levels of around 20,000 genes). However, biological features are often redundant due to underlying molecular interactions among biological entities. 15 This has been a motivation for the use of dimensionality reduction and embedding algorithms that are pervasive in bioinformatics. 16 In addition, due to the low sample to features ratio, dimensionality reduction techniques are often necessary as data pre-processing for machine learning models. 16
Non-negative matrix factorization (NMF) approaches are unsupervised algorithms that have extensively been used both as a means to integrate heterogeneous data and reduce data dimensionality. They encompass all methods that decompose a matrix, representing relational links between two sets of entities, into the product of low-dimensional, latent, positive matrices, or factors, whose sizes control the degree of dimensionality reduction. 17 Importantly, they can be used to derive an embedding in an unspecified latent space for each entity. Matrix factorization approaches have had numerous applications, including collaborative filtering 18 and biological data integration for cancer analysis.12,14 Reconstructing a matrix based on a factorization has often been used to make predictions and infer new knowledge. 12 NMF approaches have been successfully applied as pre-processing steps for downstream machine learning classifiers. 19
We propose a pan-cancer framework to uncover cancer type-specific molecular mechanisms and identify drugs that could be repurposed (Fig. 1). Our framework relies on the simultaneous integration and dimensionality reduction of various data using a joint NMF model. Our framework includes more data than the previous studies, integrating patient-specific diagnosis, gene expression, and single-nucleotide variants, (SNV) as well as generic network data on human: protein–protein interactions, protein complex associations, biological pathways, drug–target interactions, and drug chemical similarities. To integrate the wealth of data in one framework, we rely on three types of matrix factorizations: NMF, non-negative matrix tri-factorization (NMTF), and symmetric non-negative matrix tri-factorization (SNMTF). The details for each can be found in Supplementary Methods section. Data integration is achieved by jointly optimizing for multiple factorization objectives with shared factors (see Supplementary Methods section). We obtain a context-aware embedding of each entity (cancer type, patient, gene, complex, pathway, and drug) that takes into account all the input data.

Using boosted decision trees, we predict biologically relevant associations between cancer types and genes, drugs, pathways, and complexes based on the context-rich embeddings of our entities. One key insight is that the integration step, by construction, embeds the entities into three latent spaces, each associated with a different family of entities: (1) patient-related entities (i.e., patients and cancer types), (2) gene-related entities (i.e., genes, complexes, and pathways), and (3) drugs. This means that the entities in a given latent space can be substituted with each other when using a model trained to predict associations between one of these classes of entities and cancer types. In this respect, our approach is similar to zero-shot learning, 20 which aims to accurately classify at test time samples that belong to classes unseen at training time. In our case, we aim to predict the association of cancer types to unseen classes of entities at training time. Finally, our approach is able to predict patient's response to drugs, implying that our framework captures important biology that governs response to cancer drugs.
Materials and Methods
Our framework for context-aware embeddings
The core of the framework is gene information (Fig. 1a). We integrate three types of data about genes. We obtain RNA sequencing data and SNV data for 7998 patients from ICGC (see Supplementary Methods section for details) across 21 cancers. Henceforth, we refer to each cancer by its abbreviation given in Supplementary Table S1. We obtain data on gene interactions, including protein–protein interaction (PPI) network from Biogrid, protein complexes from Reactome and Corum, and biological pathways from Reactome (see Supplementary Methods section for details). These data capture physical and functional relationships between genes and are used to anchor our framework within the context of molecular interactions. The last type of gene data corresponds to drug–target interactions from DrugBank (see Supplementary Methods section for details), connecting drugs to proteins that they target. We further add drug chemical similarity information to push similar drugs closer in the latent space. We also add patient diagnosis information through which we embed cancer types and patients in a joint latent space to both push patients closer if they have the same cancer and push molecularly similar cancer types closer. This could help tailor treatments to patients by placing them within a cancer “space” since cancer is a heterogeneous disease and a given cancer type might manifest differently in different people. This may aid characterizing cancer of each patient as accurately as possible to personalize treatment options.
