
Abstract
Introduction:
The post-exertional malaise of myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) was modeled by comparing micro-RNA (miRNA) in cerebrospinal fluid from subjects who had no exercise versus submaximal exercise.
Materials and Methods:
Differentially expressed miRNAs were examined by informatics methods to predict potential targets and regulatory pathways affected by exercise.
Results:
miR-608, miR-328, miR-200a-5p, miR-93-3p, and miR-92a-3p had higher levels in subjects who rested overnight (nonexercise n=45) compared to subjects who had exercised before their lumbar punctures (n=15). The combination was examined in DIANA MiRpath v3.0, TarBase, Cytoscape, and Ingenuity software® to select the intersection of target mRNAs. DIANA found 33 targets that may be elevated after exercise, including TGFBR1, IGFR1, and CDC42. Adhesion and adherens junctions were the most frequent pathways. Ingenuity selected seven targets that had complementary mechanistic pathways involving GNAQ, ADCY3, RAP1B, and PIK3R3. Potential target cells expressing high levels of these genes included choroid plexus, neurons, and microglia.
Conclusion:
The reduction of this combination of miRNAs in cerebrospinal fluid after exercise suggested upregulation of phosphoinositol signaling pathways and altered adhesion during the post-exertional malaise of ME/CFS.
Clinical Trial Registration Nos.: NCT01291758 and NCT00810225.
Introduction
Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) is a nociceptive, interoceptive fatiguing illness that is currently defined by symptoms and exclusion of other conditions in the extensive differential diagnoses. 1 The etiology is unknown. Nociceptive refers to chronic pain and tenderness (systemic hyperalgesia and allodynia) that are controlled and regulated by spinal cord and brain processes. 2 Interoception refers to the bodily sensations from internal organs and mucosal surfaces such as throat, bronchi (e.g., dyspnea 3 ), gut, and lymph nodes.4,5 Fatigue refers to the cognitive and negative emotional strain involved in simple problem solving, loss of cognitive reserves related to insomnia and unrefreshing sleep, and influence of mitochondrial dysfunction and other mechanisms on muscular strength and stamina that combine to create a state of mental exhaustion and bodily heaviness. 6 These principles are engrained in the 1994 Center for Disease Control criteria: moderate or severe, persistent, and sustained fatigue lasting more than 6 months and causing impairment of daily activities, plus moderate or severe complaints of at least four of eight ancillary criteria: short-term memory or problems with concentration, sore throat, sore lymph nodes, myalgia, arthralgia, sleep disturbances, new-onset headaches that include migraine, and post-exertional malaise. Post-exertional malaise, also referred to as exertional exhaustion, is a unique characteristic of ME/CFS. 7
We modeled post-exertional malaise by having subjects perform two submaximal exercise stress tests on consecutive days.
8
Functional magnetic resonance imaging during a working memory task was compared between pre-exercise and post-exercise time points and demonstrated exercise-induced cognitive dysfunction. Because the neurological symptoms suggested brain pathologies, lumbar punctures were performed after exercise to sample the cerebrospinal fluid (post-exercise
miRNAs are ∼22 nucleotide-long, single-stranded RNAs transcribed from genomic DNA. 14 The primary miRNA is transcribed from intergenic DNA or introns of mRNAs. Pre-miRNAs are processed by the RNase protein DROSHA in the nucleus before export into the cytoplasm for processing by DICER protein. 15 The miRNA is loaded onto the Argonaute protein(s) to form the RNA-induced silencing complex that can then bind to complementary sequences in the 3′ untranslated region of target mRNAs to repress translation or promote mRNA degradation. miRNAs dynamically fine-tune the expression of mRNAs, their translated proteins, and hence signaling and other pathways. 16 Relatively elevated miRNA levels have greater binding to target mRNAs leading to destruction of the miRNA-mRNA duplex and loss of protein and pathway activities. Conversely, relatively diminished miRNA levels allow unobstructed mRNA translation and elevation of the protein and pathway functions. A single miRNA may target anywhere from zero to over a thousand mRNAs. Most studies investigate the effects of single miRNAs, but not the synergistic or antagonistic effects that may occur when a combination of miRNAs are modulated simultaneously.
