
Abstract
Spinal cord contusion injury results in Wallerian degeneration of spinal cord axonal tracts, which are necessary for locomotor function. Axonal swelling and loss of axonal density at the contusion site, characteristic of Wallerian degeneration, commence within hours of injury. Tempol, a superoxide dismutase mimetic, was previously shown to reduce the loss of spinal cord white matter and improve locomotor function in an experimental model of spinal cord contusion, suggesting that tempol treatment might inhibit Wallerian degeneration of spinal cord axons. Here, we report that tempol partially inhibits Wallerian degeneration, resulting in improved locomotor recovery. We previously reported that Wallerian degeneration is reduced by inhibitors of aldose reductase (AR), which converts glucose to sorbitol in the polyol pathway. We observed that tempol inhibited sorbitol production in the injured spinal cord to the same extent as the AR inhibitor, sorbinil. Tempol also prevented post-contusion upregulation of AR (AKR1B10) protein expression within degenerating axons, as previously observed for AR inhibitors. Additionally, we hypothesized that tempol inhibits axonal degeneration by preventing loss of the glutathione pool due to polyol pathway activity. Consistent with our hypothesis, tempol treatment resulted in greater glutathione content in the injured spinal cord, which was correlated with increased expression and activity of gamma glutamyl cysteine ligase (γGCL; EC 6.3.2.2), the rate-limiting enzyme for glutathione synthesis. Administration of the γGCL inhibitor buthionine sulfoximine abolished all observed effects of tempol administration. Together, these results support a pathological role for polyol pathway activation in glutathione depletion, resulting in Wallerian degeneration after spinal cord injury (SCI). Interestingly, methylprednisolone, oxandrolone, and clenbuterol, which are known to spare axonal tracts after SCI, were equally effective in inhibiting polyol pathway activation. These results suggest that prevention of AR activation is a common target of many disparate post-SCI interventions.
Introduction
Spinal cord injury (SCI) often leads to Wallerian degeneration and loss of descending and ascending axonal tracts, resulting in irreversible paralysis. 1 –3 Therefore, it is important to identify agents that can prevent axonal degeneration after SCI. Previously, we reported that administration of the superoxide dismutase mimetic tempol reduces the loss of spinal cord white matter to an extent commensurate with improved locomotor function in an experimental model of spinal cord contusion injury. 4,5 This result suggests that tempol might act by inhibiting Wallerian degeneration of spinal cord axons within white matter axonal tracts.
Wallerian degeneration, which occurs after injury in the central nervous system (CNS) as well as in the peripheral nervous system (PNS), involves the formation of foci of axonal swelling leading to axonal dysfunction, discontinuity, and fragmentation. 6 In recent studies, we found that Wallerian degeneration of spinal cord axons can be reduced by inhibitors of the aldose reductase (AR) enzyme that converts glucose to sorbitol in the polyol pathway, which is activated by SCI. 5 Post-SCI treatment with sorbinil, an AR inhibitor induced recovery of locomotor function and tissue sparing, which was similar in magnitude to tempol treatment. Combined sorbinil and tempol treatment did not further improve recovery, suggesting that these agents share a common mechanism of action. Therefore, we hypothesized that tempol might also act by inhibiting activation of the polyol pathway.
In settings of ischemia or hyperglycemia, excessive activation of polyol pathway flux is thought to be driven by increased substrate and/or oxidative stress. 7 –9 Activation of AR is thought to involve mitochondrial superoxide generation. 10 Superoxide spontaneously combines with nitric oxide to produce peroxynitrite, which is known to oxidize a regulatory AR cysteine, increasing AR catalytic activity several fold. 11 Activation of the polyol pathway is inhibited by the expression of superoxide dismutase, which reduces superoxide concentrations. 10 Tempol is a superoxide dismutase mimetic 12 that scavenges mitochondrial reactive oxygen species (ROS). 13,14 Therefore, we reasoned that tempol might prevent AR activation by inhibiting peroxynitrite generation from superoxide and nitric oxide.
AR activity also consumes NADPH, a substrate required for glutathione reductase to regenerate glutathione, 15 the predominant cellular redox buffer that counteracts oxidative stress. Glutathione is also irreversibly consumed by conjugation reactions in which glutathione adducts are formed with toxic products produced by ROS 16 generated via polyol pathway activity. 9 Glutathione is a required substrate for glutathione peroxidases such as GPX4, which reverses lipid peroxidation. 17 SCI is characterized by glutathione depletion 18,19 and extensive lipid peroxidation, 20 which undermines the structural integrity of cellular membranes and viability. Another function of glutathione is to prevent the activation of 12-lipoxygenase, 21 which promotes axonal degeneration. 22 For these reasons, we hypothesized that the ability of tempol to inhibit polyol pathway activity would preserve cellular glutathione, thereby reducing axonal degeneration.
The first goal of this study was to determine whether behavioral improvement and preservation of axonal tracts by tempol 4,5 are due to inhibition of Wallerian degeneration, as measured by swelling and loss of axons in a standard experimental animal model of spinal cord contusion injury. The second goal was to determine whether tempol inhibits post-SCI activation of the polyol pathway, 5 which contributes to Wallerian degeneration, by measuring sorbitol levels in the injured cord and AR expression in the degenerating axons. The third goal was to determine the relationship between SCI, axonal glutathione changes, and Wallerian degeneration. Therefore, we determined whether tempol treatment altered glutathione levels in the injured spinal cord. Because polyol pathway activation was observed to consume glutathione, which may be necessary to prevent axonal degeneration, we examined whether exogenously inhibiting the synthesis of glutathione with buthionine sulfoximine (BSO) blocked the ability of tempol to increase glutathione, inhibit Wallerian degeneration, and improve locomotor function.
Materials and Methods
The Institutional Animal Care and Use Committee of New York Medical College approved all procedures involving vertebrate animals. Adult female Wistar rats (∼240 g) were obtained from Charles River Breeding Laboratories and housed at a temperature-regulated (23°C) animal facility. As in our previous studies, 4,5,23 –26 the spinal cords were contused with a weight-drop apparatus at the level of T10. Prior to surgery, the rats were anesthetized with pentobarbital sodium (40 mg/kg i.p.), and the completeness of anesthesia was verified by toe, foot, and tail pinch. Laminectomy was performed aseptically from T9 to T10 to expose the spinal cord. The spinous processes at T8 and T11 were fixed with clamps to prevent movement during the contusion. Body temperature was maintained at 37°C using a temperature-controlled heating pad and a rectal thermometer. Contusion injuries were induced in rats by dropping a guided 10-g rod with a tip diameter of 2.5 mm, as described for the NYU impactor, 27 from a height of 25 mm onto the exposed dura. Following contusion, each incision was closed using surgical staples. The bladders were manually expressed twice daily for several weeks, until automaticity was achieved.