Because of the heterogeneity of our input data, our integration framework is based on joint optimization of different variants of NMF: classical NMF, NMTF, and SNMTF. Each variant, described in Supplementary Methods section, is best fitted for the decomposition of a different type of relational data. In particular, we use SNMTF to factories the PPI network and the drug similarities matrix, NMTF to factorize patient molecular data and drug–target data, and NMF for the remaining data. Each edge in Figure 1a corresponds to a subobjective of our embedding framework, that is, a specific NMF decomposition. In the joint decomposition, each group of entities is associated with a factor that is shared across all subobjectives involving that group of entities. For instance, the patient factor is shared by all subobjectives that involves patient-specific data (diagnoses, gene expressions, and somatic mutations). An entity's embedding is obtained from the factor of the associated group of entities.
Through our integrative framework, we derive embeddings for all entities (cancer types, patients, genes, pathways, complexes, and drugs) that best fit the full context of the framework, that is, the input relational data. Each entity's embedding, in one of the three latent spaces learnt by our framework, encapsulates the information from the input data that is relevant to that entity; thus, we say that this representation is context aware. Our framework has three hyperparameters, denoted by k1, k2, and k3, which correspond to the dimensionalities of the latent spaces. To find suitable values for these hyperparameters, we perform a grid search with
As the selection criterion, we measure if each patient tends to be embedded in the latent space closer to their diagnosis than to other cancer types. We quantify this with the macro-F1 score of the classifier that associates to each patient the closest cancer type in the latent space in terms of cosine distance. We found that the following hyperparameters values maximize this metric:
Each iteration of our integrative model takes around 10 seconds, depending on hyperparameters, on 8 CPUs (Intel Xeon E5-2650 v3 @ 2.30 GHz) with 16G of RAM. With n denoting the total number of entities, our matrix factorization-based framework has time complexity
Predicting cancer type associations
To extract new knowledge for each cancer type, we use our context-aware embeddings to suggest cancer–drug and cancer–gene associations. We cast the problem as a link prediction task for which we train boosted decision trees to predict known associations from our entities' embeddings. After our training step, we use the trained classifiers to predict new associations (see Supplementary Methods section). As pre-processing, we normalize all embeddings to have a unit norm. The normalization step is crucial for the transfer of a link predictor from one type of entity to another that we discuss in the next section. For each possible cancer–drug pair (or cancer–gene pair), we define the pair's representation by the concatenation of the embeddings of its components, that is, the concatenation of the cancer's embedding vector with the drug's embedding vector defines the feature vector of the pair. Finally, we use boosted decision trees for link prediction, taking as input a pair's representation and output the association's scores of its component (Fig. 1c). We choose boosted decision trees due to their simplicity and high performances in a number of competitions. 22
In the first validation step, we systematically evaluate the performance of our approach with a 10-fold cross-validation using both the area under the receiver operating characteristic (AUROC) and the area under the precision recall curve (AUPRC) and compare our results to state-of-the-art methods for link prediction. The two metrics are often used in concert as they characterize different aspects of the results. Notably, AUPRC gives robust evaluations in imbalanced settings. The splits used for the 10-fold cross-validation are performed on the set of known links and considering all nonreported links as part of the negative set of links. Note that we do not use any balancing strategy during training. Furthermore, we perform an ablation study on patient–gene data (Supplementary Fig. S6), that is, we compare the results obtained with those obtained with framework using less patient data to demonstrate the interest of considering both expression and mutation data jointly.
In the second step, we investigate the top 10 drugs and genes associated with cancer types by our methodology. Each pair is scored based on the average of the standardized scores given by 10 classifiers trained for the cross-validation (see Supplementary Methods section for details). In this step, we only consider drugs and genes that were thus far not associated with any cancer type in the ground-truth data (introduced in each subsection) to avoid trivial cases of information transfer from one cancer to another, which typically happens when one drug or one gene is associated with a majority of cancers. We perform a manual literature curation to validate the top results.