We hypothesized that exercise would significantly alter the levels of miRNAs in ME/CFS subjects, and that informatics analysis of the downstream targets would allow inferences about the mRNAs, proteins, and pathways that were modulated. The functional effects may allow inferences about potential mechanisms of post-exertional malaise. Quantitative polymerase chain reaction (qPCR) was used to measure cerebrospinal fluid miRNAs in specimens from nonexercise (
Materials and Methods
Clinical information
All subjects gave written informed consent to protocols approved by the Georgetown University Institutional Review Board (IRB 2009-229, 2013-0943, and 2015-0579) and the Human Research Protection Office of the Department of Defense Congressionally Directed Medical Research Program (HRPO A-15547 and A-18479), and listed in clinicaltrials.gov. The studies followed World Medical Association Declaration of Helsinki—Ethical Principles for Medical Research Involving Human Subjects. 18 The investigations were not considered clinical trials using the World Health Organization (WHO) definition. 19
CFS was diagnosed during history and physical examinations with inclusion according to the 1994 Center for Disease Control criteria 7 and exclusion for chronic medical and psychiatric diseases.20,21 Subjects completed the CFS Symptom Severity Questionnaire and reported the severity of their fatigue and the eight ancillary criteria of poor memory or concentration, sore throat, lymph nodes, muscle pain, joint pain, headaches, sleep, and exertional exhaustion 7 using an ordinal system with 0 for no symptom, 1 for trivial severity, 2 for mild, 3 for moderate, and 4 for severe complaints. 22
The nonexercise group rested overnight before lumbar puncture (nonexercise,
The second group had submaximal bicycle exercise stress tests on 2 consecutive days before lumbar puncture (
qPCR assay
RNA was extracted from 0.5 mL of cerebrospinal fluid, and miRNAs profiled by qPCR with a 380-miRNA panel as described previously.
9
miRNA levels were quantified from cycle thresholds (Ct) using the ΔΔCt method.23,24 Data were refined for this analysis by using a normalizer that was the average Ct from 11 miRNAs that were detected in all specimens and were not significantly different between groups. The averaged normalizer Ct value was subtracted from the Ct of each miRNA to calculate ΔCt for each individual. ΔCts were averaged for each group. ΔΔCt for each miRNA was computed as the difference between non-exercise (
Target analysis
Lists of target genes were defined using combinations of miRNAs in DIANA mirPathv3.0 (http://snf-515788.vm.okeanos.grnet.gr/) 17 and single miRNAs in Pathway Studio® (https://mammalcedfx.pathwaystudio.com/login/form), 25 Ingenuity Pathway Analysis (IPA) MicroRNA Target Filter Tool® (https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/), 26 and by literature searches (Fig. 1). The DIANA search process with genes intersection was preferred because it searched for genes that interacted with combinations of miRNAs. Single miRNAs interact with zero to thousands of mRNAs from multiple pathways. The gene union and pathway union and intersection searches from separate miRNA searches led to long lists of gene interactions, but did not provide information about potential additive or synergistic effects resulting from the combinations of miRNA changes.