Four groups of contused rats were treated with tempol (Sigma-Aldrich, St. Louis, MO, 275 mg/kg i.p. at 20 min post-injury), BSO (Sigma-Aldrich, 150 mg/kg i.p. at 24 h before injury), and tempol and BSO in combination or were untreated. Four additional groups of contused rats received sorbinil (Pfizer, Groton, CT, 65 mg/kg orally at 20 min post-injury), methylprednisolone (Upjohn, Kalamazoo, MI, 30 mg/kg iv at 20 min post-injury), oxandrolone (Steraloids Inc., Newport, RI, 4 mg/kg orally at 20 min post-injury), or clenbuterol (Chinoin Pharmaceuticals, Mexico City, 2 mg/kg i.p. at 20 min post-injury).
Behavioral analysis
Recovery of locomotor function was determined using the Basso, Beattie, and Bresnahan (BBB) locomotor scale. 28,29 One day after contusion and at 1-week intervals thereafter for up to 6 weeks post-injury, each rat was scored for locomotor function according to the BBB scale. The average score for both hindlimbs of each animal was assigned by two observers without knowledge of the treatment conditions during each 4-min session of open field testing.
Spinal cord immunohistology
Immediately following the final behavioral evaluation, the spinal cords were fixed by transcardial perfusion in anesthetized rats (pentobarbital sodium, 40 mg/kg, i.p.) with phosphate-buffered saline (PBS, pH 7.4) containing 4% paraformaldehyde, dissected, and embedded in paraffin for serial sectioning with a microtome. The spinal cords were sectioned transversely from T9 to T11, including at the contusion site, which could be visualized externally. The contusion site was transversely sectioned to a thickness of 15 μm.
Immunohistochemistry was performed using a mouse monoclonal antibody against neurofilament 200 (NF-200; N0142, Sigma-Aldrich, 1:100) or mouse polyclonal antibody against AR recombinant peptide corresponding to amino acids 76–144 of AKR1B10 (H00057016-A01, Novus Biologicals, Littleton, CO; 1:100). H00057016-A01 antibody is specific for AKR1B10 protein 30 and does not react with the AKR1B1 protein, as documented in Figure 1 in Ebert et al.’s work. 30 Both were visualized with biotinylated anti-mouse immunoglobulin G and streptavidin-conjugated horseradish peroxidase (Zymed, San Francisco, CA) in deparaffinized transverse sections at the contusion epicenter. Digitized images of four random fields (120 × 160 μm) within the ventral white matter of each rat were obtained with a 40× objective and examined using Adobe Photoshop CS (V 8.0, San Jose, CA) to determine luminance due to peroxidase staining with diaminobenzidine (DAB). Following color sampling of the DAB-stained structures, color selection was used to outline the areas occupied by DAB-stained axons. The luminosity function was used to determine axonal AR immunoreactivity. The counts and cross-sectional areas of the NF-200 immunoreactive axons were measured using particle analysis (ImageJ) within each image.
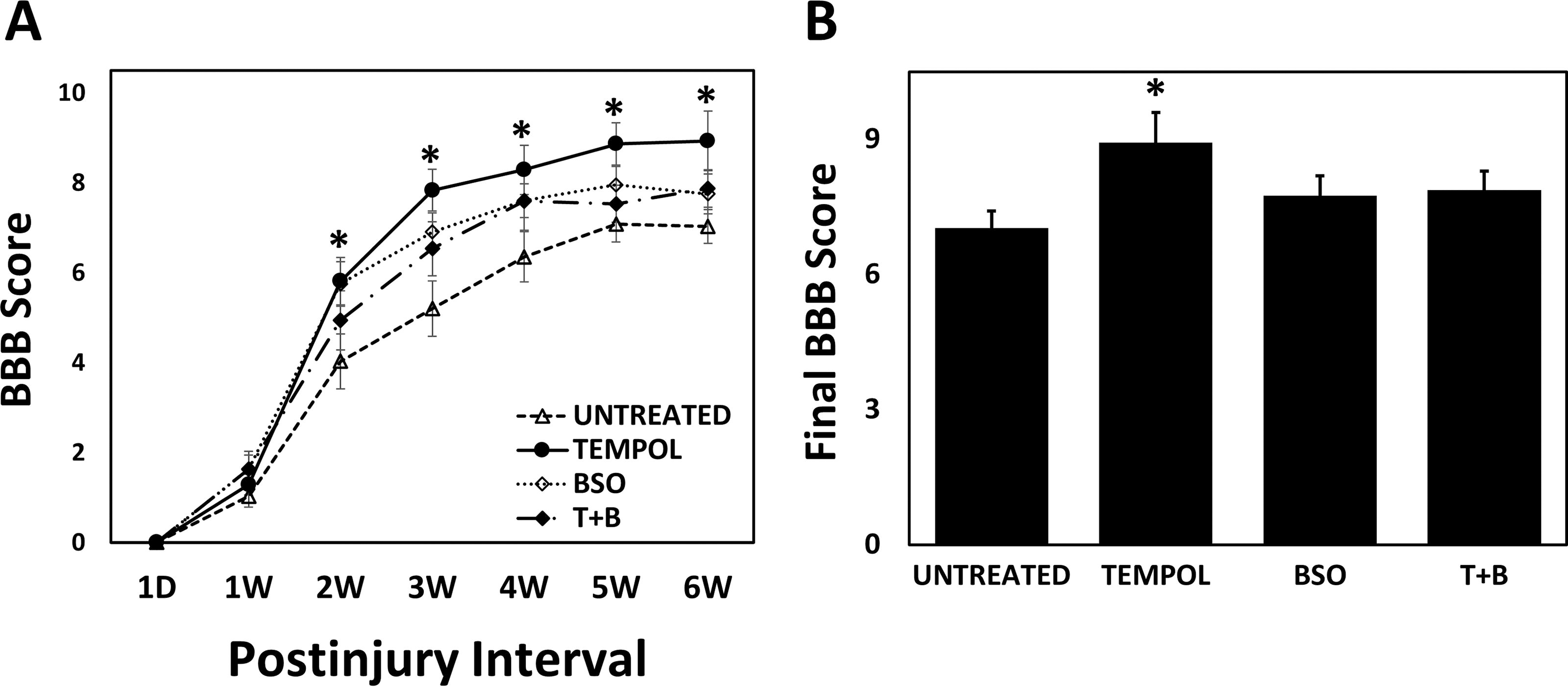
Tempol increases locomotor recovery after spinal cord contusion injury that is prevented by buthionine sulfoximine (BSO).