Results and Discussion
Patient and cancer embeddings are medically relevant
To evaluate the biomedical relevance of our joint patient and cancer embeddings, we observe that the macro-F1 score is close to 0.8 for our optimal set of hyperparameters (Fig. 2a), indicating that the majority of patients are embedded closer to their diagnoses than to other cancer types. In addition, we evaluate if patients group in the latent space with respect to either cancer type or a sampled tissue. To this end, we use hierarchical clustering with cosine distance to group patients in k groups (where k is either the number of cancers or the number of tissues) and compute the adjusted rand index (ARI) to measure the link between the clustering and the ground truth labeling (either cancer types or sampled tissues; see Fig. 2b). We observe that patients do not cluster well with respect to sampled tissues, having ARI below 0.2. However, we observe ARI 0.7 with respect to cancer type, indicating that our clusterings resemble diagnostic labeling with some discrepancies. These results are expected, as the inclusion of patient diagnosis data in the framework implies a constraint that aims to embed each patient close to their diagnosis and subsequently to other patients having the same disease. Note, however, that some patients do not fit well with the rest of their cohorts. This is an important observation, as it suggests that those patients might need different care options from the majority of their cohort and further motivates personalizing treatments to individual patients.

As an illustration, we visualize our latent space embeddings using T-distributed stochastic neighbor embedding (t-SNE). t-SNE is a machine learning algorithm for nonlinear dimensionality reduction, well suited for visualization in a two-dimensional space of high-dimensional data. 23 We observe as expected that patients tend to cluster according to cancer type, with the cancer type itself being also embedded nearby (Fig. 2c). In addition, we observe that some cancers are grouped in a meaningful way. For instance, both brain cancers, glioblastoma multiforme and lower grade glioma, form one group. The cluster in the center contains mostly squamous cell carcinomas, head and neck squamous cell carcinoma, cervical squamous cell carcinoma, and lung squamous cell carcinoma (LUSC). Both cancers that affect kidneys, kidney renal papillary cell carcinoma and kidney renal clear cell carcinoma, are also grouped. Moreover, rectum adenocarcinoma, colon adenocarcinoma, and stomach adenocarcinoma—which are cancers affecting rectum, colon, and stomach, respectively—form another cluster. We also observe that some patients having a specific type of cancer do not group with the majority of the cohort. Note that the same observations hold when using Uniform Manifold Approximation and Projection algorithm 24 instead of t-SNE (Supplementary Fig. S7).
We further investigate if our framework learns a meaningful latent space that translates into actionable representations for new unseen patients. We perform a 10-fold cross-validation in which 90% of patients of each cancer type are used to derive the embeddings of all entities in the framework. We then project the remaining 10% of patients in the derived cancer/patient latent space (see Supplementary Methods section). First, we test if these patients are placed close in space to their diagnosis (quantified as above, see Supplementary Table S5). This gives a macro-F1 score of
We also test if the unseen patients tend to be embedded closer to patients having the same diagnoses. For this, we use a k-nearest neighbor classifier with
Overall, our analysis shows that the patients/cancers latent space is consistent with known biology. Furthermore, our framework has the advantage of relaxing the hard clustering derived from patients diagnoses through patient's molecular similarity, highlighting that a patient's molecular profile can be more similar to the profiles of patients with different cancers than to the profiles of patients with the same diagnosis. This observation motivates further the need for pan-cancer perspectives in precision medicine.
Our gene latent space is biologically relevant
To evaluate the biological relevance of our genes embeddings, we cluster them in k2 group using cosine distance-based hierarchical clustering, and measure the enrichments of the clusters in terms of Gene Ontology (GO) annotations and in terms of cancer driver genes (see Supplementary Methods section for details, Supplementary Table S3). We consider all three subtypes of GO annotations: Biological Processes (GO-BP), Cellular Component (GO-CC), and Molecular Function (GO-MF), separately. The significance of the enrichments is computed with a hypergeometric test with Benjamini-Hochberg correction for multiple hypothesis testing and a significance threshold of 0.05. We observe that, regardless of the GO subtype, above 80% of clusters are significantly enriched in at least one annotation (Fig. 2c and Supplementary Fig. S2 for cluster size and number of enriched annotations).
We further evaluate the meaningfulness of our results by performing 10,000 randomized tests where clusters' sizes are held constant, but genes are randomly assigned. We obtain 0% of clusters enriched with random cluster assignment at least 9650 times out of 10,000 repeats (empirical p-values 0.035) for each type of annotation. These results show that genes with similar function are embedded closer in the latent space and thus our genes' embeddings significantly capture known biology. Interestingly, we also observe that around 10% of the clusters are enriched in cancer driver genes, indicating that cancer drivers are embedded closely, that is, clustered, in the latent space. This highlights the link between the gene latent space and the cancer context that we made a part of our framework. Furthermore, it underlines the relevance of our embeddings for the identification of putative cancer-related genes, discussed in the following section.
