
Abstract
Traumatic brain injury (TBI) affects nearly 2.5 million people each year and results in a cascade of neurometabolic effects, including prolonged inflammatory processes. Choroid plexus (ChP) swelling has been postulated to occur following TBI due to neuroinflammation. However, it is unknown if the ChP swells as a consequence of the post-TBI neuroinflammatory cascade, and it is unknown if swelling could be detectable via human volumetric imaging. Therefore, this study aims to test for the effect of TBI on ChP volume using a case–control study design. Data were acquired from two independent datasets, both of which were comprised of patients who underwent magnetic resonance imaging. First, the Alzheimer’s Disease Neuroimaging Initiative (ADNI) included 156 older adult veterans (70.1 years) with prior moderate-to-severe TBIs and 103 older adult veterans (69.1 years) without a TBI. Second, the Transforming Research and Clinical Knowledge in TBI (TRACK-TBI) included 484 participants (38.4 years) with a recent mild TBI and 92 controls (37.9 years). There were no differences found in TBI ChP volume compared with controls in either ADNI/Department of Defense with those with a history of TBI or TRACK-TBI at 2 weeks postinjury or 6 months postinjury. However, there were significant ChP differences in those with a visible pathology from head computed tomography (CT) scans compared with those without orthopedic controls. These results suggest that ChP swelling occurs only in those with CT. These findings highlight that inflammation after a TBI does manifest in volumetric increases in the ChP, but it is dependent on the pathological features of the TBI.
Introduction
Traumatic brain injury (TBI) is an epidemic affecting millions of individuals worldwide, with a higher prevalence in young and older adults. 1 As a result of primary and secondary injury mechanisms, patients report a variety of behavioral symptoms, potentially lasting for years postinjury. 2 Of particular interest, chronic inflammation within the brain is an example of a secondary injury mechanism believed to drive persistent damage and behavioral symptoms.3,4 This neuroinflammation also could lead to the swelling of specific brain regions, such as the ventricles and choroid plexus (ChP). However, it is not clear if this swelling would be detectable in the ChP by volumetric imaging in humans.
TBI leads to cell death, 5 gray matter atrophy,6,7 and cortical thinning, 8 but it is also associated with inflammatory responses. 9 These inflammatory responses have been shown to be caused by postinjury structural damage to the blood–brain barrier (BBB). 10 The BBB is a neuroprotective barrier composed of endothelial cells. These endothelial cells are connected by tight junctions that regulate the exchange of ions and proteins between the blood and brain. 11 Importantly, inflammation has been observed to lead to an increase in ventricular volume post-TBI via the BBB. 11 The ChP, a small structure next to the hippocampus, is similar to the BBB in that it also serves as a neuroprotective barrier, this time, at the junction between the brain and cerebrospinal fluid. Its structure includes epithelial cells, which regulate the passage of proteins and ions between the blood and nervous system. ChP volume has been observed to change in response to neurological diseases, such as dementia.12–15 In addition, physiological changes in the ChP have been observed following TBI, including widened epithelial intercellular spaces and increased proinflammatory mediators.16,17 Given the inflammatory response following injury, it has been hypothesized that the ChP would increase volume post-TBI. In fact, previous research in animal models has revealed ChP swelling following TBI. 18 Research has also shown inflammatory markers entering the brain through the ChP following TBI, which migrate to the site of injury. 19 Further, a rat model of TBI has suggested ChP gene expression as an initiator of biphasic injury response, triggering the production of growth regulatory proteins in response to injury. 20 These data suggest the ChP may play a critical neuromodulatory role in post-TBI inflammatory cascade in humans and perhaps similarly increase in volume, as has been observed in animal models of TBI. However, this hypothesis has yet to be studied in humans with volumetric imaging. Investigating the ChP’s role post-TBI is important for the continuation of understanding the pathophysiology of TBI in humans, which has been seen to predict outcomes21,22 and can help guide targeted treatment to vulnerable groups. Moreover, understanding the role of the ChP in TBI pathophysiology stands to have far-reaching implications for age-related cognitive decline. Specifically, post-TBI brains are estimated to be older than their chronological age, indicating a potential increased susceptibility for TBI patients to develop dementia. 23 Given ChP volume increase has been shown in age-related dementias,14,15 identifying the pathophysiological role of the ChP post-TBI is important for the short- and long-term care of patients.