Sorbitol, glutathione, and gamma glutamyl cysteine ligase activity assays
Spinal cord segments (1 cm), including the contusion region, were dissected from anesthetized rats (pentobarbital sodium, 40 mg/kg, i.p.), weighed, homogenized in ice-cold 2 M HClO4, and centrifuged (2,000 × g, 20 min) to obtain supernatants that were neutralized with 2 M KOH. Aliquots of the supernatants were enzymatically assayed for sorbitol 31 by oxidation to fructose in the presence of sorbitol dehydrogenase (SDH) and NAD+ with the formation of NADH. In the presence of diaphorase, NADH reduces iodonitrotetrazolium to formazan, which was detected at 492 nm (Megazyme, Chicago, IL).
For the glutathione assay, spinal cord segments were homogenized in ice-cold PBS (pH 7.4) containing 1 mM EDTA and centrifuged (2,000 × g, 20 min) to obtain the supernatants. Protein concentration was determined using the Bio-Rad Bradford Protein Assay Kit (Hercules, CA), with bovine serum albumin as a standard. Aliquots of the supernatants were enzymatically assayed for total glutathione (GSH + GSSG), 32 which reduces 5,5′-dithiobis(2-nitrobenzoic acid) to 5-thio2-nitrobenzoic acid (TNB), while the GSSG product is recycled by glutathione reductase and NADPH. The TNB product was measured spectrophotometrically at 412 nm (Glutathione Assay Kit CS0260, Sigma-Aldrich). The assay used a standard curve of reduced glutathione to determine the amount of GSH + GSSG in spinal cord samples.
To assay gamma glutamyl cysteine ligase (γGCL) activity, spinal cord segments were homogenized in ice-cold 10 mM Tris-HCl containing 0.32 M sucrose and 1 mM EDTA. The homogenates were centrifuged (2,000 × g for 30 min). Protein concentration was determined using the BioRad DC Protein Assay Kit with bovine serum albumin as the standard. Aliquots of the supernatants were enzymatically assayed for γGCL activity, which was determined based on ADP formation using a pyruvate kinase-lactate dehydrogenase-coupled assay. 33 Reaction mixtures were equilibrated to 37°C and, in a final volume of 1.0 mL, contained 100 mM Tris HCl buffer, pH 8.0, 25 mM l-glutamate, 25 mM l-amino butyrate, 10 mM ATP, 10 mM phosphoenolpyruvate, 30 mM MgCl2, 150 mM KCl, 2 mM EDTA, 0.2 mM NADH, 17 μg pyruvate kinase, 17 μg lactate dehydrogenase, and aliquots of supernatant extracts were added to initiate the reaction. The rate of NADH utilization, detected at 340 nm, was used as a measure of ADP formation.
Immunoblotting of GCL
Western blot analysis of spinal cord γGCL synthetase was performed with rabbit polyclonal antibody to γGCL recombinant peptide corresponding to amino acids 295–313 of rat γGCL (ab17926, Abcam, Cambridge, MA). Spinal cord segments (1 cm), including the contusion region, were dissected from anesthetized rats (pentobarbital sodium, 40 mg/kg, i.p.) and homogenized on ice in lysis buffer containing 0.15 M NaCl, 5 mM EDTA, pH 8, 1% Triton X-100, 10 mM Tris-Cl, pH 7.4, 5 mM DTT, 100 μM PMSF, and 5 mM aminocaproic acid and centrifuged (5,500 × g, 20 min) to obtain the supernatants. Protein concentration was determined using the Bio-Rad Bradford Protein Assay Kit, with bovine serum albumin as a standard. Protein samples (120 μg) were subjected to 0.1% sodium dodecyl sulfate–polyacrylamide gel electrophoresis. The proteins were then electrotransferred onto a polyvinylidene difluoride membrane (Immobilon-P IPVH00010, Millipore Sigma, Burlington, MA). The blot was incubated overnight at room temperature in PBS blocking buffer containing 3% bovine serum albumin (Fraction V) and then in blocking buffer with rabbit polyclonal anti-rat γGCL primary antibody for 1 h at room temperature. The membrane was washed 3 times with 0.05% Tween 20 in PBS and incubated with horseradish peroxidase-conjugated anti-rabbit secondary antibody in blocking buffer for 45 min. After three additional washes, chemiluminescence of the antibody conjugates was detected with the Amersham ECL kit (RPN 2106) and exposed to X-ray film. The intensity of the chemiluminescence signal on the imaged blots (ChemiImager 5500, Alpha Innotech, San Leandro, CA) was determined using Adobe Photoshop CS (V 8.0, San Jose, CA).
Statistical analysis
The statistical significance of the effect of treatments on locomotor scoring was determined by mixed-factorial analysis of variance (ANOVA) with repeated measures and weekly scores by one-way ANOVA followed by Tukey’s Honest Significant Difference (HSD) post hoc multiple range test (IBM SPSS STATISTICS 27; Chicago, IL). Between-group comparisons of axon density and cross-sectional areas, AR immunoreactivity, γGCL expression and activity, and sorbitol and glutathione concentrations were performed using one-way and two-way ANOVA and Duncan’s post hoc test. Statistical significance was set at p < 0.05. In all cases, the post hoc result was reported prior to the overall ANOVA result.
Results
Tempol treatment (275 mg/kg, 20 min post-injury) significantly improved post-SCI locomotor recovery, as measured using the BBB locomotor scale, relative to the untreated-injured group (Fig. 1A, B), as previously observed. 4,5 To determine whether the enhanced recovery of locomotor function due to administration of tempol 4,5 is glutathione dependent, the effect of the γGCL inhibitor BSO 34 on the recovery of post-SCI locomotor function was examined. Inhibition of glutathione synthesis by BSO (150 mg/kg) administered 24 h before injury blocked the ability of tempol to enhance locomotor recovery. In contrast, BSO treatment alone did not affect recovery relative to that in the untreated group.
Wallerian degeneration, which results in the loss of axonal tracts and paralysis following SCI, is more rapid and prominent in the CNS than in the PNS. 6 This mode of axonal degeneration involves the formation of foci of swelling (“beading”) along axons leading to axonal fragmentation, discontinuity, and dysfunction, 5,6 as demonstrated in Figure 3A in our previous report. 5 In order to quantify the impact of tempol and BSO on the time course and extent of Wallerian degeneration following SCI, axonal swelling and density were measured in transverse sections of ventromedial white matter (Fig. 2C) immunostained for axonal NF-200 at the contusion site, as in our previous report. 5 All determinations were made during the first week after contusion injury, when the loss of spinal cord white matter 35 and axons 36 primarily occurs. The ventromedial axonal tracts were relatively spared following spinal cord contusion; nonetheless, injury led to progressive swelling of axons (Fig. 2A), as indicated by the increased (69–102%) axonal cross-sectional area during the first post-injury week. Spinal injury also led to a progressive loss (66–81%) of axons (Fig. 2B), as indicated by decreased axonal density during the first post-injury week, consistent with the ongoing Wallerian degeneration and elimination. Tempol treatment reduced axonal swelling to 48–70% and axon loss to 54–63%. However, prior inhibition of glutathione synthesis with BSO completely blocked tempol protection against axonal swelling and loss (Fig. 2A, B).