In addition, we perform an ablation study on the gene interaction data input to investigate the effect that each dataset has on enrichment scores (Supplementary Fig. S3). For each model, all hyperparameters are selected following the same procedure outlined above. First, we observe that adding any gene data is better than not adding them from the point of biological annotation enrichment. For instance, the model without any PPI, complex, or pathway data has 40% of gene clusters enriched in GO-BP annotations, while every model with at least one data source has above 80% of gene clusters enriched for the same annotations. However, there is no clear best model among the ones with diverse combinations of the data, each scoring similar enrichment values with different models performing slightly better for different annotations. Thus, different combinations of data do not seem to lead to significantly different performances, but keeping all data in the model enables analysis of each class of entities.
Finally, as pathways and complexes are embedded in the same latent space, we investigate their positioning with respect to genes. In particular, we evaluate if a gene is embedded closer to its associated higher-order entities, that is, pathways and complexes, than to those to which it has not been associated yet. To this end, we compute the cosine distances in the latent space between a gene and its associated pathways and complexes, termed “intrapathway” and “intracomplex” distances, as well as the distances between the gene and all nonassociated pathways and complexes, termed “exo-pathway” and “exo-complex” distances. We observe that genes are embedded closer, on average, to their associated higher-order entities than they are to those that they are not associated to (Fig. 2d), with average distance below 0.5 between a gene and associated entities and above 0.9 between a gene and nonassociated entities. These results are significant according to a Mann–Whitney U statistical test (p-value
Cancer type associations
Our model predicts relevant treatment
To predict cancer–drug associations, we train decision trees to identify known associations that we collect from DrugCentral 25 (last updated October 2018). DrugCentral contains 93 associations in total between our sets of cancer types and drugs. We define our positive set with DrugCentral treatment options and consider all nonreported associations for our negative set. Our classifier takes as input the concatenation of the normalized embeddings of a drug and a cancer type and outputs their association score.
We compare our results to four baseline methods: non-negative matrix factorization reconstruction (NMFR), measure-based bidirectional random walks (MBiRW), 26 Drug Repositioning Recommendation System (DRRS), 27 and bounded nuclear norm regularization (BNNR) 28 (see Supplementary Methods section for implementation details). These methods rely on the assumption that similar diseases share treatment, and conversely, that similar drugs can treat the same disease. MBiRW uses random walks to derive repurposing hypothesis by discovering neighborhoods in similarity networks. DRRS and BNNR embed drugs and diseases in low-dimensional latent spaces with the underlying hypothesis being that missing links emerge from those latent spaces using the inner product. Our method makes similar assumptions, but takes more information into consideration, notably drug targets as well as protein interactions. Furthermore, by using boosted decision trees on the latent spaces, our model gains more flexibility and representative power over the simple inner product.