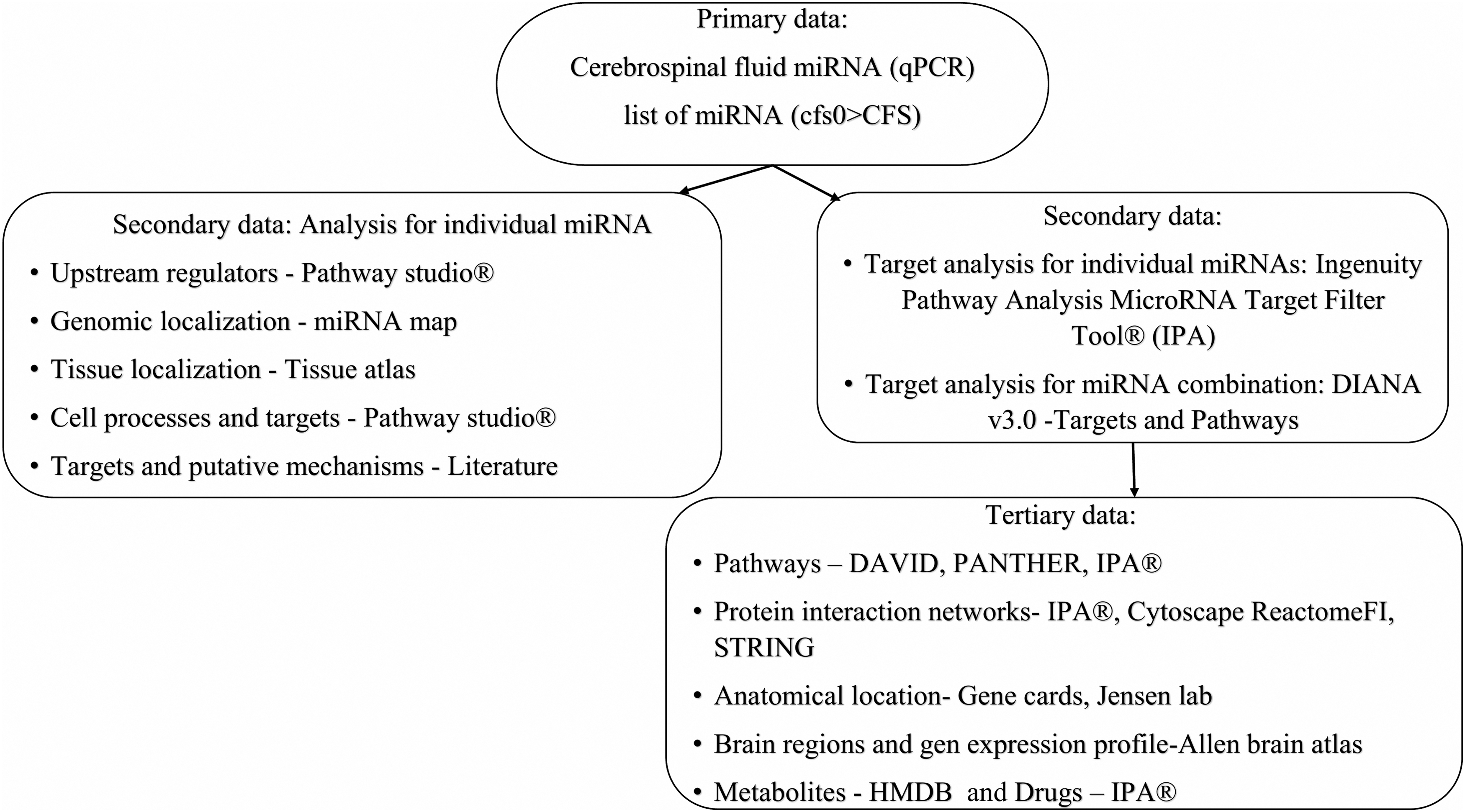
Informatics workflow explaining the flow of miRNA data from qPCR to targets and pathways. miRNA, micro-RNA; qPCR, quantitative polymerase chain reaction.
The combination of miRNAs was entered into DIANA miRpath v3.0 software (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=microT_CDS/index) 17 and DIANA-TarBase v7.0 (http://carolina.imis.athena-innovation.gr/diana_tools/web/index.php?r=tarbasev8/index) 17 database to identify the gene intersection (p<0.001) from KEGG (Kyoto Encyclopedia of Genes and Genomes; https://www.genome.jp/kegg/) 27 and GO (Gene Ontology; http://geneontology.org/) 28 databases using the highest number of miRNAs for each search. 17 Gene intersection selected genes that had interactions with multiple miRNAs. This process restricted the total number of genes selected because genes with multiple miRNA interactions were favored. By comparison, gene union provided the combination of all genes from all five miRNAs and did not preferentially select genes influenced by more than one of the five miRNAs. Pathway union and pathway intersection were not used because they also selected large numbers of genes without direct information about miRNA-mRNA interactions.
The DIANA outcomes were lists of pathways and candidate mRNAs that were targeted by combinations of miRNAs in KEGG and GO. A threshold of p<0.001 was applied to restrict our gene—miRNA interactions and pathways to those that were most likely to operate in vivo. To select candidate mRNAs that were most likely to be modulated by the combination of miRNAs, all candidates were weighted by the predicted number of miRNAs they interacted with (#miRNA) and probability (p) of the regulatory pathway provided by DIANA:
A novel MATLAB script was used to rank candidate mRNAs according to the sum of weight for each gene (Σweight) and the number of times a gene (Σhits) appeared in the different pathways (Supplementary Tables S2–S5). The weighting process selected 19 genes from KEGG and 16 from GO categories. Results were combined and reconciled to identify the final target gene list of 33 mRNAs.
The IPA MicroRNA Target Filter Tool (https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/) 26 analyzed the combination of five miRNAs simultaneously and found seven additional experimentally validated targets.
Individual miRNAs were searched in tarbase v8.0 and microTCDS from DIANA, and in Pathway Studio (https://mammalcedfx.pathwaystudio.com/login/form) 25 using its proprietary database and algorithm to identify a separate list of target genes (Supplementary Tables S11 and S12).
Literature searches for each individual miRNA found an additional 18 experimentally validated targets.