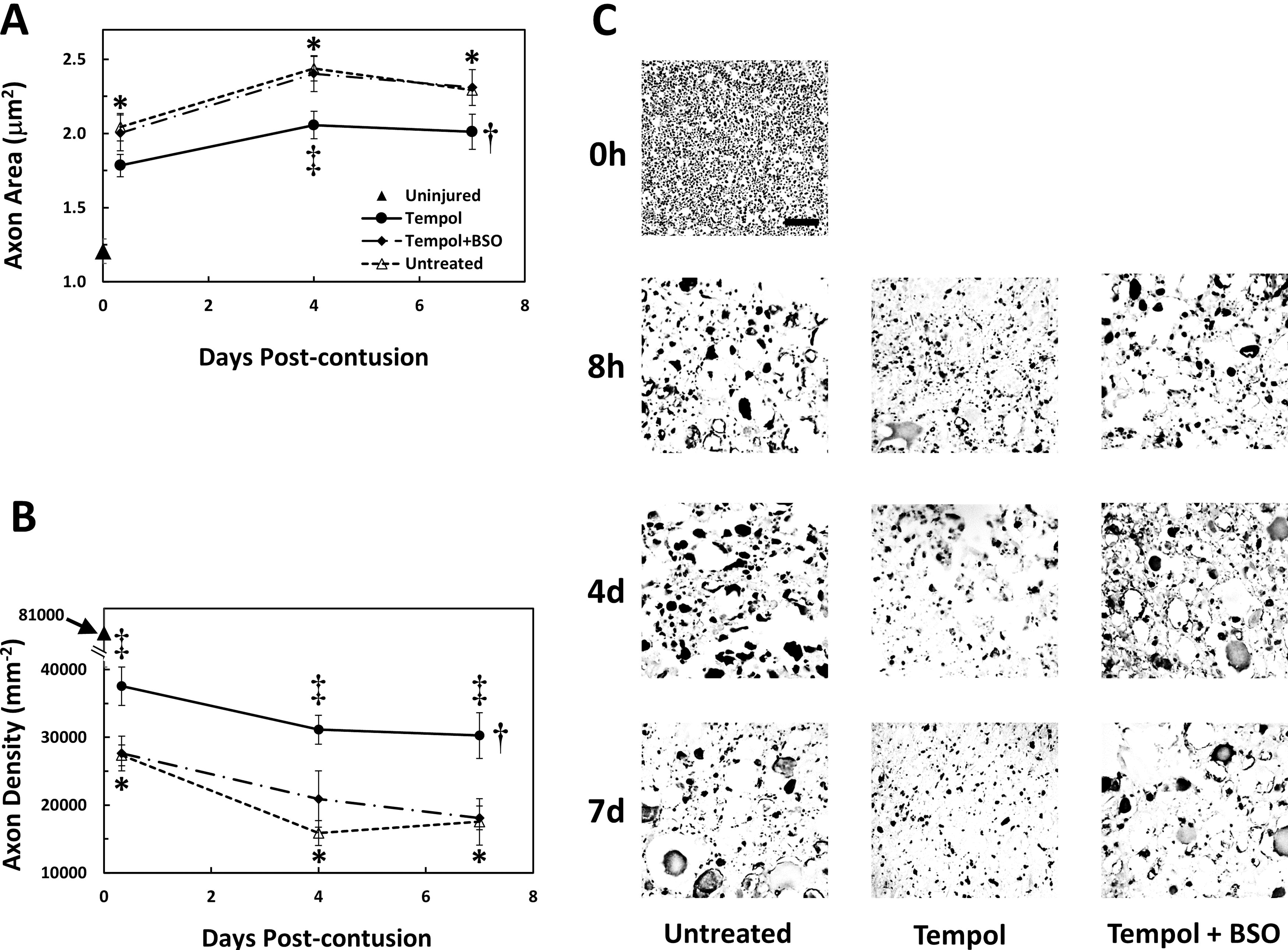
Tempol decreases Wallerian degeneration, which is prevented by BSO.
Previously, investigators have measured both spinal cord gray and white matter volume during the days and weeks after SCI at the injury epicenter as well as rostrally and caudally. 37 As opposed to gray matter, white matter did not exhibit swelling or edema and there was evidence of atrophy of white matter that occurred progressively at the epicenter. These results indicate that white matter tissue edema did not significantly affect the density of axons that were measured in our experiments.
Sorbitol determinations were performed on spinal cord segments centered at the contusion epicenter to determine whether polyol pathway activation by SCI was prevented by tempol (Fig. 3). A substantial increase (310%) in sorbitol concentration was observed at 8 h post-injury compared with laminectomized but uninjured rats. The increase in sorbitol concentration was reduced to 155% by tempol treatment (20 min post-injury), which was equal to the effect of treatment with the AR inhibitor sorbinil. These results are consistent with the ability of tempol to prevent AR activation following contusion injury.
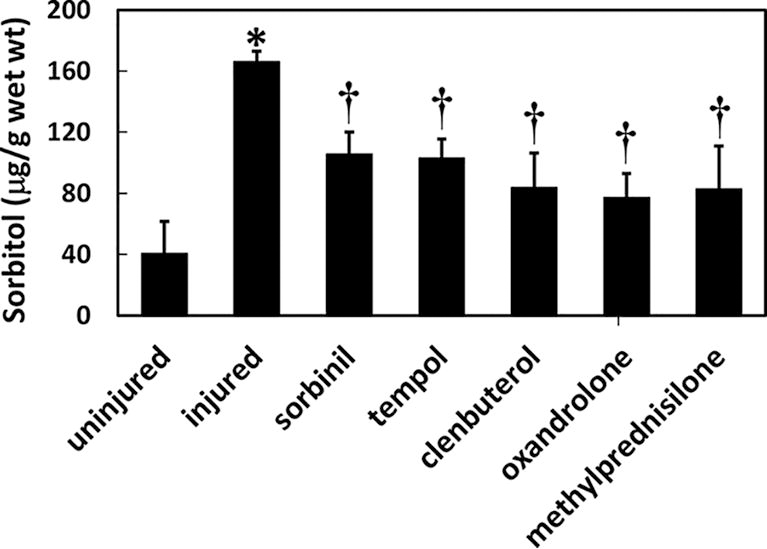
Spinal cord contusion increases sorbitol levels at the contusion site, which is reduced by tempol, sorbinil, methylprednisolone, oxandrolone, and clenbuterol. Mean values (±SE) of sorbitol concentration in 1-cm spinal cord segments centered at the contusion epicenter were measured at 0 (uninjured) or 8 h post-injury. *p < 0.05 (Duncan post hoc), significant increase in sorbitol concentration in untreated injured spinal cords at 8 h post-injury compared with uninjured spinal cords (0 h) and overall p = 0.005 (one-way ANOVA, n = 4–7). †p < 0.05 (Duncan post hoc), elevated post-injury sorbitol level was significantly reduced by treatment; p = 0.005 (one-way ANOVA, n = 4–7). ANOVA, analysis of variance; SE, standard error.
