
Abstract
Sleep
Introduction
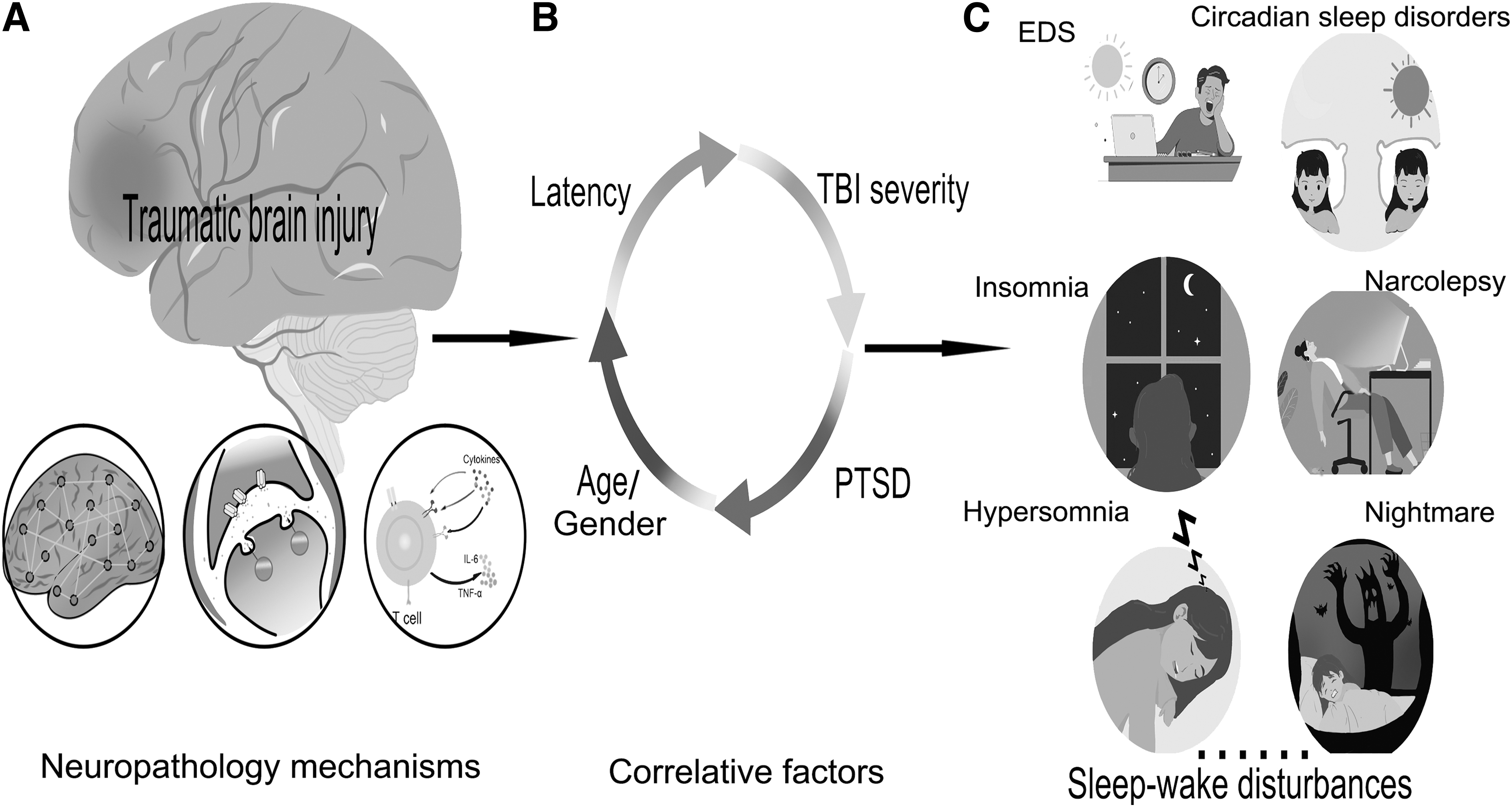
Traumatic brain injury (TBI) is one of the top three causes of injury-related death and disability. The population incidence of TBI in China is estimated to be ∼55.4–64.1 per 100,000, 1 and combined with the large population base, the total number of TBI survivors is much higher than in most other countries. External mechanical forces impact on the brain to cause primary TBI, and the subsequent secondary injuries include neuron cell death, ischemia and hypoxia, hemorrhage, blood–brain barrier (BBB) damage, and inflammatory reactions. These secondary injuries further aggravate the primary injury; induce the occurrence of functional network changes, 2 neurotransmitter alterations, 3 and inflammatory response 4 (Fig. 1A); and lead to permanent changes in neurological function. 5 Most TBI survivors report post-traumatic sequelae such as abnormal emotional behavior, cognitive impairment, sensory disorder, and sleep–wake disturbances (SWDs). 6
Neural mechanisms and characteristics of sleep
SWDs occur in 30–70% of the TBI population, 7 irrespective of the severity, and can be manifested in different periods post-injury, with some patients experiencing persistent SWDs in the chronic phase. The occurrence of SWDs in TBI patients will hinder the process of rehabilitation, which can affect the quality of life, cause interpersonal barriers, reduce work efficiency, and induce diseases such as those with metabolic, cardiovascular and cerebrovascular, and neuropsychiatric symptoms in severe cases. 6,7 More evidence suggests that SWDs may have a detrimental impact on the neurological recovery and long-term neurodegeneration of TBI patients.
Based on the abovementioned points, a scoping review of the features and neural mechanisms of post-TBI SWDs is warranted. It is hoped that through this review of TBI, SWDs will receive greater recognition by clinicians, medical insurance departments, and society. In addition, by understanding the current research progress on the neuropathological mechanism of post-TBI SWDs, it provides a reference for future research directions and the opening of new treatment methods, with the aim of fundamentally improving the quality of life of TBI survivors.
Correlative Factors of Post-TBI SWDs
To date, there have been clinical reports on post-TBI SWDs and potential correlative factors in individuals with TBI. Several studies have assessed factors associated with SWDs after TBI, including timing of injury, severity, age, and post-traumatic stress disorder (PTSD), but there is considerable heterogeneity among these studies in terms of population and assessment (Fig. 1B).