Pathway analysis
In some cases, the DIANA outcomes selected general terms like “organelle” (GO:0043226, 16 genes), “nucleoplasm” (GO:0005654, 8 genes), “Transcriptional misregulation in cancer (hsa05202),” “Adherens junction,” and “Chronic myeloid leukemia (hsa05220)” that provided few mechanistic insights. Therefore, we used an iterative refinement strategy to search for more specific mechanisms and pathways using other databases. DAVID Bioinformatics Resources 6.8 (https://david.ncifcrf.gov/summary.jsp) 29 (Enrichment Score >2, Benjamini Hochberg p<0.05) and Reactome (https://reactome.org/) 30 generated lists of pathways that were significantly enriched for the target genes (p<0.001). Pathway Studio 25 provided upstream regulators and the top 5 biological processes. Panther (www.pantherdb.org/) 31 gave the top enriched GO terms for the target genes (p<0.05). IPA software®26 generated a protein interaction network map with linkers, and a list of enriched pathways. ReactomeFI app (https://reactome.org/tools/reactome-fiviz) 32 was downloaded into Cytoscape (https://cytoscape.org/) 33 and used to cluster the targets into modules according to pathway enrichment. Modules were ranked by the largest number of clustered gene targets. The top pathway was identified for each module. The protein–protein association network was drawn by STRING v:11.0. (https://string-db.org/) 34 The iterative process honed our outcomes from general terms into smaller segments of large pathways and subsets of general GO terms.
Cellular and tissue localization
Genomic localization and parent genes for miRNAs were obtained using miRNA Map (http://mirnamap.mbc.nctu.edu.tw/), 35 UCSC genome browser (https://genome.ucsc.edu/cgi-bin/hgGateway), 36 and Entrez Gene (https://www.ncbi.nlm.nih.gov/gene) 37 databases.
Tissue Atlas for miRNA (https://ccb-web.cs.uni-saarland.de/tissueatlas/) 38 found tissues that were enriched for each individual miRNA.
The Allen Institute Brain Atlas (http://human.brain-map.org/) mapped the anatomical distribution of the brain transcriptome mRNAs detected by in situ hybridization to six normal ex vivo post-mortem brains. 39 mRNA levels were compared to the overall brain average to define brain regions where each mRNA was significantly upregulated or downregulated, or equivalent to the rest of the brain.40,41
We previously found the cerebrospinal fluid miRNA levels were equivalent for nonexercise control and
Results
Demographics
At baseline, the nonexercise (
Demographics
There were no significant differences in demographics, fibromyalgia, or severity of CFS complaints (average±SD) between the non-exercise (
CFS, chronic fatigue syndrome.
Cerebrospinal fluid miRNAs and target genes
miR-608, miR-328, miR-200a-5p, miR-93-3p, and miR-92a-3p had higher levels in cerebrospinal fluid in the nonexercise
Relative miRNA Levels in cfs0>CFS After Normalization of Quantitative Polymerase Chain Reaction Using 11 miRNAs (Mean±SD)
Differences were significant if ANOVA p<0.05, Tukey HSD <0.05, FDR <0.05, and ROC asymptotic significance <0.05 (Supplementary Fig. S1).
ANOVA, analysis of variance; AUC, area under the curve; FDR, false discovery rate; HSD, honest significant difference; miR, micro-RNA; ROC, receiver operating characteristics.
Genomic localization of miRNA
Genomic localization of the five miRNAs and their parent genes was relevant for understanding miRNA expression and regulation. miR-328 and miR-608 are located in introns of ELMO3 (engulfment and cell motility protein 3, chromosome 16q22.2) and SEMA4G (semaphorin-4G gene, 10q24.31), respectively. miR-93-3p was unusual because it was clustered with miR-106 and miR-25 in intron 13 of the MCM7 (minichromosome maintenance complex component 7, 7q22.1) gene indicating tight regulation between miR-93-3p and MCM7. 46 miR-92a-3p (13q31.3) is hosted by the MIR-17-92a-1 Cluster Host Gene. miR-200a-5p (1p36.33) is intergenic.
Upstream regulators for the miRNAs
Pathway Studio 25 found complex regulation of miRNA expression. miR-93 was elevated by HDAC and MYC, diminished by LEP and AKT1, and modulated in unknown manner by TGFB1 and DICER. miR-200a was diminished by TGFB1, elevated by DICER, and also influenced by LEP and HDACs. miR-92a was modulated by TGFB1, AKT1, MAPK, and MYC. Additional interactions have been proposed, but effects were not documented (Supplementary Table S9).
Target genes
DIANA miRpath
The combination of five miRNAs was used to search for gene targets using TarBase v7.0 and DIANA v3.0 and gene intersection. The intersection of the five miRNAs was used because individual miRNAs identified many target genes ranging from 1970 (miR-92a-3p), 883 (miR-93-3p), 175 (miR-200a-5p), and 49 (miR-608) to 0 (miR-328). The union of these genes was not manageable for defining the effects of the co-expressed miRNAs and may have identified targets and pathways that occurred only once out of the hundreds of targets found in the searches. KEGG and GO databases were searched using the highest number of miRNAs possible to select pathways with p<0.001. The significant KEGG pathways found using four miRNAs were Adherens junction (hsa04520) and Transcriptional Misregulation in Cancer (hsa05202). Each pathway contained seven target genes. Search of the GO category database with four miRNAs identified organelle (GO:0043226, 16 genes) and nucleoplasm (GO:0005654, 8 genes) (Supplementary Tables S6 and S7). Potential target genes were weighted (Supplementary Methods) and the KEGG and GO lists reconciled to identify 33 targets. Initial inspection was noteworthy for the receptors TGFBR1, IGF1R, and SCARB2, cell cycle proteins CCND2, CCNE2, and CDC42, and transcription factors ELK4, TCF3, and ZNF703 (Table 3). For clarity, the DIANA gene targets are shown in italics.