Older adults are at an increased risk of long-term deficits and developing dementia after a TBI. 24 Similar to TBI pathology, dementia is associated with atrophy and cell death 25 but also increased volume of the ventricles and ChP, possibly as a result of chronic inflammatory processes. 26 The ChP plays an important role in protein clearance; thus, dysfunction can lead to protein accumulation, which is a hallmark feature observed in Alzheimer’s disease (AD). 27 The development of AD and poor TBI outcomes shares a common genetic risk factor, apolipoprotein E4 (APOE4). APOE4 impairs BBB repair and amyloid beta protein clearance following TBI28,29 and has been specifically associated with increased mortality, 30 prolonged coma, 31 and poor prognosis 32 following a TBI. 33 Moreover, evidence suggests that APOE4 links TBI to an increased risk of developing AD. 34 For example, after an acute TBI, APOE4 carriers have higher levels of amyloid beta protein than those without. 35 Prior work has shown increased accumulation of APOE4 in the ChP in AD, suggesting a potential relationship between APOE4 status and ChP volume. 36 However, not all studies demonstrate this relationship between TBI and AD due to APOE4; thus, the field remains ambiguous on the relationship between TBI and AD.37–39
Evidence-based guidelines call for the use of CT scan examinations as a part of the diagnosis for mTBI. 40 However, prior work suggests that magnetic resonance imaging (MRI) has better diagnostic sensitivity for nonhemorrhagic contusions and shear-strain injuries, 41 with CT missing up to 30% of abnormalities detected by MRI. 42 However, clinical readings of imaging, specifically CT scans, can inform if neurosurgery is required and detect abnormalities such as cerebral contusions and subdural and epidural hematomas. These injuries are often associated with worse outcomes following a TBI. 43
This study aims to test whether differences in ChP between individuals with and without a TBI may implicate the structure as a candidate player involved in the mechanism responsible for post-TBI neurological changes. This study aims to test whether volumetric differences in the ChP could indicate the structure’s mechanistic role in the neurological changes observed in patients post-TBI. Volumetric differences were assessed using MRI between individuals with and without TBI for both acute and chronic TBI in older adults. We hypothesized that ChP volume would be higher in TBI in both acute and chronic TBI compared with control participants without a TBI. Furthermore, we hypothesized that those APOE4 carriers with a prior TBI would have increased ChP volume.
Methods
Participants
This study utilized two open access datasets as described below and in a prior publication: 44
For the current study, we included male veterans with and without a history of nonpenetrating TBI. Participant characteristics for ADNI/DoD are presented in Table 1. Participants were classified as having either a prior TBI or not using the RECTBIINJ.csv file (TBI Injury Questionnaire). These questionnaires determine when the injury occurred, its severity, and the total number of injuries. The TBI was classified as moderate to severe based on the Veteran's Associations (VA)/DoD criteria for TBI with a loss of consciousness ≥24 h, posttraumatic amentia >24 h, or alteration of consciousness ≥24 h. Moderate-to-severe TBIs occurred at a mean age of 27 years, an average of 25 years ago. Depression and posttraumatic stress disorder (PTSD) were also characterized using the Geriatric Depression Scale (GDS) and the Clinician-Administered PTSD Scale. GDS scores of 0–4 are considered normal, 5–8 indicate mild depression, 9–11 indicate moderate depression, and 12–15 indicate severe depression, with interpretations dependent on age, education, and complaints. Current and lifetime PTSD was dichotomized with a score of 50 or above indicating PTSD. APOE4 status was determined by direct sequencing. 44 Out of 260 participants from ADNI/DoD that had MRI scans, 259 had usable scans after preprocessing due to excess motion, comprising 156 participants with TBI and 103 controls. Participant characteristics for ADNI/DoD are presented in Table 1.
ADNI/DoD Participant Characteristics
Percentage of participants who had depression, taken from the Geriatric Depression Scale.
Percentage of participants who currently have PTSD.
Percentage of participants who have had PTSD in their lifetime.
MMSE to check for cognitive impairment.
Percentage of participants who have at least one APOE4 allele, indicating a risk of Alzheimer’s disease.