Temporal relationship between TBI and SWDs
Studying the development trajectory of post-TBI SWDs has great significance for early treatment and monitoring of the dynamic development of the disease. In the acute phase of moderate-to-severe TBI, it is difficult for patients to maintain a 24-h sleep–wake cycle. Sleep is often fragmented (the patient is unable to maintain longer continuous bouts of sleep), sleep efficiency is low, and the average night-time sleep is only 5.7 h 8 Sleep efficiency was lower for moderate than for severe TBI at 1 month (66.7% vs. 73.5%) and at 3 months (72.1% vs. 76.2%) post-injury. As patients reintegrated into the community, sleep efficiency showed improvement (reaching 82%), but did not reach a normal threshold (e.g., ≥85%). Variations in sleep efficiency across studies may be attributed to the use of different assessment methods. 9 When Baumann and coworkers evaluated sleep in patients 6 months post-TBI, they found that there were still different forms of SWDs, excessive daytime sleepiness (EDS) (25%), hypersomnia (22%), and insomnia (5%). 10 In a prospective follow-up study, at 18 months post-TBI, objective sleep examination in patients with TBI showed increased sleep needs, >60% of patients showed EDS. 11 In another follow-up study, the authors performed sleep examinations on 51 patients 3 years post-TBI, and 67% of patients continued to experience symptoms, including fatigue (35%), lethargy (27%), EDS (12%), and insomnia (10%). 12
Although some studies suggest that there is no significant correlation between the occurrence of SWDs and recovery time post-TBI, 12 more studies suggest that SWDs caused by TBI are chronic and that they increase over time. 13 The reasons for this inconsistent conclusion may be related to several factors: patients with mild-TBI (mTBI) often have transient loss of consciousness, whereas the disturbance of consciousness in moderate-to-severe TBI can last for several days or even longer, which has a great troubling impact on the objective evaluation of sleep. Different research centers have different definitions and standards for SWDs. TBI patients also have other symptoms, such as pain, depression, and cognitive impairment, which factors are also related to the occurrence of SWDs. However, the consistent conclusion is that the main complaint of SWDs varies with time post-TBI. Insomnia is predominant in the subacute phase, with difficulty initiating and maintaining sleep, whereas EDS is the predominant symptom in the chronic phase. 11,12,14 In conjunction with the aforementioned delineations, vigilant monitoring of the evolution of post-TBI SWDs is imperative. The early detection and timely intervention of such disruptions are of considerable consequence for the affected demographic.
TBI severity and SWDs
The impact of TBI severity on the occurrence of SWDs is still under investigation. It has been reported that mTBI patients represent a high proportion of the population, accounting for 70–90% of the total TBI population, who have a higher incidence of SWDs than do moderate-to-severe patients. 7 Further, it is pertinent to acknowledge that the severity of the injury exerts a discernible influence on the typology of SWDs. 13 Studies by Masson and coworkers have shown that patients with mTBI are more likely to experience EDS and insomnia, whereas patients with severe impairment are more likely to experience hypersomnia. 15 And more studies suggest that regardless of the severity of the brain injury, TBI patients are generally susceptible to SWDs. 12
Recent findings suggest that sleep macrostructure, including sleep onset, sleep duration, and sleep cycle, is preserved in mTBI. Microstructural features of non-rapid eye movement (NREM) sleep in patients with moderate-to-severe TBI, including electroencephalogram (EEG) spectral power, slow wave activity (SWA), and sleep spindles, may be affected. 16 However, when the sleep quality of 34 mTBI patients was evaluated by polysomnography (PSG) and the Pittsburgh sleep quality index (PSQI), the results showed that the subjective sleep quality of mTBI patients was significantly worse than the objective assessment. During NREM sleep, there was a significant increase in β-wave power in the occipital lobe, whereas no between-group differences were found in the characteristics of slow waves and spindle waves. The increase of β-wave spectrum power during NREM sleep may explain the subjective report of some patients with poor night-time sleep quality and the feeling of being awake. The mechanism responsible for the increase in β waves during NREM sleep is unclear and may be attributed to TBI itself or may be related to coexisting psychiatric symptoms such as pain and anxiety. 17
Most mTBI cases show no obvious abnormalities on conventional imaging, but can be detected on high-resolution magnetic resonance imaging (MRI), revealing compromised perivascular spaces, which are often associated with sleep disturbances, 18 including increased sleep need and poor sleep quality. 19 Although moderate-to-severe TBI is mostly accompanied by intracranial hemorrhage, brain structures have varying degrees of damage, especially the cerebral cortex. This different severity of anatomical damage can explain the corresponding functional changes, which may be the neuroanatomical mechanism of different severities of TBI inducing different forms of SWDs.
SWDs in children with TBI
The main presentations of post-TBI SWDs in children are difficulty falling asleep or maintaining asleep, sleeping longer or shorter than usual, or difficulty keeping alert during the day. These SWDs may persist for years after the initial TBI. 20 In a study of 15 children 0.5–6 years post-TBI, Sumpter and colleagues 21 found that children with moderate-to-severe TBI had more sleep disturbances than sibling controls and that these patients had lower sleep efficiency and longer sleep onset latencies. The possibility of developing post-TBI SWDs increases with the age of children, and older children have a shorter latency to develop SWDs than younger ones. Children with severe TBI had a shorter latency of SWDs, whereas those with mild or moderate TBI had comparable onset. 22
Evidence suggests that the occurrence of SWDs predicts deficits in executive functioning, increased emotional and behavioral disturbances, poor communication and self-care skills, and decreased participation in family, school, and community activities. 23 In view of the particularity of children patients, the occurrence of SWDs should be closely monitored post-TBI. In the mTBI child population, the incidence of SWDs in children reported by parents was higher than that self-reported by children or than objective monitoring. 24 The guardian's opinion should be more inclined when evaluating SWDs, and objective sleep examination should be performed when necessary. Because of the importance of sleep for learning, memory, and neuroplasticity in this special population, it is necessary to actively explore the relevant mechanisms leading to the occurrence of post-TBI SWDs.