We observe that our approach significantly outperforms the competing methods (Fig. 3a). BNNR achieves slightly better AUROC scores (
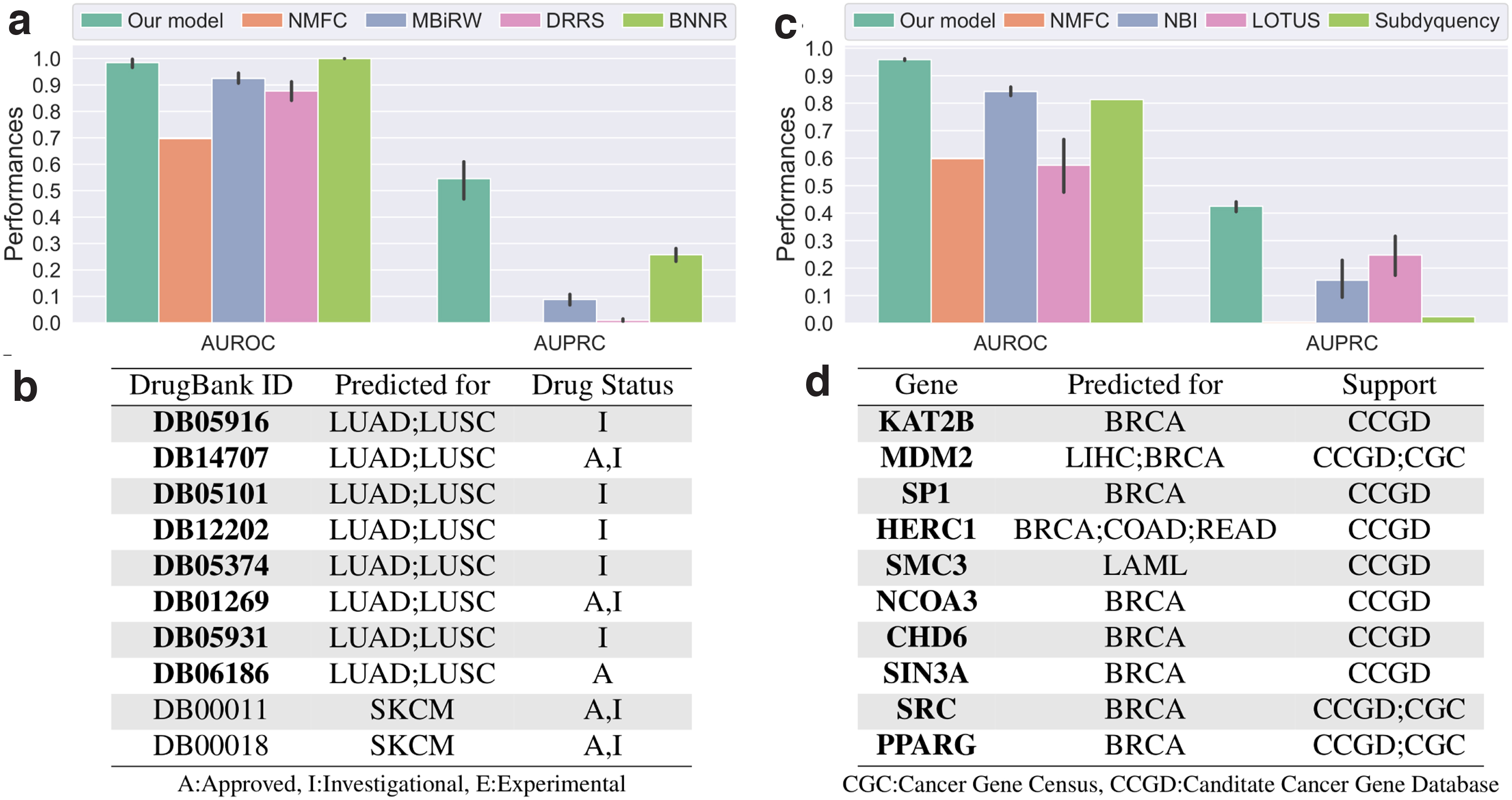
Performances of our cancer–drug association predictor (left column) and cancer–gene association predictor (right column). The bar charts give the performances of our classifiers measured with 10-fold cross-validation in terms of AUROC and AUPRC. The tables give the top 10 associations between cancers and drugs
Among the top 10 drugs that are the most associated to cancer types by our classifiers (Fig. 3b), a majority is recorded in DrugBank as investigational or approved for the treatment of some cancers. The approved predicted drugs either are not present in DrugCentral, as their approval postdates the DrugCentral release, or target a cancer type not considered in this study. We discuss below supporting information for our top 3 predicted drugs. We provide validations of all of our predictions in the Supplementary Data.
DB05916 (CT-011) targets gene PDCD1, which has immunomodulating and antitumor activities. CT-011 is currently being investigated for the treatment of tumors and unspecified cancers. 29 DB14707 (Cemiplimab) is an FDA-approved drug for the treatment of advanced cutaneous squamous cell carcinoma. 29 Our classifier suggests that it could be used to treat lung cancer and notably LUSC. DB05101 (Matuzumab) is an investigational drug that targets EGFR gene, which is often associated with cancers, including lung cancers. 30
The manual literature curation highlights that our predicted drugs are often investigated or approved for the treatment of forms of cancer and that their targets, or mechanisms of actions, can be linked to the specific cancer types we predict. Overall, the analysis strongly supports our methodology.