ADNI, Alzheimer’s Disease Neuroimaging Initiative; APOE4, apolipoprotein E4; DoD, Department of Defense; MMSE, Mini-Mental State Examination; PTSD, posttraumatic stress disorder; SD, standard deviation; TBI, traumatic brain injury.
TRACK-TBI Participant Characteristics
Neurological assessment to measure a patient’s level of consciousness after a TBI by scoring responses to stimuli. Scores range from 3 to 15 with 15 being the best. Categorization for mild TBI is considered 13–15. This was taken 24 h after injury.
Clinician-rating scale that measures a patient’s functional outcome after a TBI. Scores range 0–8 with 8 indicating no impairments.
Those who demonstrated positive CT scans, according to the Marshall classification with visible pathology.
CT, computed tomography; SD, standard deviation; TBI, traumatic brain injury; TRACK-TBI, Transforming Research and Clinical Knowledge in Traumatic Brain Injury.
Imaging data acquisition
MRI scans from TRACK-TBI were acquired across 11 centers using 13 3T scanners. Sagittal three-dimensional T1-weighted images were acquired. Scans were performed within 2 weeks and 6 months of injury. All TRACK-TBI participants received a head CT within 24 h of their clinical evaluation for TBI. Head CTs were read and coded by a central board-certified neuroradiologist blinded to characteristics in accordance with the National Institute of Neurological Disorders and Stroke (NINDS) Common Data Elements (CDEs) version 1 for neuroimaging. 49 Marshall CT classification was utilized for scoring. 50
MRI acquisition details for ADNI/DoD are previously described. 51 Briefly, MRI scans were acquired using 18 3T scanners and included a three-dimensional, T1-weighted scan (voxel size = 1.2 × 1.0 × 1.0 mm3).
ChP segmentation
ChP segmentation was performed using a previously established pipeline 52 that has shown better segmentation results in clinical populations (Fig. 1). Briefly, an automated segmentation of the lateral ventricles was performed using the FreeSurfer software. 53 Then, a lightweight segmentation algorithm based on Gaussian mixture modeling was performed within the lateral ventricles to tease apart cerebrospinal fluid (CSF), ventricular wall, and ChP voxels. This method has been validated in clinical populations and healthy participants against manual segmentation.13,52 This code is freely available on GitHub (https://github.com/EhsanTadayon/choroid-plexus-segmentation).

ChP from GMM in blue; ChP from original FreeSurfer mask in yellow. ChP, choroid plexus; GMM, Gaussian mixture modeling.
Statistical analysis
All statistics were performed using R package “stats.” 54 Due to heteroskedastic data, ChP volume was log transformed for all analyses to meet assumptions of linear regressions. All p values were corrected for multiple comparisons using Benjamini and Hochberg’s false discovery rate with a q level of 0.05 after pooling all p values from each model. To test if there were differences in ChP volume in those with a history of TBI compared with those without, we performed a bivariate linear regression controlling for age, race, site, Socioeconomic Status (SES), years since injury, number of TBIs, total intracranial volume, and PTSD status using ADNI/DoD. Lastly, to test if there were ChP volume differences in older adults with a history of TBI and APOE+ and history of TBI APOE−, we separated those with a history of TBI into two groups, those with at least one APOE4 allele (APOE4+), and those without an APOE4 allele (APOE4−). We performed a linear regression, controlling for age, race, site, SES, years since injury, and PTSD status using ADNI/DoD. To test if there was a difference in ChP volume in acute mTBI, we performed a linear regression, controlling for age, sex, race, site, total intracranial volume, and SES using TRACK-TBI. To assess if there was a difference in ChP volume between 2 weeks and 6 months, we performed a linear mixed effect model with participant-specific random intercept and slope controlling for the same covariates using TRACK-TBI. This analysis was only performed on the TBI sample. To test if there was an effect of CT outcomes on ChP volume, we performed an analysis of variance (ANOVA) controlling for all covariates. Following the initial ANOVA, post hoc analyses were conducted to identify specific differences among groups. Tukey’s honest significant difference test was applied to adjust for multiple comparisons and to assess pairwise differences between CT+, CT−, and orthopedic controls.