Gender effects on SWDs post-TBI
In a military-related mTBI study, findings consistently demonstrate that females experience a higher incidence of poor sleep quality than males. 25 Another study aims to explore the impact of gender on the prevalence of sleep disturbances in older adults (≥ 60 years of age) who have had a TBI. Results indicate that females experience a more pronounced effect of TBI on sleep disturbances than males, 26 and that females have a higher prevalence (44%) of excessive daytime sleepiness, in contrast to only 13% of male participants. 27 In sport-related concussion patients, females are more susceptible to sleep disturbances and may require extended time for full recovery post-concussion. The disparity in concussion sequelae between the sexes is likely attributable to an interplay of biomechanical differences, hormonal variations, and social factors. 28
In an investigation involving a mouse model with diffuse TBI, notable sex-specific differences in inflammatory and sleep responses were observed post-TBI. Female mice exhibited interleukin-6 (IL-6) levels that were 77–124% higher than those in male mice. Conversely, male mice that sustained TBI demonstrated an increased neutrophil count in their blood relative to female mice. Further, sleep analysis revealed that male TBI mice experienced a 11–17% increase in total sleep duration compared with female TBI mice. 29 A mouse model of mTBI has revealed that female mice augmented the amplitude of post-hyperpolarization following action potentials in orexin neurons, an effect not observed in male mice. Conversely, decreased the action potential threshold exclusively in male mice, with no corresponding change detected in females. Moreover, mTBI was found to dampen afferent excitatory input while enhancing afferent inhibitory input to orexin neurons. 30 These findings highlight the potential sex differences in sleep-related outcomes following TBI and underscore the necessity for tailored approaches in the assessment and management of sleep disturbances in this population.
PTSD and SWDs
There is a correlation between SWDs and PTSD post-TBI. 31 Neuroendocrine abnormalities post-TBI may be the associated factors of PTSD and SWDs. Compared with healthy controls, PTSD patients had increased number of nocturnal awakenings, which was positively correlated with adrenocorticotropic hormone (ACTH) levels. The levels of ACTH and cortisol are negatively correlated with slow-wave sleep, and ACTH can be used as an independent predictor of slow-wave sleep disturbance in PTSD patients. Elevated ACTH in PTSD patients suppresses slow-wave sleep, revealing the phenomenon of fragmented sleep in PTSD patients. 32
Other concomitant symptoms of TBI, such as depression and pain, also have a significant impact on the sleep of patients. Suboptimal self-reported sleep quality and insomnia were found to be robustly associated with heightened depressive and/or anxiety symptomatology. However, these associations were not evident when sleep quality was assessed through objective metrics in TBI patients. Longitudinal investigations noted that the exacerbation of depressive manifestations over time correlated with persistent insomnia and ongoing sleep disturbances among TBI patients. 33 Additionally, interventional research indicated that amelioration of sleep-related issues led to a notable improvement in the symptoms of depression and anxiety. In patients with mTBI accompanied by pain, diminished sleep quality exhibited a significant correlation with elevated fast β and δ EEG oscillations across frontal, central, and occipital regions, as detected in EEG readings throughout all stages of sleep. 34 Notably, mTBI patients experiencing pain demonstrated a pronounced augmentation in rapid EEG frequency bands, predominantly during REM sleep, as well as an increase in β activity during NREM sleep stages when compared with mTBI patients. 35 Pain is intimately linked with suboptimal sleep quality and emerges as a pivotal element in the management of post-concussive symptomatology. Moreover, the concomitance of pain and sleep dysregulation appears to prognosticate the most adverse long-term cognitive and affective sequelae.
Sleep Structure Characteristics of TBI-related SWDs
When evaluating the SWDs of patients post-TBI, the objective PSG examination and the subjective questionnaire scale are consistent in reflecting the changes in sleep structure, which are manifested as affected sleep continuity, increased night-time wakefulness, frequent awakenings, and sleep efficiency decreased. For a more detailed analysis of the macroscopic and microscopic sleep structures of SWDs patients, PSG should be the first method to be considered. In the acute phase of moderate-to-severe TBI, PSG examination showed no significant change in the N1 phase of NREM sleep, a decrease in the proportion of N2 phase, an increase in slow-wave sleep, a shortened REM sleep latency, and a decrease in REM sleep. In the chronic phase, the proportion of slow-wave sleep was still relatively high. 16,36 In patients with mTBI, however, PSG examination showed that the average duration of nocturnal awakenings was longer, N2 sleep and REM sleep were shorter than normal, and the changes in slow-wave sleep were not statistically different. 37 In the penetrating brain injury model of rats, it was found that EEG recordings 24 h after injury showed increased slow-wave sleep and spindle waves, and decreased REM sleep; correspondingly, structural reorganization was found around the injured cortex, such as axonal sprouting and synaptic plasticity. These morphological changes play an important role in neuroplasticity or regeneration. 38 Combined with the EEG changes and morphological changes in the acute sleep phase post-TBI, we have reason to believe that the increase in slow-wave sleep in the acute phase is a positive response of the brain to trauma and is conducive to the recovery of neurological function. This view has indeed been confirmed by studies showing that TBI patients with increased slow-wave sleep during the acute phase had significantly improved cognitive function at 6 months compared with patients with no changes. 16 However, there are few studies on the significance and mechanism of increased slow-wave sleep in the chronic phase in TBI patients, and further research is needed.
There are also characteristic changes in different frequency brain waves on the EEG of SWDs patients post-TBI. Rao and colleagues performed spectral analysis on the EEG frequency bands of TBI patients. They noticed that compared with healthy controls, several frequency bands of the NREM sleep (lower δ, higher α and β power) and REM sleep (lower β and higher δ power) appear abnormal. 39 Another study reported similar findings in 24 mTBI patients who had higher β and γ wave activity (often associated with arousal from sleep) during NREM sleep than 18 healthy controls, whereas REM sleep periods have lower δ bands (often associated with deep sleep). 35 Arbour and colleagues 17 showed that mTBI athletes had increased δ wave activity and decreased α wave activity during wakefulness, which may make sleep inertia (a groggy and disoriented state experienced immediately upon awakening from sleep) slower diffuse in mTBI patients. Combining the distribution changes of the EEG spectrum in TBI patients when awake and during NREM sleep and REM sleep, we can draw the following inferences: the increase in the activity of δ waves in the wake period may be the cause of drowsiness and executive ability decline in mTBI patients during the day. The increase of β wave activity during NREM sleep may make it difficult for patients to maintain sleep continuity and cause them to wake up frequently. Whether the changes in brain waves on the EEG post-TBI can be used as a marker of treatment effect still needs to be verified with a large sample.