We also examined the possible role of AR overexpression 5 in axonal swelling and degeneration after contusion injury. Transverse sections of ventromedial white matter at the contusion site were examined for AR immunoreactivity. Axons at different stages of swelling and degeneration were immunopositive for AKR1B10 (Fig. 4B), a member of the aldo-keto reductase AKR1B subfamily. During the first week following injury, 615–720% increases in axonal AKR1B10 immunoreactivity were observed between 8 h and 7 days post-injury (Fig. 4A) compared with that in laminectomized but uninjured rats. Tempol treatment reduced axonal AKR1B10 immunoreactivity due to contusion to 264–400% above the uninjured level. Prior inhibition of glutathione synthesis by BSO abolished the ability of tempol to reduce AKR1B10 expression relative to the untreated-injured group (Fig. 4A, B).
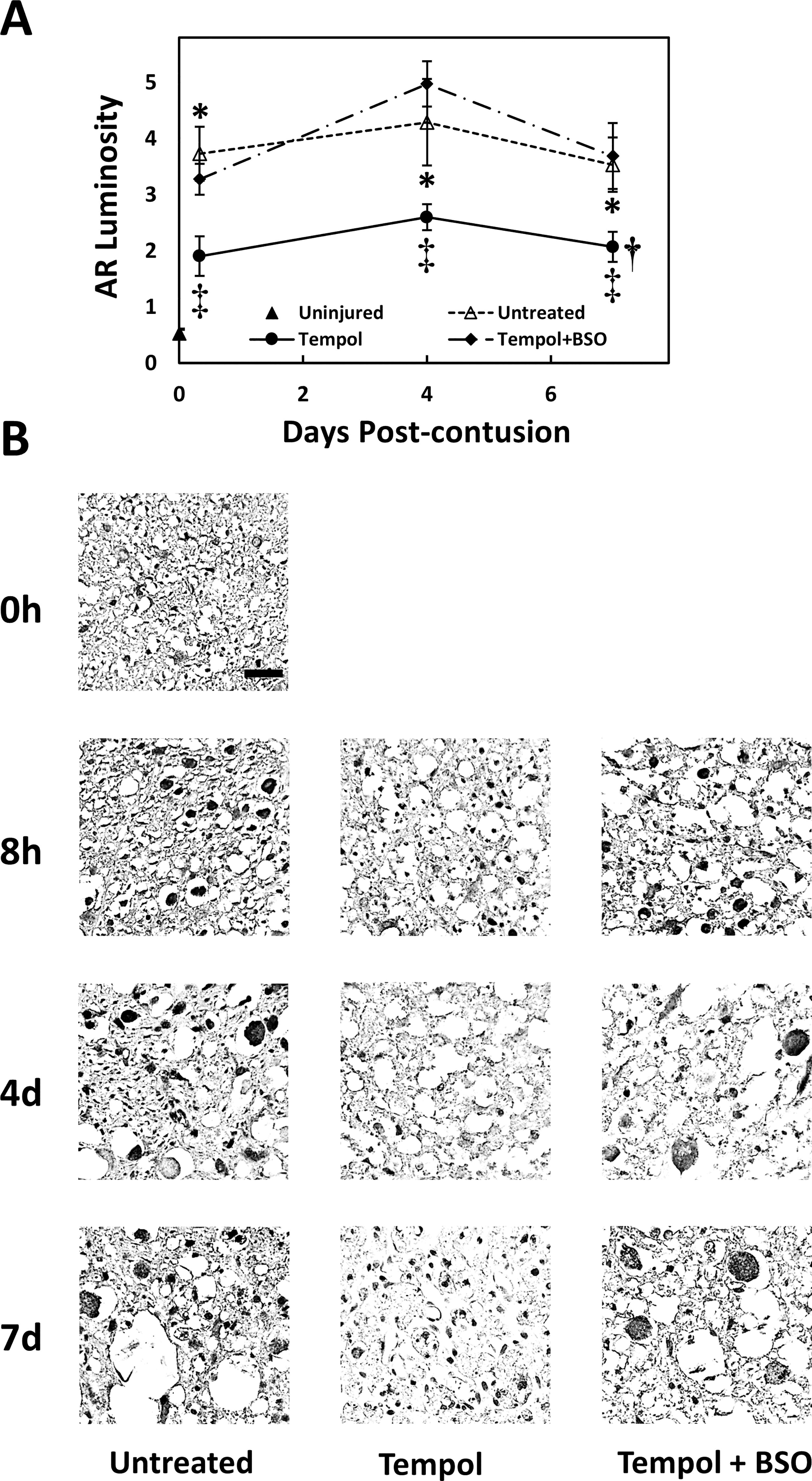
Tempol inhibits increases in post-injury AKR1B10 immunoreactivity at the contusion site, which is prevented by BSO.