Target Gene List (n=33) for the Intersection of Five miRNAs Using DIANA mirPath v3.0 for the
The tarbase v8.0 tool 47 was used for separate searches with each of the five miRNAs to find the miRNA-gene interactions. Using prediction scores=1, 18 gene interactions were found for the five miRNAs (Supplementary Table S11). These genes were searched in DAVID and identified positive regulation of ubiquitin protein transferase activity (p=0.019), protein binding (p=0.026), DNA damage (p=0.033), and phosphoprotein (p=0.035). ELK4, HNRNPC, IGF1R, and SETD7 matched between the tarbase v8.0 and DIANA miRpath targets. DKK3, ITGA5, and MAP2K4 matched between the tarbase and IPA lists. None matched the literature search list.
The microTCDS tool 48 in DIANA tools examined individual miRNA interactions. Using a miTG score >0.95 revealed 153 gene targets, 17 targets for miR-328, 48 targets for miR-608, 27 targets for miR-200a-5p, 30 targets for miR-92a-3p, and 49 targets for miR-93-3p (Supplementary Table S12). However, none of these targets matched with the DIANA miRpath list. ADRB1, ITGA5, and MAP2K4 matched the IPA list. None matched the literature search list. Pathways found with DAVID were transcription factor activity (p=0.022), DNA binding (p=0.037), zinc finger (p=0.018), and nucleus (p=0.038).
Pathway Studio
The five miRNAs were searched in individual manner using Pathway Studio and its proprietary text mining and database methods. The union of the five searches identified 42 target genes that did not overlap with the DIANA list (Table 4).
MicroRNA Targets from Pathway Studio
Positive regulation indicated the target genes were elevated by the miRNA. Negative regulation suggested direct inhibition of the mRNA to diminish the target protein expression. Interactions with unknown outcomes may increase or decrease target mRNA levels.
PI3K was predicted to be elevated by miR-200a and miR-92a (positive, indirect effect), with unknown effects for miR-608 and miR-93. IL6 was reported to be elevated by miR-200a, but inhibited by miR-608 and miR-92a (negative, probably direct inhibition of IL6 target mRNA). VIM was inhibited by miR-200a and miR-93, with an unknown effect by miR-328. CD36 was elevated by miR-200a, but diminished by miR-328. Collagens were affected in opposite directions. TNF was elevated by miR-200a and diminished by miR-92a. Additional targets were selected by individual miRNAs. However, it is not always apparent if the miRNAs bound directly to the 3′ UTR of these genes. The Pathway Studio union list did not overlap the list selected using intersection of pathways and miRNAs in DIANA. Therefore, the target list from DIANA was used for further analysis. Pathway Studio outcomes were considered to be parallel or complimentary findings.
Literature searches identified 22 potential targets with evidence of direct miRNA binding to 3′ UTRs. Only CDC42 was shared with the DIANA list. General categories were adhesion and extracellular matrix (MMP16, CD44, CDC42, COL5A1, ZEB1, ZEB2, CTNNB, ADAMTS4, and ADAMTS5). miR-200a-5p modulated PTEN, which recycles inositol-trisphosphate for the PIK3R3-AKT pathway (Table 5).49–65
Literature Search Results for Individual miRNAs and Downregulated Target Genes
IPA MicroRNA Target Filter
The five miRNAs were searched as a group with IPA MicroRNA Target Filter tool. Experimentally validated and highly predicted targets were found for miR-92a-3p (n=191) and 200a-5p (n=40). The mRNA targets and predicted pathways were redundant with, for example, 13 genes shared between cardiac hypertrophy signaling and molecular mechanisms of cancer and 7 with Wnt signaling. Conversely, PIK3R3 was the most prevalent mRNA as it was matched with 175 pathways. Many of these pathways were named for upstream mediators and their receptors that used PIK3R3 for downstream intracellular signaling. The lists were consolidated to seven targets (Table 6) that were fed into DAVID for further iterative pathway analysis (Supplementary Table S8). The IPA Network showed that EZH2, a histone methyl transferase, and AGO2 had the most protein interactions (connections) (Fig. 2). CCNE2 was shared with the DIANA list.

IPA MicroRNA Target Filter® Network formed with three miRNAs miR-92a-3p, miR-328-3p, and miR-200a-5p.