Presentation Types of TBI-induced SWDs
A systematic review found sleep disturbances reached in 30–70% of patients with TBI. 7 SWDs were even more prevalent (87%) in war veterans with TBI. 40 Common SWDs associated with TBI mainly include insomnia, EDS, hypersomnia, sleep-related breathing disorders (such as OSA), narcolepsy, and circadian rhythm disturbances (Table 1). Other sleep disorders (such as sleep-related movement disorders) may also be present, but there is little information about them. In this section, we mainly describe the characteristics of common post-TBI SWDs (Fig. 1C).
Characteristics of Common SWDs Post-TBI
EDS
EDS is characterized by daily or near-daily episodes of an irrepressible need to sleep, or unintentional lapses into sleep at potentially inappropriate times, and is one of the most common post-TBI arousal disorders. The diagnosis of EDS can be determined by subjective or objective criteria. Among the 71 patients who underwent sleep examination 60 months post-TBI, 47% had a mean Multiple Sleep Latency Test (MSLT) sleep latency (typically >10 min) of ≤10 min and 18% had a sleep latency of ≤5 min. It is worth noting that 30% of the patients scored as having EDS on the Epworth Sleepiness Scale (ESS) had scores of ≥10 points, whereas the normal range is typically ≤8 points. However, their PSG showed normal respiration and a normal periodic limb movement index. 41 In another prospective methodological study, 42 it was confirmed that EDS diagnosed by MSLT was more prevalent than subjective EDS measured by ESS. The authors believe that some patients may underestimate post-traumatic EDS. The correlation between ESS scores and MSLT examination results is poor; therefore, the diagnostic rate of subjective rating scales may be lower than the actual occurrence. Clinicians must keep this fact in mind when treating TBI survivors who are engaged in high-risk occupations, and must make early warning guidance key in the treatment plan.
The occurrence of EDS post-TBI is the result of many factors. Recent studies have shown that the presence of OSA is a major cause of exacerbated post-TBI EDS, 43 attributable to poor night-time sleep quality and increased daytime sleep propensity. Neuroendocrine abnormalities are common complications post-TBI, and lower basal cortisol levels are associated with the development of fatigue, depression, and anxiety, which can also exacerbate EDS. 44 The information suggests that in the treatment of EDS, it is crucial to first identify any comorbid conditions that may impact EDS. By addressing and treating these coexisting diseases, significant relief of EDS symptoms can potentially be achieved.
Hypersomnia
Hypersomnia is another major problem post-TBI and is often referred to as post-traumatic lethargy. Increased sleep requirements post-TBI may represent a debilitating symptom, and affected patients face difficulties with social, family, and work-related activities. 45 In a follow-up study specifically characterizing post-TBI SWDs, hypersomnia was present in 58% of the patients. These patients do not have subjective EDS, but sleep duration is prolonged by 1–2 h for every 24 h, mainly slow-wave sleep duration. 46 These patients may underestimate how much sleep they actually need, and report significantly shorter sleep times per 24 h on sleep diaries than those measured by actigraphy. 47 Additionally, patients with post-TBI hypersomnia are at risk for secondary EDS, which may be a compensatory effect of sleep deprivation. 42
Insomnia
Insomnia is a sleep disorder in which people may have difficulty falling asleep or staying asleep as long as desired. The prevalence of insomnia post-TBI varies because of differences in diagnostic methods and definitions. In a recent prospective cohort study of 2022 TBI patients, 61.6% reported persistent mild insomnia symptoms that gradually resolved over time; 4.5% reported persistent severe insomnia symptoms. In the same study, insomnia symptoms were relieved in 2.2% of patients after 12 months, whereas 0.7% of patients had no insomnia symptoms initially, but developed severe insomnia symptoms after 12 months. Multivariate regression analysis showed that female gender, psychiatric symptoms, and imaging abnormalities were related factors for the occurrence of insomnia. 48 In another systematic review report, the incidence of insomnia in TBI patients ranged from 30% to 70%. 49 In prospective studies using objective measures, insomnia was found in only 5% of patients. 10 The reason for such a large discrepancy between subjective scales and objective diagnostic methods is that patients with TBI may overestimate insomnia symptoms. Compared with subjective, questionnaire-based measures of insomnia, PSG showed a lower incidence of sleep disturbance. 50 Another factor contributing to the variation in the incidence of insomnia post-TBI among studies may be that some studies did not consider the effect of psychiatric symptoms on insomnia. Pain, for example, is a common complication of TBI and a common cause of SWDs. In a retrospective study, 45% of TBI survivors reported insomnia, with pain being the main underlying contributing factor. Other contributing factors include dizziness, anxiety, and depression, 51 and the occurrence of insomnia post-TBI can not only impede recovery but further exacerbate these symptoms.
Circadian sleep disorders
Circadian rhythm sleep disorders, a family of sleep disorders that affect the timing of sleep, can be caused either by dysfunction in the biological clock system, or by misalignment between endogenous oscillators and externally imposed cues. There are a few studies on circadian post-TBI SWDs, probably because body temperature and melatonin levels were not measured in most studies, and there is a lack of objective data for diagnosing circadian SWDs. Therefore, the incidence of circadian post-TBI SWDs is difficult to assess. In a cohort study evaluating SWDs in 42 TBI patients using actigraphy, salivary melatonin measurements, and body temperature measurements, circadian SWDs were found in 15 patients. 52 In addition, Shekleton and colleagues 53 monitored the nocturnal melatonin levels of 23 TBI patients 1 year after injury, which showed that the melatonin level of TBI patients was significantly lower than that of age- and sex-matched healthy controls. Likewise, in a rat model of blast-induced TBI, decreased melatonin levels in plasma and cerebrospinal fluid (CSF) were significantly associated with circadian SWDs. 54 These clinical and preclinical studies point the occurrence of circadian post-TBI SWDs to impaired melatonin synthesis.
Narcolepsy
The occurrence of narcolepsy post-TBI may be the result of direct damage to orexin-producing neurons in the hypothalamus, which leads to narcolepsy symptoms, including EDS, cataplexy, narcolepsy hallucinations, and sleep paralysis. Clinical studies have found that acute TBI can lead to severe but transient deficiency of orexin in cerebrospinal fluid. 55 Post-mortem autopsy results of patients with severe TBI also show that orexin-producing neurons have be damaged to varying degrees. 56 However, TBI does not specifically damage the posterior hypothalamus, but does damage other sleep–wake regulating hypothalamic cell populations, including neurons that produce melanin-concentrating hormone or histamine. 56,57 This is also reflected in the clinical features, which show that most patients with so-called post-TBI narcolepsy do not have typical cataplexy, but often have hypersomnia (as opposed to the normal amount of sleep per 24 h in narcolepsy patients). Even with CSF orexin deficiency and positive MSLT results, it is recommended to describe the symptoms of such patients as narcolepsy-like post-TBI. Therefore, the existence of true post-TBI narcolepsy needs to be questioned.