Results
ChP in chronic moderate/severe TBI: ADNI/DoD
Using ADNI/DoD, there was no difference in ChP volume in those with a history of TBI compared with controls in right ChP (β = −0.06, R2 = 0.14, 95% confidence interval [CI]: [−0.19, 0.08], p = 0.41), left ChP (β = −0.09, R2 = 0.16, 95% CI: [−0.24, 0.06], p = 0.22), or bilateral (β = −0.23, R2 = 0.19, 95% CI: [−0.57, 0.11], p = 0.18) (Fig. 2).

Moderate-to-severe TBI and controls right and left ChP volume from ADNI/DoD.
In addition, there was no difference in ChP volume in those with TBI+APOE+ compared with TBI+APOE− in (β = −0.003, R2 = 0.19, 95% CI: [−0.09, 0.10], p = 0.87) or left (β = −0.03, R2 = 0.08, 95% CI: [−0.087, 0.14], p = 0.64), or bilateral (β = 0.54, R2 = 0.16, 95% CI: [−0.18, 2.94], p = 0.64) (Fig. 3).
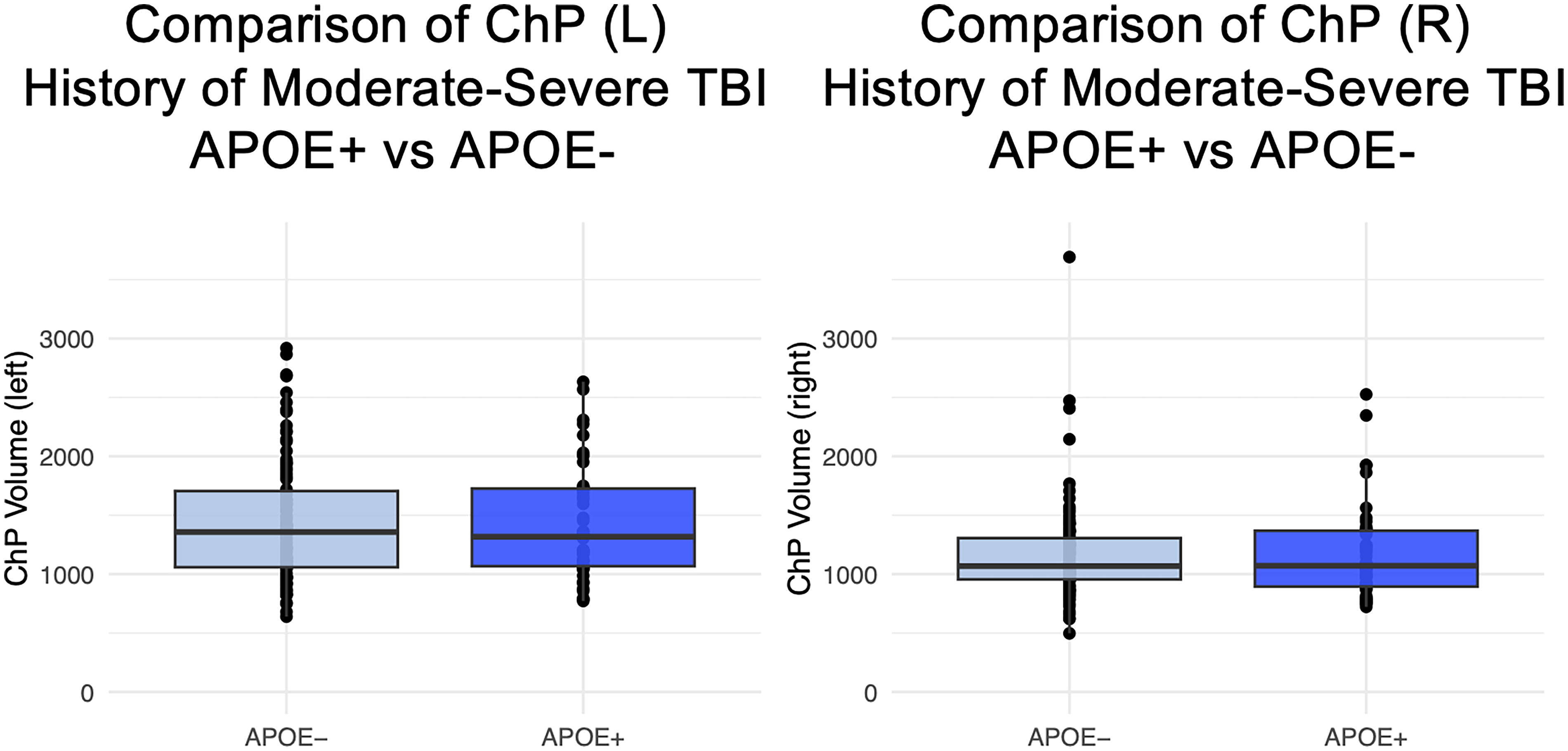
TBI+APOE+ versus TBI+APOE− (moderate to severe) in the left and right ChP from ADNI/DoD. (a) Box plots demonstrating the no difference between ChP volume between those at least one APOE4 allele and those with no APOE4 allele in each hemisphere in just the TBI group. ADNI, Alzheimer’s Disease Neuroimaging Initiative; APOE, apolipoprotein E; ChP, choroid plexus; DoD, Department of Defense; TBI, traumatic brain injury.