Our framework identifies genes relevant to cancer types
Based on known cancer genes from IntOGen, 31 we train classifiers to identify associations between genes and cancer types. In total, IntOGen reports 1129 associations between our sets of cancer types and genes. These interactions constitute our positives set. All nonreported associations are considered part of our negatives set. As above, a decision tree takes as input the concatenation of the normalized embeddings of a gene and a cancer type and outputs their association score.
We compare the performance of our method with the following state-of-the-art methods: NMFR, network-based integration (NBI), 32 LOTUS, 33 and Subdyquency. 34 The methods were developed to predict cancer-related genes in slightly different contexts and are adapted to our problem here (see Supplementary Methods section for details). The baselines rely on the “guilt-by-associations” where genes that are linked to a disease, but are not necessarily mutated or differentially expressed, can be identified in biological networks through links to known disease genes. NBI and Subdyquency use random walks to exploit proximities on networks to identify such genes. LOTUS uses kernels to capture similarities between cancers and between genes, combining the two kernels to feed to a Support Vector Machine algorithm. Our approach draws from similar ideas, combining both factorization to augment entity proximities through latent space embedding and machine learning model. Furthermore, our approach can integrate additional protein interaction data, notably functional protein interaction (pathways).
We observe that our approach outperforms the competing methods (Fig. 3c) in terms of AUROC, which is over 0.9 for our method compared to below 0.8 for the other approaches, and in terms of AUPRC, which are around 0.4 for our approach compared to below 0.25 for the other methods. We further evaluate if our approach accurately captures known cancer-related genes that are reported in Candidate Cancer Gene Database (CCGD), but not in IntOGen. We perform this analysis both globally and on a per cancer basis (note that 14 cancer types have data for this test). We observe that our method ranks highly associations between genes and cancer types in CCGD, notably giving AUROC scores above 0.59 (p-values
We observe that our top 10 scoring genes are listed in either the CCGD or the Cancer Gene Census (CGC) as linked to at least one form of cancer. Furthermore, the pairs MDM2–LIHC, HERC1–COAD, HERC1–READ, SMC3–LAML, NCOA3–BRCA, and CHD6–BRCA are associated in CCGD. In addition, KAT2B (PCAF) activity has been linked to cancers, and in particular, to breast cancers, in the literature.37–39 MDM2 has also been associated with breast cancer. 40 SP1 expression has been linked to breast cancer in multiple prior studies.41–43
For each of these three genes, we stratify our breast invasive carcinoma (BRCA) cohort into two groups: patients having higher than average expression of the gene and patients having lower than average expression of the gene. We compute a logrank statistical test (with 0.05 cutoff) and observe that for each of the three genes, the patient groups have significantly different survival rates with p-values 0.002 for KAT2B, 0.026 for MDM2, and 0.039 for SP1 (see Supplementary Fig. S5a–c for Kaplan-Meir plots). For each of the three genes, higher expressions are associated with lower survival rates. We provide validations for the remaining predictions in the Supplementary Data. Note that BRCA is overrepresented in the results. However, the global validation results (Supplementary Fig. S4) do not indicate a bias toward BRCA and a possible explanation for this observation is that BRCA is associated with more driver genes than the other cancer types in this study (96 driver genes compared to no more than 74 for other cancer types).
The literature curation highlights that each gene identified through our approach is relevant to the associated cancer type, with support through existing research and databases, as well as statistical evidence for a connection between gene expression level and patient prognosis. Thus, this supports our methodology.
Repurposing boosted decision trees
Links between types of entities are not always known or available (e.g., associations between cancer type and protein complexes or associations between patients and drugs), which prevents us from using the same methodology to derive new knowledge. However, our framework allows for extrapolating those links from known associations between other types of entities. Our approach relies on the previously observed fact that, by design, some entities are embedded in the same latent spaces (e.g., genes, pathways, and complexes or cancer types and diseases). We have further shown, in the first section, that the relative location in the latent space of related entities was biologically consistent, that is, related entities are closer to each other than nonrelated entities. Based on these observations, we postulate that a classifier trained from the embeddings of a given type of entities can be repurposed to predict from the embeddings of another type of entities. For instance, decision trees that learnt to associate genes to cancer types can be used to predict which biological pathways or protein complexes could be associated with which cancer types. This could effectively provide insights into the impact of cancers onto cells by identifying affected higher-order cellular structures and functions. We focus below on the analysis of top associations between biological pathways and cancer types. Similar results can be obtained for protein complexes and are discussed in Supplementary Results section and Supplementary Table S6.