IPA Top Ranked Targets
Pathway analysis
Pathways predicted for DIANA target genes
The 33 targets from DIANA (Table 3) were iteratively fed into other search engines to refine the selection of relevant pathways.
Pathway enrichment in DAVID identified Adherens junction, RNA binding, and Nucleosome-RNA polymerase complex mechanisms. 29
In PANTHER, 31 the target genes were enriched for GO terms related to epidermal growth factor responses, histone methylation, dendritic spine organization, and viral RNA genome replication.
IPA using the IPA Core analysis tool®26 selected Molecular Mechanisms of Cancer (p=0.0000016), Epithelial Adherens Junction Signaling (p=0.0000025), Cyclins and Cell Cycle Regulation (p=0.00024), and TGF Signaling (p=0.0081) as its top hits.
Protein interaction networks
The gene targets from DIANA were organized into protein interaction networks using IPA, Cytoscape ReactomeFI, and STRING. The top IPA network contained 14 target genes plus linkers that matched the Reactome pathway PKMTs methylate histone lysine (R-HSA-3214841) (Fig. 3A). The second IPA network matched Signaling by Receptor Tyrosine Kinases (R-HSA-9006934).

Target protein interaction networks. The top networks that included linker proteins were drawn with
The Cytoscape Pathway Enrichment Algorithm and ReactomeFI app organized the DIANA target genes into five modules by incorporating linker proteins: Adherens junction (hsa04520), Beta integrin cell surface interactions (NCI pathway interaction database), Processing of capped intron-containing pre-mRNA (R-HSA-72203), and Cell cycle (hsa04110) (Fig. 3B and Table 7).
Cytoscape Modules of Target Proteins with Linkers using ReactomeFI App
STRING showed the interactions between the target proteins without linkers. The result was a long-extended network with several unconnected genes. STRING identified Adherens junction (hsa04520) with a subnetwork centered by CDC42 and ACTB (ACTB, CDC42, FYN, IGF1R, IQGAP1, PTPRJ, and TGFBR1) (Fig. 3C upper left), Lysine degradation (hsa00310: ASH1L, KMT2D, SETD7, and SUV420H1) (Fig. 3C lower right), and Transcriptional misregulation in cancer (hsa05202: CCND2, ELK4, EWSR1, IGF1R, SIN3A, amd TCF3).
Pathway analysis from targets of IPA MicroRNA Target Filter
The seven targets from IPA MicroRNA Target Filter were searched in DAVID and the top KEGG pathways were found. The KEGG pathway diagrams were inspected and cross-referenced for redundant proteins in signaling pathways. This process identified potential upstream mediators, seven transmembrane G protein-coupled receptors and their downstream signaling mechanisms. Acetylcholine was one prominent mediator because its muscarinic and nicotinic receptors acted through several signaling pathways (hsa04725 Cholinergic synapse) (Fig. 4).

Compilation of KEGG pathways from DIANA and IPA® target lists. Targets found by searching through DIANA were in white boxes, while IPA targets were highlighted in light yellow. Representative members of PLC, PKC, cAMP, PIK3R3–AKT, Adherens junction, and classical MAPK and MAPK–JNK were interconnected. Cytoscape classified 15 other DIANA targets to adhesion and integrin pathways (Table 6). KEGG, Kyoto Encyclopedia of Genes and Genomes.
GNAQ was one hub that linked muscarinic receptors M1, M3, and M5 (CHRM1/3/5) and many other mediators and receptors to activation of phospholipase C (PLC), diacylglycerol, and inositol trisphosphate production, endoplasmic reticulum calcium release, activation of calmodulin and CAMK2A, protein kinase C, and the classical MAPK pathway (MAPK signaling pathway hsa04010). GNAQ was activated by other agonists, including angiotensin, cholecystokinin, gonadotrophin-releasing hormone, thrombin, trypsin, oxytocin, adenosine, lysophosphatidic acid, cysteinyl leukotrienes, adrenaline, and noradrenaline that acted by binding to their seven transmembrane receptors, including LPAR1 through 5, ADORA2B, ADORA2A, F2R, F2RL3, P2RY1, FPR1, PTGFR, OXTR, GRPR, GRM5, CYSLTR1, CYSLTR2, CCKAR, and CCKBR. The PLC pathway was also activated by IGF1R.
Acetylcholine also acted through nicotinic receptors to activate four other signaling mechanisms. Adenylyl cyclase 3 (ADCY3) generates cAMP that activates PKA and RAP1B (RAP1 signaling pathway hsa04010) followed by MAPK and JNK pathway (JNK and p38 MAP kinase pathway hsa04010). Calcium flux through the voltage-gated calcium channel pathways activates CAMK2A. Activation of FYN and JNK stimulates the PIK3R3 and AKT pathway (PI3K and AKT pathway hsa04151).