ChP in acute mTBI: TRACK-TBI
Using TRACK-TBI, there was no difference in ChP volume at 2 weeks postinjury compared with orthopedic controls in the right (β = −0.04, R2 = 0.18, 95% CI: [−0.02, 0.11], p = 0.21), left (β = −0.04, R2 = 0.19, 95% CI: [−0.03, 0.11], p = 0.30), or bilateral (β = 0.55, R2 = 0.29, 95% CI: [−0.80, 0.02], p = 0.42) (Fig. 4).

Left and right volume at 2 weeks postmild TBI from TRACK-TBI.
There was no difference in ChP volume at 6 months postinjury compared with orthopedic controls in the right (β = 0.05, R2 = 0.22, 95% CI: [−0.007, 0.11], p = 0.09), left (β = 0.02, R2 = 0.24, 95% CI: [−0.05, 0.08], p = 0.56), or bilateral (β = −0.34, R2 = 0.27, 95% CI: [−0.89, 0.02], p = 0.29) (Fig. 5). Finally, TBI recovery had no effect on ChP volume in the right ChP (β = −0.13, 95% CI: [−0.36, 0.094], p = 0.24), left (β = −0.21, 95% CI: [−0.49, 0.64], p = 0.13), or bilateral (β = −0.35, 95% CI: [−0.75, 0.05], p = 0.08) (Fig. 5).

ChP volume at 2 weeks and 6 months in the left and right ChP in mild TBI from TRACK-TBI. (a) Box plots demonstrating the no change in ChP volume between those with a mild TBI between 2 weeks and 6 months postinjury. ChP, choroid plexus; TBI, traumatic brain injury; TRACK-TBI, Transforming Research and Clinical Knowledge in Traumatic Brain Injury.
Results showed no effect of sex on ChP volume at 2 weeks postinjury in the right (β = −0.009, R2 = 0.19, 95% CI: [−0.08, 0.07], p = 0.803), left (β = 0.03, R2 = 0.22, 95% CI: [−0.06, 0.12], p = 0.54), or bilateral (β = −0.53, R2 = 0.29, 95% CI: [−0.18, 7.25], p = 0.40).
Results from the ANOVA revealed an effect of group (CT+, CT−, orthopedic controls) on the right (F = 10.17, p < 0.001, η2 = 0.04), left (F = 11.91, p < 0.001, η2 = 0.04), and bilateral ChP at 2 weeks (F = 10.78, p < 0.001, η2 = 0.04). Post hoc results showed that there were differences in ChP between CT+ and CT− in the right (95% CI: [55.76, 178.31], p < 0.001), left (95% CI: [81.27, 232.64], p < 0.001), and bilateral hemisphere (95% CI: [117.36, 358.19], p < 0.001). Post hoc results also revealed differences in ChP volume between CT+ and orthopedic controls in the right (95% CI: [−175.16, −10.58], p < 0.001), left (95% CI: [−218.05, −14.77], p = 0.02), and bilateral hemisphere (95% CI: [−327.42, −3.28], p = 0.04). Results from the ANOVA revealed an effect of group (CT+, CT−, orthopedic controls) on right (F = 8.39, p < 0.001, η2 = 0.03), left (F = 9.53, p < 0.001, η2 = 0.03), and bilateral ChP at 6 months (F = 11.52, p < 0.001, η2 = 0.04). Post hoc results showed that there were differences in ChP volume at 6 months between CT+ and CT− in the right (95% CI: [115.43, 338.16], p < 0.001), left (95% CI: [59.09, 197.01], p < 0.001), and bilateral hemisphere (95% CI: [115.43, 338.16], p < 0.001). Post hoc results also revealed differences in ChP volume at 6 months between CT+ and orthopedic controls in the right (95% CI: [−170.16, −16.60], p = 0.01) and bilateral hemisphere (95% CI: [−321.39, −27.63], p = 0.01) but not the left (95% CI: [−34.13, 127.98], p = 0.09) (Fig. 6).