The association score between a cancer type and a biological pathway is obtained by simply feeding the concatenation of the normalized embeddings of both entities to the 10 decision trees trained to predict cancer–gene associations. The average of the standardized scores across all decision trees gives the final association score.
Comparative Toxicogenomics Database (CTD) database
44
gives associations between diseases and pathways based on shared associated genes and can be used for global validation. We achieve an AUROC score of
Top 10 biological pathways associated to cancer types
BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; GBM, glioblastoma multiforme; LAML, acute myeloid leukemia; LGG, low grade glioma; LUSC, lung squamous cell carcinoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; UCEC, uterine corpus endometrial carcinoma.
MAPK1 (ERK2) activation pathway (R-HSA-112411) and MAPK3 (ERK1) activation pathway (R-HSA-110056) have been linked to numerous cancers, such as breast cancer, as discussed in the previous section, and colorectal cancer. 45 The ERK MAPK pathway is critical for cell proliferation and thus is naturally often connected to cancers. Cellular responses to stress pathway (R-HSA-2262752) is a subpathway of the cellular responses to external stimuli pathway (R-HSA-8953897). 46 Anticancer treatments are often successful when able to induce apoptosis through external stimuli that induce cellular stress. 47 For instance, tumor suppressor gene P53 can be stimulated through cellular stress. 48 Thus, perturbation to those pathways might lead to cancer onset and resilience to treatment.
The literature review highlights the ability of our repurposing approach to identify associations between biological pathways and cancer types that are supported by the existing literature. Thus, this analysis underlines the ability of our framework to extract biological pathways associated with cancer.
Predicting patients' responses to cancer drugs
We collect data on patient responses to cancer drugs from TCGA. 4 We only consider patients and drugs that are present in our dataset. The task corresponds to a binary classification where we predict if a patient's response to a drug is positive or negative. A response is considered positive if TCGA reports a complete response of the patient, and negative otherwise. As we are interested in finding drugs that improve the state rather than maintain a status quo, or degrade the state, we believe it is reasonable to use a binary variable for this task. We further discard 100 entries corresponding to combinations of drugs as our model is not suitable for the analysis of these data. From the remaining data, we only consider drugs that have both positive and negative response. After processing, we have 2589 patient–drug pairs. We split these data in train, validation, and test sets with a 70%/10%/20% partition, repeating the experiment 10 times and measuring AUROC and AUPRC scores.
We train a boosted decision tree model to predict patients' responses to drugs. As above, the input to the model is the concatenation of the normalized embedding of a patient and a drug. The output can be interpreted as the success probability of the treatment. Our approach performs well, achieving AUROC score of
To analyze this claim further, we investigate which features the models use most to predict response. First, we compute the gain, that is, the relative importance, associated with each feature in each 1 of the 10 models trained. For each feature, we take the average gain across models as a final feature importance score. Note that we have both patient and drug features; thus, we have two vectors of feature importance scores,
Top 5 genes and biological pathways associated to drug response prediction-based feature importance in both drug and patient spaces
Interestingly, eight of the genes identified in Table 2 have been linked to cancer response to general, or specific treatments (HSP90AA1, 49 PIK3CA, 50 EGFR, 51 PTEN, 52 PRKACA, 53 KRT19, 54 CLDN4, 55 and AGR256), and the remaining two have been associated to cancer prognosis (KRT857 and KRT1858). Put together, the results indicate that our model assigns meaningful importance to features. This is further corroborated when investigating the predicted pathways. We observe that most pathways are linked to external signaling, notably G protein-coupled receptors signaling. Signal transduction is naturally critical to drug response, as it is the route through which drugs interact with a cell.59,60 The state of the extracellular matrix also plays an important role, as it can prevent the penetration of small molecules into the cell, thus impairing pharmacologic treatments. 61 Thus, our model learns to weigh meaningful features that relate to biological processes involved in drug mechanisms of actions.