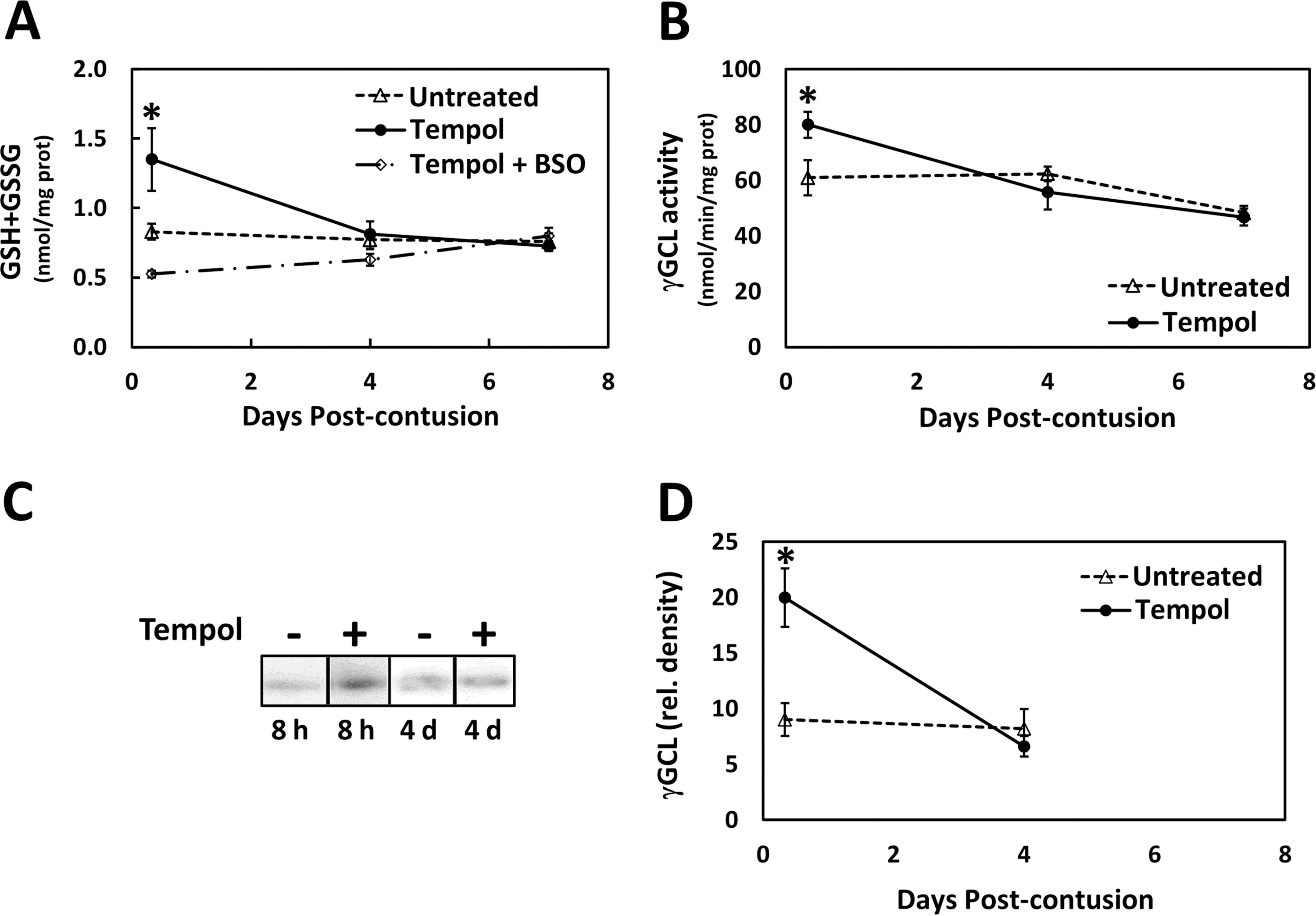
Tempol might inhibit axonal degeneration by preventing depletion of the glutathione pool, which can be reduced as a consequence of polyol pathway activity. Consistent with this hypothesis, we observed that a single post-injury tempol treatment (20 min post-injury) led to 63% greater glutathione content in the injured spinal cord at 8 h post-injury but not at 4 or 7 days post-injury (Fig. 5A). It is likely that the tempol had washed out between 8 h and 4 days. Administration of the γGCL inhibitor BSO prevented the increase in glutathione levels due to tempol administration. The increase in glutathione content at 8 h post-injury was accompanied by greater activity (32%, Fig. 5B) and expression (121%, Fig. 5C, D) of γGCL (EC 6.3.2.2). These results indicated that tempol increased the de novo synthesis of glutathione by increasing the abundance and activity of γGCL, the rate-limiting enzyme for glutathione synthesis.
Finally, we assayed the impact of drugs that spare axonal tracts and restore behavior after SCI but have putatively distinct targets on the tissue levels of post-SCI sorbitol. We found that methylprednisolone, 38 oxandrolone, 25 or clenbuterol 24,26 was significantly effective in reducing the increase in sorbitol concentration (Fig. 3), with efficacies similar to those of sorbinil and tempol. These results suggest that inhibition of the polyol pathway may be a common mechanism of action for all of these SCI protective agents.
Discussion
The main findings of this study were that following contusive SCI (1) tempol treatment improved the recovery of locomotor function, which was prevented by BSO (Fig. 1). (2) Tempol protects axons from post-SCI swelling and loss, characteristic of Wallerian degeneration, which is also prevented by BSO (Fig. 2). (3) Sorbinil and tempol had similar inhibitory efficacies against post-SCI polyol pathway activation (Fig. 3). (4) Tempol treatment inhibits the increased expression of AR (AKR1B10) in degenerating axons (Fig. 4), which was prevented by BSO treatment. (5) Tempol treatment led to greater glutathione content in the injured spinal cord, which was accompanied by increased expression and activity of γGCL (Fig. 5). Tempol-induced increases in glutathione content were prevented by BSO treatment. (6) We also found that methylprednisolone, oxandrolone, and clenbuterol reduced sorbitol levels in post-SCI tissues to a similar degree (Fig. 3).
Figure 6 summarizes a mechanistic model of spinal cord contusion-induced injury that incorporates our current and prior observations of post-SCI responses. Contusion injury to the spinal cord causes vasoconstriction and vasospasm in vessels supplying blood to the cord, resulting in (1) ischemia, as previously shown. 39 Subsequently, (2) hyperglucosis, that is, increased tissue levels of glucose, occurs within the spinal cord, 5 which may be due to greater sodium glucose cotransporter and glucose transporter 1 activity in response to hypoxia. 40 Hyperglucosis, in turn, provides excess substrate for the polyol pathway enzymes (3) AR and (4) SDH, which can generate O2 −, ROS, and oxidative stress, in part, via increased NADH/NAD+ and (6) NOX activity. 7,9 Increased polyol pathway activity also activates (5) NF-κB, 41 which induces (10) iNOS 42 and NO production, causing reactive nitrogen species (RNS) formation and further oxidative stress. ROS/RNS generation causes redox activation of (3) AR 8 and induces AR expression, 43 further increasing polyol pathway activity. NF-κB activation also opposes (7) Nrf2 activation 44 so that (9) γGCL expression 45 is suppressed, leading to (8) glutathione depletion 18,19 and further oxidative stress. Glutathione is necessary for glutathione reductase and peroxidase activity, 17 which opposes (11) oxidative stress as well as preventing 12-LOX activation 21 and the subsequent NOX activation 46 –48 necessary for (12) Wallerian degeneration. 22,45 (13) Paralysis and locomotor dysfunction result from degeneration of the ascending and descending spinal cord axonal tracts.