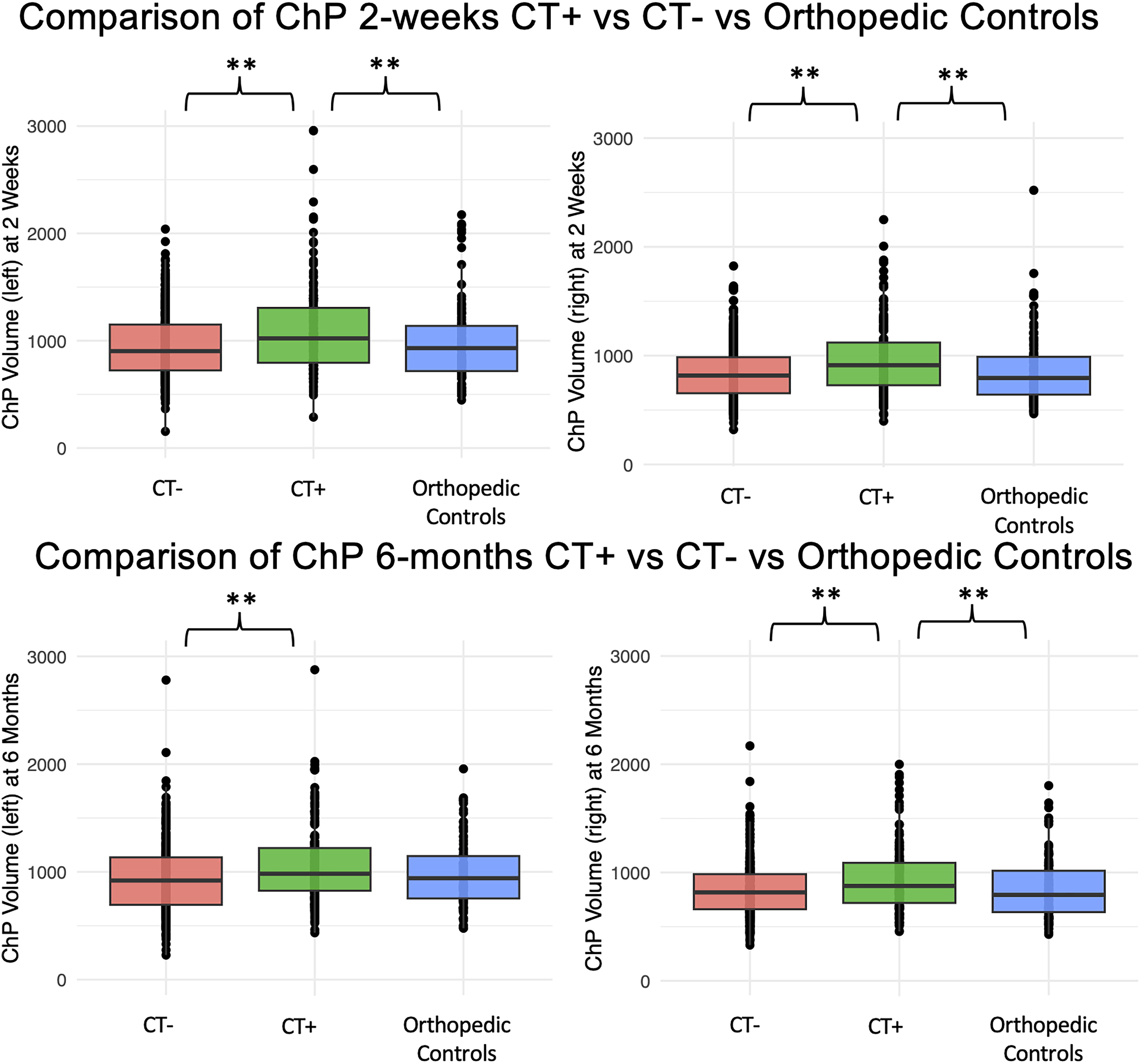
ChP volume at 2 weeks and 6 months in the left and right ChP from TRACK-TBI in mild TBI CT+, mild TBI CT−, and orthopedic controls. Box plots demonstrating post hoc comparisons with higher ChP volume in the right and left hemisphere at 2 weeks in CT+ compared with CT− and orthopedic controls. This relationship exists at 6 months for the left and right hemisphere for CT+ compared with CT− but only for the right hemisphere when comparing CT+ to orthopedic controls. ChP, choroid plexus; CT, computed tomography; TBI, traumatic brain injury; TRACK-TBI, Transforming Research and Clinical Knowledge in Traumatic Brain Injury.
Discussion
Using two open access datasets with varied injury types, our results suggest that ChP volume is not affected by mild or moderate-to-severe TBI in either the acute or chronic stages of injury. There were no differences in ChP volume in those with a history of moderate/severe TBI compared with controls. To examine the effect of APOE4 carrier status, we split the TBI group into APOE4+ and APOE4− and saw no effect on APOE4 status on ChP volume. Moreover, there were no differences in ChP volume between mTBI and orthopedic controls at 2 weeks postinjury, as shown in the TRACK-TBI dataset. Likewise, there were no differences in ChP volume between mTBI and orthopedic controls at 6 months postinjury. However, there was an effect of CT status on ChP volume at both 2 weeks and 6 months postinjury, with increased ChP volume in those with a positive head CT diagnosis.
There was no effect of TBI on ChP volume in those with a history of TBI compared with controls. Prior work has shown that having a history of a TBI may lead to negative outcomes with age, such as dementia and cognitive impairment.24,55 The exact mechanisms behind this have yet to be discovered; however, some propose that inflammatory markers that are associated with TBI may contribute to these processes.38,56 ChP has been seen to increase with age and with dementia,13–15,52 thus providing a reasonable target to test if this mechanism of dementia and the inflammatory processes may initiate as a function of TBI shared common variance. However, no difference in ChP volume was observed in those with a history of TBI. Further, a well-known dementia risk factor, APOE4, did not influence ChP volume in TBI, despite prior work showing a relationship between ChP volume and protein levels in healthy older adults and dementia patients. 13