Another study by Poryazova and colleagues shows that, 58 in the survey of 37 narcolepsy patients, 7 patients had a history of TBI, and all of these patients had definite cataplexy. Latency between history of TBI and onset of narcolepsy was ≤2 years in all but one patient. The high prevalence of TBI in narcolepsy patients (19%) may be related to the partial loss of orexin neurons caused by TBI, leading to the apparent narcolepsy in susceptible patients; but it is also possible that the fact that these patients experience cataplexy narcolepsy puts them at higher risk for TBI.
Sleep-related breathing disorders
Sleep-related breathing disorder is defined as abnormally shallow breathing or slow respiratory rate while sleeping. OSA is the most common sleep-related breathing disorder post-TBI. In a prospective study, only 11% of TBI patients had OSA, 10 whereas other studies reported a higher incidence, ranging from 23% to 36%. 59,60 When comprehensively assessing pre-injury sleep behaviors in TBI patients, OSA was found to emerge as a new post-TBI feature in the majority of TBI survivors. In an analysis of 116 patients with war-related TBI, it was found that blunt trauma was more likely to lead to the development of OSA. 61
There is a bidirectional relationship between OSA and TBI. EDS caused by OSA syndrome increases the risk of TBI, especially in those performing high-risk jobs. Therefore, OSA may be a risk factor for TBI rather than a post-traumatic consequence in many patients. On the other hand, complex medical conditions post-TBI, such as physical disability, depression, pain-related decreased activity, and medication for post-traumatic epilepsy or mood disorders, may lead to weight gain that increases the risk of OSA syndrome. 62 Therefore, it can be seen that there is mutual influence between TBI and OSA, and aggressive treatment of OSA may be significant in terminating this vicious circle.
Nightmare disorders
Nightmare disorders in TBI patients depend on the time since TBI, the presence of comorbidities, and the memory of the moment before TBI. Guilleminault and colleagues 51 reported that up to 41% of TBI patients had nightmares. In the acute phase post-TBI, dream recall appears to be reduced, and may even disappear completely, despite normal REM sleep. 63 In contrast, acute TBI patients with disturbance of consciousness appear to be prone to nightmares. 61 During recovery, TBI survivors tend to have longer and more consolidated sleep, and although dream recall is restored, patients' dreams are less vivid. 64 However, among TBI survivors who experience nightmares, the traumatic content of dreams is so common that patients are awakened from sleep. 65 This frequent occurrence leads to disrupted sleep continuity, which in turn exacerbates post-traumatic SWDs.
Neuropathological Mechanisms of SWDs
Primary TBI refers to immediate mechanical damage to the brain during rapid acceleration, deceleration, direct impact, penetration, and shockwaves. Diffuse axonal injury is the most common form of primary tissue damage, resulting in dynamic shearing of white matter tracts and tensile and compressive strains. 66 Following the primary injury, a series of secondary injuries occur, including excitotoxicity, inflammation, free radical production, oxidative stress, lipid peroxidation, hyperglycemia, gene activation, neurotransmitter release, and calcium-mediated damage. 5 The primary and secondary injuries of TBI are the initiating factors leading to the occurrence of SWDs, and the possible neural mechanisms involved are the focus of this section.
Neural structures injury and post-TBI SWDs
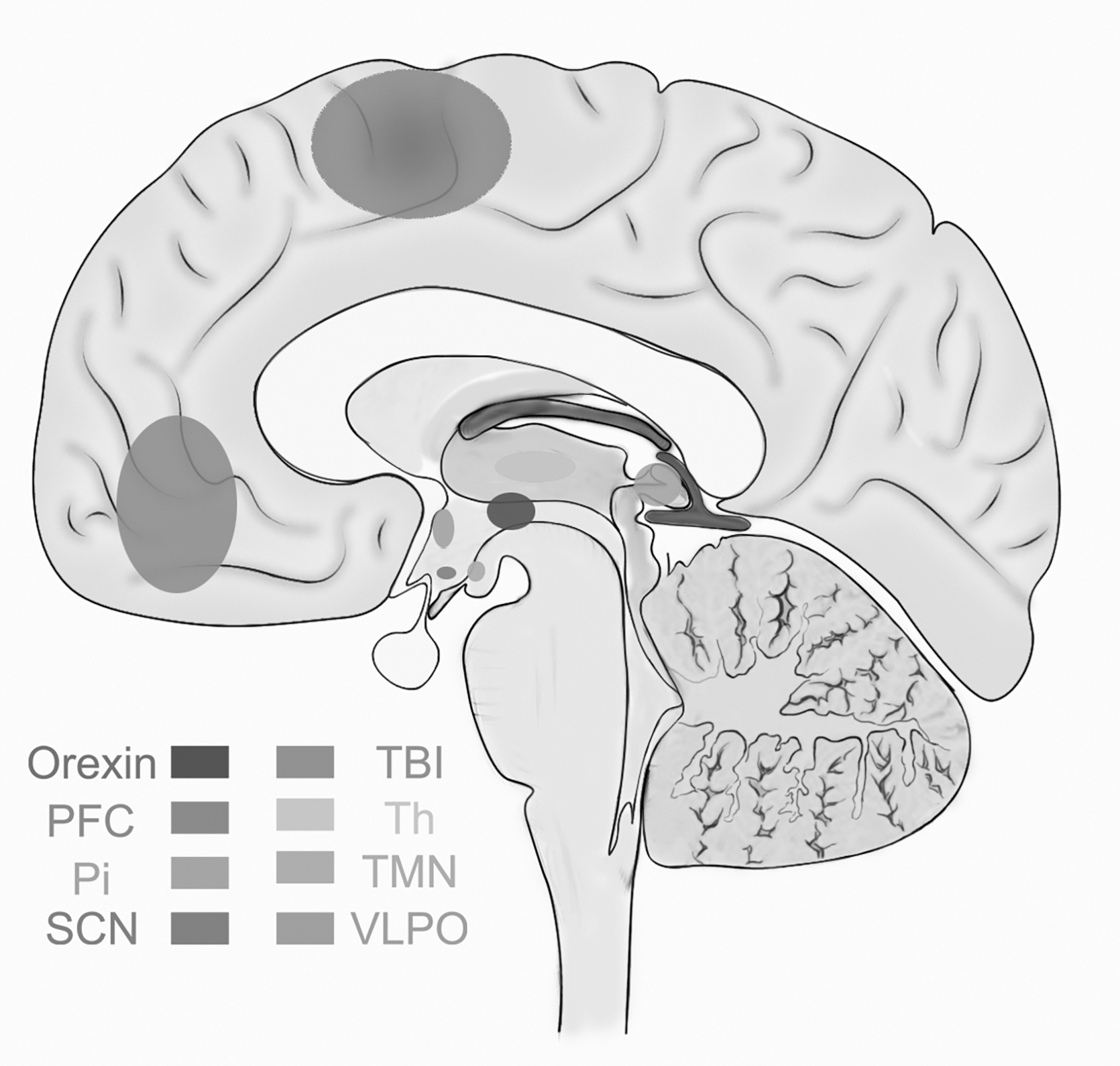
The occurrence of post-TBI SWDs appears to be associated with neuroanatomical damage, attributed to impairment of the brain's central regulation networks of sleep and wakefulness. These networks are functionally mutually inhibitory in the healthy physiological state, and in the case of brain injury, the disorder of the networks leads to the occurrence of SWDs. 67 The hypothalamus ventrolateral preoptic nucleus (VLPO) and median preoptic nucleus (MNPO) contain sleep-promoting neurons that are active during NREM sleep but inactive during wakefulness. 68 During sleep, VLPO and MNPO are thought to promote and maintain sleep by inhibiting wake regions, and this reciprocal inhibition functions like a trigger switch. However, because of varying degrees of damage to neurons on both sides of the switch post-TBI, the result may be a decrease in the stability of the switch inhibitory mechanism, leading to more transitions between sleep and wakefulness. 69