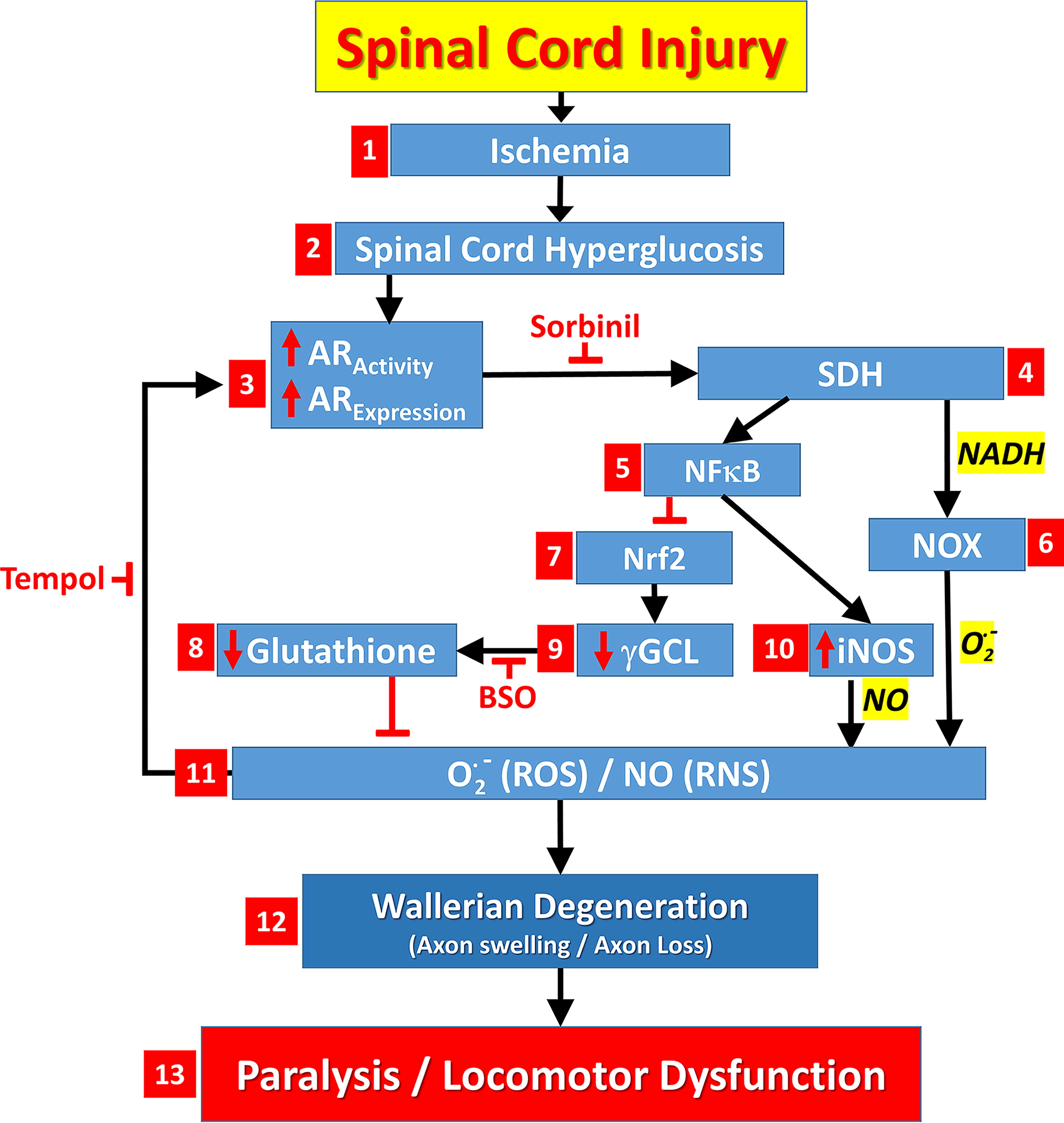
A mechanistic model in which spinal cord contusion-induced injury leads to ischemia/hypoxia and spinal cord hyperglycemia that activates the polyol pathway causing oxidative stress, glutathione depletion, Wallerian degeneration, paralysis, and locomotor dysfunction. For an explanation of this figure, see the Discussion section. (The Discussion section refers to numbered features within this figure using the notation: Fig. 6.x, where x equals the feature number).
Based upon our earlier studies, 4,5 more fully described in the Introduction section, we hypothesized that tempol might act by inhibiting Wallerian degeneration of spinal cord axons within the white matter axonal tracts. Consistent with this hypothesis, we observed that axonal swelling and loss of axonal density at the contusion site, characteristic of Wallerian degeneration, were partially inhibited by tempol (Fig. 2) and improved locomotor recovery (Fig. 1). Axonal preservation by tempol after SCI was expected, by analogy to our recent report on the inhibition of SCI-induced Wallerian degeneration by sorbinil, which correlated with improved functional recovery.
Inhibition of Wallerian degeneration and the associated enhancement of locomotor function by tempol were incomplete. However, the extent of improvement was functionally significant, since tempol treatment led to a transition from unsupported hindlimb movement to weight-supported standing and stepping. 5 It is well documented that recovery of locomotor function after SCI is positively correlated with the number of remaining axons. 49
How does tempol protect against axonal loss? Polyol pathway activation results in the generation of ROS and RNS, as described above with Figure 6 (features 4–10). Polyol pathway activation also results in the loss of antioxidant protection, specifically glutathione (Figs. 5 and 6.8). Tempol is a radical scavenger that protects against ROS and RNS production and glutathione loss.
BSO inhibition of glutathione synthesis 34 (Fig. 5) completely abolished the locomotor improvement caused by tempol (Fig. 1). Similarly, the critical importance of glutathione in inhibiting Wallerian degeneration was demonstrated by the complete BSO blockade of the tempol-induced reduction of axonal swelling and loss due to SCI (Fig. 2). These results suggest that Wallerian degeneration is a direct metabolic consequence of axonal glutathione loss. This is similar to the glutathione dependence of Wallerian degeneration observed in the PNS. 50
We reported previously that Wallerian degeneration of spinal cord axons can be reduced by AR inhibitors. 5 AR converts glucose to sorbitol via the polyol pathway (Fig. 6.3), which is activated by SCI. Superoxide (generated by excessive mitochondrial activity 10 ) combines spontaneously with nitric oxide to produce peroxynitrite, which oxidizes a regulatory cysteine in AR thereby increasing its catalytic activity by about threefold (Fig. 6.3). 11 The radical scavenger tempol can prevent redox-mediated stimulation of AR enzymatic activity, thereby preventing the post-SCI increase in sorbitol. We observed that tempol inhibited sorbitol production in the injured spinal cord to the same extent as the AR inhibitor sorbinil (Fig. 3). It is likely that activation of AR was decreased by tempol scavenging of superoxide, similar to the prevention of AR activation by superoxide radical scavenger observed in cardiac tissue. 11
Reduced glutathione is the predominant cellular redox buffer that counteracts oxidative stress. 15 Glutathione is irreversibly consumed by conjugation reactions, in which glutathione adducts are formed with toxic products produced by ROS 16 generated via polyol pathway activity. 9 Glutathione is also a substrate for glutathione peroxidases such as GPX4, which reverses lipid peroxidation that contributes to Wallerian axonal degeneration. 17 SCI is characterized by the depletion of glutathione 18,19 and greatly increased rates of lipid peroxidation, which are inhibited by tempol, 51 thereby protecting axonal tracts.