These findings support the hypothesis that ChP enlarges after TBI, with enlargement dependent on the neuropathology observed in CT scans. Lack of changes to ChP at 2 weeks postinjury in mTBI could be due to not enough time since injury to allow for volumetric changes. Prior work has shown in moderate-to-severe TBI, volumetric change was not detectable until 3 months postinjury. 57 However, between-group differences were also not detected at 6 months postinjury in this analysis. Several limitations could speak to this null result, as the control group was significantly smaller than the TBI group. Moreover, the control group consisted of orthopedic controls, with a prior orthopedic injury that could have indirectly influenced brain structure. However, there was no longitudinal change in ChP volume in the mTBI group, suggesting that mTBI does not impact ChP volume at 2 weeks or at 6 months after injury. However, the window of ChP enlargement may have been missed, given that scans were acquired 6 months apart. It may take longer than 2 weeks to develop and return to baseline within 6 months. Prior animal work shows increased neuroinflammatory markers in the ChP after a TBI,19,58,59 suggesting that these results might be dependent on if one experiences neuroinflammation after a TBI. Differences in ChP volume dependent on CT status suggest that not all patients will experience ChP swelling with a TBI, but that those with CT+ will show manifestations of possibly as a result of neuroinflammation in the ChP. There are mixed results on the utility of CT scans predicting outcomes and the use for treatment, with some studies showing CT does not predict outcomes. 60 However, other work shows that CT outcomes are associated with adverse outcomes after mTBI but only when differentiated into different pathological features. 61 Together, this work highlights the heterogeneity of injury mechanisms in TBI patients.