The frontal lobe is the brain region most vulnerable to TBI, and mechanical shear forces can cause damage to cortical and subcortical brain regions. 70 A study of transcranial magnetic stimulation in patients with mild-to-moderate TBI found that the excitability of the cerebral cortex was reduced in patients with EDS, 71 possibly reflecting a disturbance of the excitatory hypothalamic orexin neurotransmitter system. However, transcranial magnetic stimulation studies in the rat TBI model showed that the inhibitory effect of the cerebral cortex was gradually lost, and that the cortex was in an excitatory state 2 weeks after injury. This feature is accompanied by a loss of parvalbumin (PV) immunopositively inhibitory interneurons around the injured cortex. Before PV cell loss, the perineurial network (PNN), a specialized extracellular structure that wraps PV neurons, degrades. Decreased inhibitory tone caused by these changes is the pathological basis leading to post-TBI SWDs. 72 Heterogeneous changes in cerebral cortical excitability post-TBI may reflect the underlying mechanism of SWDs with different phenotypes, and in the future, further studies on cerebral cortical excitability should be conducted on SWDs with different presentations (Fig. 2).
Sleep
It was found that post-TBI, patients showed changes in frontal cortical thickness and gray matter volume on cranial MRI; continuous supplementation of growth hormone (GH) therapy for 1 year can alleviate the related post-TBI SWDs. 73 The dorsal-medial pre-frontal cortex is involved in regulating activity in sublimbic regions (e.g., the amygdala) during REM sleep, and post-TBI nightmares may be caused by decreased activity in the dorsal-medial pre-frontal cortex. 74 On MRI brain scans of military personnel with TBI experience, the fractional anisotropy of the right uncinate bundle was reduced in patients with SWDs. 75 The right uncinate tract is a key regulatory structure regulating limbic function, and damage to these fibers by TBI may be the mechanism by which sleep disturbance occurs. Damage to the corpus callosum fibers following axonal injury may affect the synchronization of interhemispheric connections during REM and NREM sleep, 76 ultimately disrupting sleep–wake processes. When analyzing brain MRI data from patients with TBI, a correlation between SWDs and abnormalities in para-hippocampal white matter was noted, 77 but the exact mechanism remains unclear. Until now, it has been difficult to link the development of post-TBI SWDs to specific injury sites or networks, but a wealth of research evidence suggests that these neuropathological changes may at least partially explain the development of SWDs.
Neurotransmitter change and SWDs
The reduction of wake-promoting neurotransmitters is an important reason for the occurrence of post-TBI SWDs. In clinical studies, it has been observed that the wake-promoting neurotransmitters in different brain regions are reduced to varying degrees post-TBI, including orexin 21%, 56 histamine 41%, 56 5-HT 17%, 78 and norepinephrine 29%. 78 Orexin is produced by a unique group of neurons in the posterior hypothalamus and plays a crucial role in maintaining wakefulness and regulating the transition between sleep and wake states. It has been reported that orexin levels in the CSF of patients in the acute and chronic phases are reduced post-TBI, 57 and the reduction of orexin levels in the CSF 6 months post-TBI is associated with EDS. In animal models of TBI, mice and rats with TBI have difficulty maintaining sustained wakefulness, have increased fragmented activity patterns during the dark period, and increased the number of transitions from wakefulness to sleep, these presentations are associated with reduced orexin concentrations in the hypothalamus. 79
In a rat model of TBI, closed diffuse brain injury resulted in increased sleep requirements 1 month after trauma, which was associated with a decrease in the number of histamine-immunoreactive cells in the tuberomammillary nucleus. 80 The decrease in histamine neurons may be a direct consequence of TBI or may be associated with increased neuroinflammatory responses. However, in this study, the monoaminergic and orexinergic neurotransmitter systems in the rostral brainstem and hypothalamus were not affected. 80 These results indicate that the mechanism of post-TBI SWDs is complex and variable, and the damage of specific neurons is not the specific change of post-TBI SWDs. Although animal TBI models can partially mimic human post-traumatic SWDs and related histopathological changes, they are insufficient to reflect the complex clinical features of TBI.