Conclusion
We introduce a two-step framework to perform data integration, feature reduction, and classification to uncover cancer-related knowledge. First, we develop an integrative NMF model to jointly embed entities in multiple connected latent spaces based on heterogeneous, diverse relational data between those entities. Note, that due to the wide range of data incorporated in our framework and the different levels of noise present in each, balancing strategies of the diverse objective functions should be investigated to improve the results further. Our model can easily be modified to accommodate such an approach.
We show that relative positions of entities in our latent spaces are consistent with what we know about them. For instance, we show that genes group in functional domains and are close to associated higher-order molecular structures (pathways and complexes) embedded in the same latent space. Patients tend to be closer to other patients having the same diagnosis and to the diagnosis itself. By taking a pan-cancer approach, we are able to identify groups of patients with similar molecular manifestations spanning various cancers, confirming that cancer classification may need to be rethought on a global scale and the need for initiatives such as PCAWG. 7
Based on known drug indications for the treatment of each cancer type and known cancer type driver genes, we train decision trees through which we can predict relevant new associations for each cancer type. Due to the joint embedding of different entities in the same latent space, we hypothesized that decision trees trained to identify associations with one type of entities could be repurposed to derive associations with other, less-studied, entities. In this way, we can uncover biological mechanisms affected by each cancer type, such as genes whose expressions are significantly correlated with patient's survival (Supplementary Fig. S5). We also identify pathways that can be linked to cancer in the literature. Furthermore, our results suggest that cellular response to stress (pathway R-HSA-2262752) plays an important role in breast cancer. Similarly, we find that FGFR signaling mediated by SHC (pathways R-HSA-5654688, R-HSA-5654699, and R-HSA-5654719) is implicated with multiple cancer types; the precise role of SHC in these pathways has not been elucidated yet.
Interestingly, our work opens the door for actionable precision medicine. Through the joint embedding of cancers and patients, decision trees trained on high-level knowledge about cancer types can be repurposed to help identify patient-specific information, such as potential drug treatment. Furthermore, our model is able to capture the underlying information relevant to the characterization of patients' response to drug treatment. However, as the biological validation of such predictions is difficult, requiring cell line experiments or clinical trials, we leave it for future work.
Our framework is general and flexible and can accommodate additional and different data. New families of entities can be naturally added to the framework and could be illustrated by new nodes in Figure 1a. For instance, microbiomics, which is known to impact response to drugs, 62 can be easily included in the general framework by adding a new node representing microbes in Figure 1a. Additional data connections between entities already in the framework, such as gene methylation data or gene copy number variation data, would add subobjectives to the optimization problem (represented by edges in Fig. 1a). While we focus on cancer in this study, our work paves the way for general cross-disease analysis, which could be useful to identify treatment repurposing based on molecular similarities among medical conditions. Alternatively, tasks such as drug side effects or drug combination synergy prediction can be addressed using the embedding framework as a basis with a specific machine learning model (e.g., boosted decision trees), taking the embedding as inputs.
Data Availability
The data are available at https://life.bsc.es/iconbi/context_aware_embeddings/index.html
Code Availability
The c++ library for the joint factorization is available at github.com/tgaudelet/nmfif
The analysis code, including baselines methods, is available at https://life.bsc.es/iconbi/context_aware_embeddings/index.html
Footnotes
Authors' Contributions
T.G. contributed to developing the computational concepts, implementation, data analysis, result interpretation, and article writing. N.M.D. contributed to developing the computational concepts, result interpretation, and article writing. N.P. conceived, directed, and supervised the project, and contributed to result interpretation and article writing.
All co-authors have reviewed and approved the article before submission.
This article has been submitted solely to this journal and is not published, in press, or submitted elsewhere.
Author Disclosure Statements
No competing financial interests exist.
Funding Information
This work was supported by the European Research Council (ERC) Consolidator Grant 770827, the Spanish State Research Agency AEI 10.13039/501100011033 grant number PID2019-105500GB-I00, and UCL Computer Science Department.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.