Cholinergic CHRM1/3/5 receptors also are linked to the G protein GNG12 that activates CDC42 and provides signaling links to IQGAP1 and Adherens junction, actin and cytoskeleton, and MAPK and JNK pathways. The target lists and pathways found using the DIANA search and IPA miRNA Target Filter were different, but complementary for identifying pathways modified by GNAQ, IGF1R–PLC–MAPK pathways, ADCY3–RAP1B–MAPK JNK pathways, FYN–PIK3R3–AKT, and CDC42–IQGAP1–Adherens junction interactions.
In the

Proposed modulation of miR-93-3p, MCM7, SMAD7, and TGFBR1 by exercise (
Anatomical location
The quantile-normalized expression data from Tissue Atlas for miRNA 38 showed that miR-200a-5p is abundant in cerebral cortex and white matter.
Brain
Brain regions containing the targets derived from DIANA (Table 3) were assayed using the ENRICHR tool 40 and Jensen tissue database. 42
The tissue that was most enriched with the 33 target genes was corpus callosum (p=0.00029). The relatively upregulated genes were CCNE2, ELK4, HNRNPC, IQGAP1, MCM7, NCL, SCARB2, SCD, SETD7, SPOPL, and TCF3. This list was interrogated in Pathway Studio, which identified the biological processes Eat me signal: apoptotic cell induces phagocytosis, SIRT7 signaling in aging, Adherens junction assembly, Transcytosis, Cell cycle, and Metabolism of Glycerophospholipids and Ether Lipids.
Choroid plexus
Because up to 70% of cerebrospinal fluid is produced in choroid plexus by ultrafiltration and secretion, 65 we proposed that choroid plexus epithelial cells may be a major source of miRNAs in cerebrospinal fluid. The Allen brain atlas adult human brain tissue gene expression profile data set was reviewed, and a choroid plexus database was extracted and searched to find proteins that were upregulated or downregulated relative to whole brain (p<0.05). These agreed with a lateral ventricle database extracted from Jensen tissue atlas 42 and text mining.
Targets that were overexpressed were BAG6, EWSR1, IGF1R, IQGAP1, NUF1P2, PCBP2, PRPF8, PTPRJ, SIN3A, and TCF3. DAVID identified EWSR1, TCF3, IGF1, and IGF1R in KEGG transcriptional misregulation in cancer. KEGG Adherens junction included IQGAP1, IGF1R, and PTPRJ. GO term RNA binding included EWSR1, NUFIP2, PCBP2, PRPF8, and SIN3A. Pathway Studio identified IGF1R as an important signaling mechanism in choroid plexus epithelium. These mechanisms may be accentuated in choroid plexus during the post-exercise
mRNAs that were downregulated (p≤0.05) in choroid plexus were CCND2 (cell cycle), CDC42 (adhesion), FASN, DYNC1H1, FYN, HNRNPC, SCD, and SPOPL. DAVID identified downregulated GO:0016020 membrane with CDC42, FASN, DYNC1H1, HNRNPC, and SCD. Focal adhesion (hsa 04510) was indicated by CDC42, FYN, and CCND2. Pathway Studio predicted downregulation of leukocyte activation, and cell processes related to Adherens junctions. Downregulated targets may be relevant to the cfs0 condition when miRNAs were elevated and mRNAs and proteins would be anticipated to be repressed.
Targets that were equivalent between choroid plexus and whole brain were TGFBR1, MCM7, ACTB, ASH1L, CCNE2, DCP2, DYRK1A, ELK4, H3F3B, KMT2D, NCL, SCARB2, SETD7, SUV420H1, and ZNF703.
Brain cell types
Only 7 of 33 DIANA targets were matched to transcriptomes of individual brain cell types using Genecards.
44
IGF1R, TGFBR1, DYNC1H1, and IQGAP1 were matched to microglia. All seven IPA targets were localized to neurons, with six out of seven in astrocytes (RAP1B not identified). The seven targets were widely expressed in amygdala, corpus callosum, and cerebral cortex. Microglia contained ADCY3, MAP2K4, and RAP2A. The targets were anticipated to be downregulated in
Subsets of our target genes were matched to the previously defined brain cell transcriptome modules CD1-neuron (ACTB, BAG6, DYNC1H1, FASN, and PRPF8) and CD5-neuron (CCND2, CDC42, and DYNC1L1). 66
Metabolomics
Metabolomic alterations have been suggested in CFS.67,68 The list of target genes was searched in Human Metabolite Data Base (HMDB) using Metaboanalyst, 69 which identified S-adenosylhomocysteine (HMDB00939), S-adenosylmethionine (HMDB01185), and manganese (HMDB01333) that participate in methane, glyoxylate, and amino acid metabolism pathways (p<0.025).