There are several limitations to this study. A history of TBI in ADNI/DoD was self-reported, potentially leading to incorrect classification or missed TBIs from the control group. Moreover, the ADNI/DoD sample only consisted of male veterans, and TRACK-TBI contained both men and women. The ChP has been shown in prior literature to vary based on sex, and sex differences in ChP volume are further exacerbated with age. 55 We attempted to address this by including sex as a covariate in our models, and we saw no effect of sex on ChP volume in TRACK-TBI. The ADNI/DoD sample consisted of veterans with PTSD due to the nature of the ADNI/DoD study. PTSD may be a potential bias due to the mixed effects of TBI and PTSD. However, PTSD was used as a covariate, and there were no PTSD differences between groups. The TRACK-TBI sample did not have measures of PTSD; hence, this bias could not be addressed here. Moreover, the ADNI/DoD sample does not obtain objective measures of neuroinflammation or CT scans. Lastly, an effect from scanner type may have influenced the sequences and imaging. However, we controlled for site and scanner and tested if the results replicated in just one scanner type (Supplementary Data S1).
Conclusion
This study investigated the impact of TBI on ChP volume as assessed using volumetric imaging in two separate TBI populations. The findings demonstrate that pathology from a TBI, as detected by a CT scan, manifests as increased ChP volume. However, this finding is only in those with a CT pathology, suggesting that not all TBIs manifest in ChP swelling. These findings highlight some discrepancies between clinical and preclinical models of TBI and emphasize the need to continue investigating translational research. Importantly, this work shows the heterogeneity in TBI, stressing the importance that not all TBIs manifest in the same injury profiles.
Footnotes
Acknowledgments
The authors thank the Alzheimer’s Disease Neuroimaging Initiative (ADNI) and Transforming Research and Clinical Knowledge in Traumatic brain injury investigators for use and availability of the data. Data collection and sharing for this project were funded by the ADNI (National Institutes of Health [NIH] Grant U01 AG024904) and Department of Defense (DoD) ADNI (award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the NIH (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. Data used in preparation of this article were obtained from the ADNI database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: ![]() . Other data and/or research tools used in the preparation of this article were obtained and analyzed from the controlled access datasets distributed from the DoD- and NIH-supported Federal Interagency Traumatic Brain Injury Research (FITBIR) Informatics Systems. FITBIR is a collaborative biomedical informatics system created by the DoD and the NIH to provide a national resource to support and accelerate research in TBI. Dataset identifier: NCT01565551. TRACT-TBI was funded by the U.S. NIH, National Institute of Neurological Disorders and Stroke (grant U01 NS1365885) and supported by the U.S. DoD (grant W81XWH-14-2-0176). This article reflects the views of the authors and may not reflect the opinions or views of the DOD, NIH, or the Submitters submitting original data to FITBIR Informatics System. In addition, this work was completed in part using the Discovery cluster, supported by Northeastern University’s Research Computing team.
. Other data and/or research tools used in the preparation of this article were obtained and analyzed from the controlled access datasets distributed from the DoD- and NIH-supported Federal Interagency Traumatic Brain Injury Research (FITBIR) Informatics Systems. FITBIR is a collaborative biomedical informatics system created by the DoD and the NIH to provide a national resource to support and accelerate research in TBI. Dataset identifier: NCT01565551. TRACT-TBI was funded by the U.S. NIH, National Institute of Neurological Disorders and Stroke (grant U01 NS1365885) and supported by the U.S. DoD (grant W81XWH-14-2-0176). This article reflects the views of the authors and may not reflect the opinions or views of the DOD, NIH, or the Submitters submitting original data to FITBIR Informatics System. In addition, this work was completed in part using the Discovery cluster, supported by Northeastern University’s Research Computing team.
Data Availability
The data that support the findings of this study are available in FITBIR and LONI. These data were derived from the following resources available in the public domain: ida.loni.usc.edu and fitbir.nih.gov.
Author Disclosure Statement
The authors declare no conflict of interest.
Funding Information
No funding was received for this work.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.