The suprachiasmatic nucleus (SCN) is located in the hypothalamus and is the body's master clock. The SCN may be susceptible to damage during TBI, and damage to the retinal-hypothalamic tract and projection fibers between the SCN and the pineal gland can disrupt circadian physiology. 53 Additionally, in a retrospective analysis of brain MRI scans of 34 patients with mTBI, the occurrence of diurnal SWDs was found to be associated with increased tentorial length and narrowing of the tentorial angle. 81 We can speculate that the impact of TBI on the tentorium may damage the pineal gland, causing impaired synthesis of melatonin, leading to sleep disturbance. In fact, melatonin secretion is reduced in both the acute and chronic phases post-TBI 82 and melatonin receptors are downregulated in the cortex and hippocampus, 83 and melatonin supplementation significantly improves diurnal SWDs. 82 Therefore, the relationship between the disturbance of melatonin synthesis post-TBI and the occurrence of diurnal SWDs is relatively clear.
Immune response, inflammatory response and cytokines associate with SWDs
TBI can activate immune system-related inflammatory responses and cytokine production, which participate in the regulation of SWDs after trauma. In an animal model of TBI with controlled cortical injury, an increase in reactive microglia was found in the thalamic region starting at 4 weeks post-injury, and delayed reactive astrocytes were evidently increased in the reticular nucleus of the thalamus, and these cellular changes preceded the onset of sleep disruption. 84 Resolvins RvE1, an anti-inflammatory mediator derived from omega-3 fatty acids. TBI mice treated with Resolvins RvE1 showed increased multi-branched microglia and decreased rod microglia in the cortex, corresponding to improvement in SWDs in the mice. 85 These findings suggest that damage to the cortex may trigger immune and inflammatory responses in deeper structures, disrupting the thalamocortical network that regulates sleep/wake patterns, resulting in the sleep disruption observed post-TBI. Modulating this inflammatory response could alleviate the post-TBI SWDs.
Analysis of patients' plasma IL-6 levels and neurological symptoms 2 years after moderate-to-severe TBI found that the occurrence of sleep disturbances was positively correlated with IL-6 levels. 86 Another study showed that in the chronic phase of TBI the levels of IL-10 were also associated with the occurrence of SWDs. 87 These results suggest that chronic inflammatory response to TBI is closely related to the persistence of neurological symptoms. In a mouse model of diffuse brain injury, elevated levels of the peripheral inflammatory cytokine IL-6 were associated with increased sleep time during the first photoperiod. 88 In a mouse model of mTBI, a chronic increase in complement C1q expression in the corticothalamic system coexists with neuronal loss and chronic inflammation, and is associated with sleep spindles; and blockade of complement C1q counteracts these results. 89 TBI-induced SWDs were consistent with increased levels of tumor necrosis factor (TNF), and blockade of the inflammatory response was associated with a corresponding decrease in sleep. 90 A variety of inflammatory factors can promote sleep, and abnormally elevated inflammatory factors post-TBI, as sleep regulators, will prolong sleep duration and increase sleep tendency; vice versa, inhibition of these cytokines will reduce spontaneous sleep, and anti-inflammatory therapy can be used as a potential target for TBI rehabilitation.
SWDs also lead to increased oxidative stress and neuroinflammation. Oxidative stress damages intracellular organelles, whereas neuroinflammation leads to increased cytokines that can disrupt vascular function and increase the risk of ischemic stroke and neurodegeneration. 91 Sleep disturbance following TBI also affects neurological function by increasing levels of the inflammatory cytokine TNF-α and IL-10. Protein synthesis can be altered post-TBI by activation of the unfolded protein response (UPR), 92 which is impaired in SWDs, resulting in a reduced ability to eliminate misfolded and damaged proteins and exacerbating neuropsychiatric symptoms in TBI patients. 93
There is increasing evidence that dysregulation of brain-derived growth factor (BDNF) is associated with post-TBI SWDs. Animal studies have shown that exploratory behavior while awake correlates with the extent to which BDNF is induced and the extent of slow-wave sleep responses in sleep homeostasis. 94 SWA is a sensitive marker of sleep demand and NREM sleep intensity, and exogenous application of BDNF is associated with increased SWA during NREM sleep. It is not difficult to understand why patients with insomnia have decreased BDNF post-TBI, because the downregulation of BDNF post-TBI may reduce the drive for sleep initiation and the intensity of maintaining SWA sleep.
Endocrine alterations with TBI and sleep
The intricate anatomical configuration of the hypothalamic–pituitary axis renders it particularly vulnerable to TBI. Perturbations in the homeostatic control of hypothalamic–pituitary endocrine axes are known to precipitate hormonal dysregulation in the aftermath of TBI. Interactions between the hypothalamic–pituitary system and sleep–wake processes are bidirectional. Sleep exerts a substantial impact on hormonal secretion dynamics, with specific sleep phases being intricately linked to the release of distinct hormones. 95 Consequently, it stands to reason that the alteration of sleep architecture subsequent to TBI may play a contributory role in the ensuing hormonal imbalance.
Pituitary dysfunction post-TBI causes insufficient secretion of GH, which is associated with the occurrence of SWDs. 73 A survey study analyzing 119 patients 1 year post-TBI revealed significant endocrine dysfunctions: 65% of the cohort exhibited moderate-to-severe GH deficiency; adrenal insufficiency, indicated by low fasting cortisol levels, was present in 64% of cases; central hypothyroidism was diagnosed in 12%; and testosterone deficiency was identified in 15% of the male participants. 96 These endocrine abnormalities demonstrated a strong association with fatigue, further exacerbating sleep disturbances.
Damage to the hypothalamus arising from TBI can precipitate endocrine dysfunction and consequent alterations in hormone levels and can also lead to a spectrum of functional effects that extend beyond sleep–wake disturbances to include cognitive impairments and psychological disorders. Prompt assessment for endocrine anomalies in patients with TBI is justified. Hormone replacement therapy emerges as a viable therapeutic option to address sleep–wake disturbances in TBI patients who are also contending with concurrent endocrinopathies.
Vascular and gliovascular changes post-TBI
More and more studies have recognized the importance of timely diagnosis and treatment of post-TBI SWDs. The occurrence of post-TBI SWDs not only affects the performance of daytime daily functions 97 and prolongs the time needed for recovery, but also increases the risk of neuropsychiatric diseases (depression, anxiety, apathy). 98 Considerable research has been done on the hypothetical mechanisms by which SWDs contribute to poor prognosis in TBI patients. We describe the mechanisms by which sleep disturbances have negative effects on TBI patients.