Treatment type
Drugs for the target proteins were identified through analysis in Ingenuity pathway software®26 (Supplementary Table S10). Oncology drugs targeted FYN, IGF1R, TGFBR1, and FASN.
Discussion
The informatics search tools were based on literature text mining using proprietary algorithms. There was a surprising lack of overlap between the search in IPA, Pathway Studio, Cytoscape, and other tools. We placed priority on the DIANA and IPA searches that used combinations of miRNAs to identify gene intersection, proteins, and their pathways. The rationale was that single miRNAs can modulate hundreds of mRNAs. Therefore, we searched for targets that were regulated in unison by several of the miRNAs in the
miR-200a-5p was elevated in the nonexercise specimens (
The miRNA data were derived from cerebrospinal fluid expression patterns, which may have the greatest impact on gene regulation in choroid plexus, epithelial lining of the cerebral ventricles and associated immune cells, superficial subventricular gray and white matter, and floor of the fourth ventricle. Microglia and neurons were implicated in exercise-induced alterations of miRNAs and their patterns of target expression using GeneCards.
The high expression of the target genes in the corpus callosum was of interest because CFS patients have decreased white matter volumes by voxel-based morphometry.71,72 The affected white matter tracts extend from the inferior frontal lobe, dorsal right prefrontal lobe, bilateral internal and external capsules, and anterior midbrain, to the bilateral pons.
These outcomes are limited to the effects of the physiological stressor of exercise between two groups of ME/CFS subjects. The analysis was based on the exercise-induced changes in cerebrospinal fluid miRNAs in ME/CFS (
A novel weighting strategy was used to find targets that were recognized in parallel by the combination of miRNAs (genes intersection). The combinations may have reduced false positive rates compared to conducting searches with individual miRNAs (gene union). The weighted approach may have excluded some targets that were selected by single miRNAs that had large effects in brain cells. Future studies are required to investigate in vitro and in vivo effects of both single miRNAs and the combination.
The literature searches for individual miRNAs did not reflect the complexity of the in vivo interactions between combinations of miRNAs, multiple gene targets, and pathways. The literature search did not verify the miRNA-mRNA interactions found using combinations of miRNAs with DIANA or IPA miRNA filter, suggesting that our weighting strategy had advantages over assessments with single overexpressed miRNAs in model systems.49,51,52,55,56,60–62,64
The categories of GO were often general and vague (e.g., organelle) and did not indicate more specific mechanisms. Therefore, secondary analysis of these clusters of targets was needed in other software such as DAVID.
IPA and Cytoscape used proprietary software tools and databases to find their lists of targets, but these also did not overlap with the DIANA or literature searches. The outcomes of these tools suggested not only direct inhibitory actions of the miRNAs on the 3′ UTR of mRNAs but also “positive” effects with increased expression of some mRNAs (Table 4) that may be the result of modulation of other regulatory mRNAs, transcription factors, target proteins, and their pathways (Fig. 4). These search strategies did not provide consistent probability estimates that could be used for additional connectivity and mechanistic investigations. 73
Therapeutic implications were limited because many of the available drugs are indicated for cancer treatments, and have not been studied in neurological or other diseases. However, the proposed mechanisms (Fig. 4) may offer new insights for pathogenic mechanisms, experimental verification, and future drug treatments.
The miRNAs were measured in cerebrospinal fluid, and we do not have histological or single cell RNA evidence of their origins. Similarly, the means of secretion in exosomes or other microvesicles and target cells, and kinetics of intercellular regulation are not known. The postulated interactions of the combinations of miRNAs and effects on their targets and pathways will require confirmation in vitro. Future studies may use post-mortem human tissues or animal models to study miRNA production, transport, and the kinetics of effector functions in the baseline state and after exercise to better understand ME/CFS and post-exertional malaise.
Footnotes
Authors' Contributions
J.N.B. organized the studies. N.S. performed the qPCR in blinded manner. V.N. performed the informatics analysis. J.N.B. and V.N. wrote the article. All the co-authors have reviewed and approved the article.
Acknowledgment
We thank Dr. Sona Vasudevan, Director, Systems Medicine program, Georgetown University, for assistance with pathway analysis and for comments that greatly improved the article.
Disclaimer
This article has been submitted solely to the journal of systems medicine.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The study was supported by funding from The Sergeant Sullivan Circle, Barbara Cottone, Dean Clarke Bridge Prize, Department of Defense Congressionally Directed Medical Research Program (CDMRP) W81XWH-15-1-0679 and W81-XWH-09-1-0526, and the National Institute of Neurological Disorders and Stroke R21NS088138 and RO1NS085131.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.