Tempol treatment led to greater glutathione content in the injured spinal cord (Fig. 5A). This is due to increased glutathione synthesis by γGCL (Fig. 5B), a result of increased γGCL enzyme abundance (Figs. 5C and 6.9). Increased γGCL expression is stimulated by Nrf2 activation (Fig. 6.7), a transcription factor activated in response to oxidative stress. 45 Opposing this, polyol pathway activity activates the transcription factor NF-κB (Fig. 6.5), 41 which antagonizes Nrf2-dependent transcription. 44 Tempol, by inhibiting the polyol pathway, could allow increased Nrf2-dependent transcription, promoting γGCL expression and resulting in greater glutathione synthesis. Consistent with this hypothesis, tempol has been shown to decrease NF-κB activation and increase Nrf2 activation. 52
Tempol treatment also reduced expression of AKR1B10 in the degenerating axons (Fig. 4). The SCI-associated increase in AKR1B10 abundance (Fig. 6.3) might be attributed to the upregulation of AR gene expression by nitric oxide (NO), which has been reported for AKR1B10 43 and AKR1B1. 53 Increased NO production by iNOS (Fig. 6.10) is a consequence of NF-κB induction (Fig. 6.5), 42 which in turn is stimulated the polyol pathway enzyme SDH (Fig. 6.4). Thus, tempol reduces sorbitol levels by preventing increases in both AR expression (Fig. 4) and AR activation (Fig. 3). Although some studies have found that the glucose reductase activity of AKR1B10 is lower than that of AKR1B1, 54 –56 experiments with live cells are consistent with the equivalent glucose reductase activity of these two enzymes. 57 Clarification of the relative roles of AKR1B10 and AKR1B1 in SCI and recovery promises to be an interesting topic for further research.
Finally, we examined whether other agents that promote behavioral recovery and spare axonal tracts after SCI such as methylprednisolone, 38 oxandrolone, 25 or clenbuterol 24,26 inhibit polyol pathway activity. We found that these agents were equally effective in inhibiting polyol pathway activity (Fig. 3), suggesting the involvement of a common pathway. Methylprednisolone or clenbuterol might reduce hypoxic stress by restoring normal perfusion at the injury site, 39,58 thereby preventing hypoxia (Fig. 6.1), excess glucose uptake, 5 AR activation, and loss of glutathione (as in this report). Alternatively, methylprednisolone 20 can act as an antioxidant that protects axonal tracts in a manner similar to tempol. 51 In addition to our current observations, several other agents that spared spinal cord axonal tracts and improved locomotor function such as resveratrol, 59 cannabidiol, 60 curcumin, 23 or quercetin 61 are direct AR enzyme (Fig. 6.3) inhibitors 62 –64 consistent with a common mechanism of action for many neuroprotective agents.
Conclusions
This study demonstrates that treatment with the SOD mimetic tempol improved the recovery of locomotor function following spinal cord contusion injury by inhibiting Wallerian degeneration of spinal cord axonal tracts. Previously, we found that post-SCI Wallerian degeneration can be reduced by inhibitors of AR, which converts glucose to sorbitol via the polyol pathway. We now report that tempol inhibited sorbitol production in the injured spinal cord to the same extent as the AR inhibitor sorbinil and prevented the increased expression of AR (AKR1B10) within degenerating axons. Therefore, we propose a positive feedback loop, in which increased AR cycling generates excess ROS/RNS, resulting in increased AR-specific activity and protein expression. Tempol treatment led to greater glutathione content in the injured spinal cord, which was accompanied by increased expression and activity of γGCL, interrupting the feedback loop at a different point than sorbinil (feature 11 instead of feature 3 in Fig. 6). Conversely, administration of the γGCL inhibitor BSO abolished all the effects of tempol administration (by depleting glutathione independently of AR-dependent ROS/RNS production). Together, these results support a role for polyol pathway activity in depleting glutathione, resulting in Wallerian degeneration following SCI. Other agents known to spare axonal tracts, such as methylprednisolone, oxandrolone, and clenbuterol, reduce sorbitol levels in post-SCI tissues, suggesting that preventing AR activation is a common consequence of disparate post-SCI interventions.
Transparency, Rigor and Reproducibility Summary
A priori power analysis was performed to determine the number of rats required for each experiment. A sample size of 16 rats per group was planned based on an expected effect size calculated to yield 80% power to detect significant behavioral recovery using mixed-factorial ANOVA with repeated measures with a p-value of <0.05. For histological and biochemical measurements, a sample size of seven rats per group and, for western blots, a group size of six rats were planned based on expected effect sizes calculated to yield 80% power to detect significant histological and biochemical outcomes using two-factor ANOVA with p < 0.05. Five animals died due to post-injury complications and were excluded from the study. The rats were randomly assigned to each experimental group. The rats were tested between 0900 and 1400 in a fed state. The investigators who conducted the outcome assessments were blinded to the assignment of the experimental groups. The statistical tests used were based on the hypothesized normality of the distributions and the sample sizes indicated the number of independent measurements. Homogeneity of variance between experimental groups was determined using the Levene and Brown–Forsythe tests. Correction for multiple comparisons between experimental groups was performed using Tukey’s (HSD) and Duncan’s post hoc multiple range tests. The experimental injury model and primary outcome measures were established as standards in the field. The validation of the methodology, which uses a mixed-factorial ANOVA with repeated measures, is included in Basso, Beattie, and Bresnahan and Basso et al. 28,29 Data from this study will be accessible in the FAIR data repository upon contacting the corresponding author. The article will be published using the Mary Ann Liebert, Inc. ‘‘Open Access’’ option, under an appropriate license. Correspondence and requests for additional materials should be addressed to R.J.Z.
Footnotes
Authors’ Contributions
Conception and design: R.J.Z. and J.D.E. Acquisition of data: X.W. and N.O. Data analysis and interpretation: R.J.Z., J.D.E., and A.M.B. Drafting the article: R.J.Z. and A.M.B. Reviewed submitted version of the article: All authors. Approved the final version of the article on behalf of all authors: R.J.Z. Statistical analysis: R.J.Z. Study supervision: R.J.Z.
Author Disclosure Statement
The authors report no conflicts of interest concerning the materials or methods used in this study or the findings specified in this article.
Funding Information
This work was supported by grants (to R.J.Z.) from the National Institutes of Health (