Vasodilation is a response to increased neuronal activity, which is necessary to increase blood flow and provide adequate nutrition to maintain neuronal responses. This hemodynamic response is impaired following sleep disturbance, with blunting of vascular responses exacerbated by sleep disorders. 99 Studies in rodents have shown that cerebrovascular endothelial dysfunction is related to the nitric oxide (NO) synthase pathway, and both sleep disturbances and TBI can disrupt vascular endothelial function, leading to severe cerebral vasospasm. 100 Good sleep is an important activity necessary for neurological recovery in patients with TBI, but the occurrence of SWDs may damage the vascular system, which in turn causes the brain to be under glucose metabolism, thereby further damaging the injured brain. 101 Metabolic theory suggests that sleep is essential for maintaining proper adenosine triphosphate (ATP) production as well as restoring the immune system and regulating inflammation. 102 SWDs can lead to altered brain metabolism through epigenetic changes, increasing susceptibility to metabolic derangements in TBI patients. 103 Sleep disturbances can cause cerebral vasospasm, or the uneven constriction of blood vessels, which limits the delivery of glucose and oxygen to nerve cells. These results suggest that sleep plays an integral role in vascular integrity and metabolic homeostasis after injury.
Recent findings suggest that sleep deprivation impairs BBB function. REM sleep deprivation in rodents resulted in increased BBB tracer permeability and decreased expression of vasoactive substances, and several tight junction proteins were disrupted in brain micro-vessels. 104 Elevated levels of brain-specific proteins in human serum post-TBI, including neuron-specific enolase (NSE) and S100 calcium binding protein B (S100B), indicate neuronal damage and BBB dysfunction. 105 These neuronal injury-specific proteins were more elevated after SWDs occurred. It is speculated that the occurrence of post-TBI SWDs exacerbates BBB dysfunction that allows blood-borne substances to enter the brain and induce excitotoxic events of neuronal cell death. Additionally, in a mouse model of TBI, impairment of aquaporin-4 (AQP-4) was found to be associated with BBB impairment, and AQP-4 is also required for the glymphatic system to maintain normal function. 106 Thus, the negative effects of SWDs on the BBB and the glymphatic system of the brain work together with the deleterious effects of TBI to exacerbate neuronal damage and death.
The glymphatic system theory of sleep suggests that during sleep, the glymphatic system is activated, allowing for greater removal of waste products produced during waking. This clearance mechanism appears to be even more important following TBI, clearing a dramatic increase of metabolites and toxins that are deleterious to neurons. 107 The occurrence of post-TBI SWDs may impair perivascular spaces, even in mTBI patients, which probability inhibits the clearance of waste products such as amyloid-β (Aβ) and tau by the glymphatic system, and induces the development of neurodegenerative diseases. 18,108 The clearance of Aβ protein and other metabolites from the brain primarily depends on the activation of the glymphatic system during sleep. Some scholars believe that the decline in the function of the glymphatic system post-TBI leads to insufficient clearance the harmful substances, and that TBI itself will lead to the accumulation of harmful substances in a short period of time. 109 It is possible that increased sleep demand to activate the glymphatic system may be required to remove these excess harmful substances to aid in neuronal recovery. However, the reasons for the persistent increase in sleep demand in the chronic phase of TBI are unclear.
Sleep is also an important activity for brain plasticity. Studies have shown that sleep disruption suppresses long-term potentiation, resulting in impaired synaptic plasticity and neurogenesis. 110 Structural differences exist in the hippocampus of patients with SWDs compared with controls, mainly manifested by reduced hippocampal volume. 111 Non-specific loss of neurons in the hippocampus of sleep-deprived rats has also been found in animal studies. 112 These changes in the hippocampus, a key region of the brain for learning and memory, may explain the apparent cognitive deficits seen in patients with post-TBI SWDs.
Broadly speaking, the negative effects of post-TBI SWDs is mainly through the mechanisms of causing metabolic disorder, glymphatic system dysfunction, inducing inflammatory response, increasing the vulnerability to stress response, causing vascular regulatory dysfunction, leading to the destruction of the BBB and neuronal death and apoptosis, which impede neurological recovery in TBI patients and increase the risk of neuropsychiatric and cognitive impairment in these patients. The occurrence of sleep disorders can also increase the occurrence of diseases such as hypertension, cardiovascular disease, and obesity, 113 but the pathological role of sleep disorders in the development of these diseases is still unclear. Future research should focus on the causal relationship between the occurrence of post-TBI SWDs and health outcomes, and possible interventions.
Conclusion
The increasing prevalence and detrimental effects on quality of life have elevated post-TBI SWDs to being a focal point of scientific inquiry. Despite their significance, the precise pathophysiological underpinnings of these disorders are yet to be fully elucidated. Consequently, this gap in understanding has resulted in a deficiency of efficacious therapeutic strategies.
Future research endeavors must persist in the quest to decode the intricate mechanisms of post-TBI SWDs, as well as to comprehend how SWDs adversely influence health outcomes. There is an imperative to develop tailored regulatory interventions that align with the diverse pathological mechanisms at play. This endeavor mandates the undertaking of multi-center clinical prospective studies, which meticulously factor in the intrinsic characteristics of TBI, including injury severity, injury type, and the timing of patient presentations.
The complexity of clinical TBI manifestations presents a considerable challenge, as current animal models fall short in fully replicating the clinical features and pathophysiological progression of post-TBI SWDs. This limitation curtails our investigative reach into the pathological mechanisms. Therefore, a critical issue that requires deliberation is the strategic utilization of available animal models to design methodologically sound experimental studies that can yield meaningful insights into post-TBI SWDs.
Transparency, Rigor, and Reproducibility Summary
No new data were generated for this review. Therefore, no human or animal subjects were used. A complete list of reviewed and referenced works can be found in our References section. Further, the authors agree to be available for contact by parties interested in discussing this review.
Footnotes
Authors' Contributions
Conceptualization, investigation, methodology, writing—review and editing: Guizhong Yan, Yuan Wei, Dengfeng Wang, Dong Wang, Haijun Ren, and Boru Hou; funding acquisition, review and editing: Guizhong Yan, Dengfeng Wang, and Boru Hou. All authors have read and agreed to the published version of the manuscript.
Funding Information
This work was supported by the Gansu Province Youth Science and Technology Fund program (22JR5RA1019), the Scientific Research Project of Lanzhou Chengguan District Science and Technology Bureau (2021-9-5), Gansu Provincial Health Industry Scientific Research Program (GSWSQN2021-005), and the National Natural Science Foundation of Gansu (22JR5RA970).
Author Disclosure Statement
No competing financial interests exist.