
Abstract
Metabolic uncoupling has been well-characterized during the first minutes-to-days after a traumatic brain injury (TBI), yet mitochondrial bioenergetics during the weeks-to-months after a brain injury is poorly defined, particularly after a mild TBI. We hypothesized that a closed head injury (CHI) would be associated with deficits in mitochondrial bioenergetics at one month after the injury. A significant decrease in state-III (adenosine triphosphate production) and state-V (complex-I) driven mitochondrial respiration was found at one month post-injury in adult C57Bl/6J mice. Isolation of synaptic mitochondria demonstrated that the deficit in state-III and state-V was primarily neuronal. Injured mice had a temporally consistent deficit in memory recall at one month post-injury. Using proton magnetic resonance spectroscopy (1H MRS) at 7-Tesla, we found significant decreases in phosphocreatine, N-Acetylaspartic acid, and total choline. We also found regional variations in cerebral blood flow, including both hypo- and hyperperfusion, as measured by a pseudocontinuous arterial spin labeling MR sequence. Our results highlight a chronic deficit in mitochondrial bioenergetics associated with a CHI that may lead toward a novel approach for neurorestoration after a mild TBI. MRS provides a potential biomarker for assessing the efficacy of candidate treatments targeted at improving mitochondrial bioenergetics.
Introduction
M
The commonalities of mitochondrial dysfunction after TBI and in neurodegenerative diseases suggest a potential mechanism whereby TBI induces a premature aging of mitochondria—particularly synaptic mitochondria. Using a mouse model of mild TBI, induced by a closed head injury (CHI), we found strong support for our hypothesis that a mild TBI would result in a premature aging of synaptic mitochondria. Specifically, we found a decrease in mitochondrial bioenergetics at one month post-injury, particularly in neuronal synaptic mitochondrial bioenergetics, which corresponded temporally with deficits in spatial memory as measured in the radial arm water maze.
To extend the translational potential of our findings, we tested in our mouse mild TBI model two surrogate radiological measures for bioenergetics, which could be used in head-injured patients. The field of neuroimaging is far more mature in humans than in rodent models. Multi-modal magnetic resonance (MR) changes in humans after a mild TBI have been well-described, 6 while there is a paucity of studies in mild TBI models in rodents. Technological improvements, such as high-field strength scanners (7–11 Tesla), are providing the spatial and/or temporal resolution necessary to image the rodent brain after a mild TBI.
We sought to reverse translate two advanced neuroimaging modalities, which have shown utility clinically in mild TBI 7 –11 to a mouse model of mild TBI. The MR methods included pseudo-continuous arterial spin labeling (pCASL) to measure cerebral blood flow and proton magnetic resonance spectroscopy ( 1 H-MRS). The current results demonstrate that 1 H-MRS deficits in phosphocreatine, a surrogate radiological biomarker of mitochondrial dysfunction, occur at the same chronic time point where deficits in mitochondrial bioenergetics were confirmed in isolated mitochondria. Our results provide strong support for a novel mechanism of chronic mitochondrial deficits after TBI and a translationally relevant radiological method to monitor this mechanism.
Methods
Animals
All procedures were approved by the Institutional Animal Care and Use Committee of the University of Kentucky, and that were conducted in accordance with the standards of proper experimentation in the Guide for the Care and Use of Laboratory Animals and ARRIVE guidelines. Experiments used four-month-old C57BL/6J mice and included an equal ratio of male and female for all end points. This project used a total of 204 mice: 36 mice were used for mitochondrial isolation; 19 mice were used for behavioral assays; 37 mice were used for pCASL and 1 H-MRS experiment 1; 112 mice were used for 1 H-MRS experiment 2; 22 of the 204 mice were not included in the data analysis for reasons described in the following method sections for each end point. The final number mice used for each end point are indicated in the figure or figure legend.
Mice were randomized and assigned groups before the start of the experiment. The study was completed with multiple batches of mice using a block experimental design, with each batch including all experimental groups; the order of the group was randomized for each block, so that the order of sham and CHI surgery varied for each cage. Each cage of mice included more than one experimental group. Each subject was given a unique identification number, which does not identify the experimental group. The persons conducting the mitochondrial bioenergetics (HV), behavior (TM), and neuroimaging (DNL, VB) were blinded to the treatment conditions and were not involved on the surgery or postoperative care of the mice, which could potentially lead to unblinding of the experimental groups.
CHI
The CHI was performed as described previously. 12,13 Briefly, mice were anesthetized with isoflurane (3–5%). The head was stabilized in a astereotaxic frame before a midline sagittal incision was made. A 1 mL latex pipette bulb filled with water was placed under the head. The stereotaxic electromagnetic impactor 14 used a 5.0 mm flat steel tip (Leica Biosystems). It delivered a closed-skull midline impact (coordinates: mediolateral, 0.0 mm; anteroposterior, 1.5 mm) 1.0 mm deep with a controlled velocity (5.0 ± 0.2 m/sec) and a dwell time of (100 msec). No mice were eliminated because of surgery complications. Mice were randomized to a group (sham or injured), with each group containing approximately 50% males and females. Sham mice received the incision, but not the impact and were included for the three day post-injury and 28 day post-injury time points.
Immunohistochemistry (IHC)
Mice were deep anesthetized with 5% isoflurane before transcardial perfusion with ice-cold phosphate buffered saline (PBS) for 5 min. The brains were rapidly removed, dissected, processed for IHC end points as described previously. 12,13 Primary antibodies used included: rabbit anti-βAPP (LifeTechnologies cat# 51–2700 (1:2000)) and rabbit anti-glial fibrillary acidic protein (GFAP) (Dako Cat# Z0334; (1:10,000). Aperio ScanScope XT digital slidescanner was used to image the entire stained slide at 20x magnification to create a single high-resolution digital image.
Mitochondrial bioenergetics
Total mitochondria were isolated from the cortex and hippocampus as described previously. 15,16 Synaptic and nonsynaptic mitochondria were isolated from the cortex as described previously. 3 Mitochondrial bioenergetics were measured using the Seahorse XF-24 Flux Analyzer as described previously. 16 One mitochondria sample from the cortex was lost because of technical error.
Six-arm radial arm water maze
A modified four-day version of the previously reported radial arm water maze (RAWM) 13,17 was used as described previously. 12,13 The RAWM protocol consisted of four days of training, which occurred on nonconsecutive days (that is, day 14, 15, 16, and 21 post-injury). The total trials/day were 15, and each trial was up to 60 sec long. EthoVision (Noldus Information Technology) was used for video recording and scoring behavior. A memory retention 60 sec probe trial was performed seven days after the last training day (28 days post-injury). Time spent in the goal arm and the percentage of entries in the goal were recorded.
MR imaging
MR imaging was performed on a 7-Tesla Clinscan scanner (Bruker, Billerica, MA) using a 2 × 2 Bruker brain surface coil or a Bruker quadrature, cryocoil at the University of Kentucky Magnetic Resonance Imaging and Spectroscopy Center, as described previously. 18 Heart rate (90–130 bpm), respiration rate, and rectal temperature (37 ± 0.5°C) were monitored continuously. A water bath with circulating water at 45–50°C was used to maintain the body temperature. The order of mice scanned was randomized between sham and CHI groups for each acquisition period.
T2-weighted structural images were acquired with field of view (FOV) = 18 × 18 mm2, matrix = 256 × 256; slice thickness = 1 mm, six slices, repetition time (TR) = 1500 msec, and echo time (TE) = 35 msec.
Quantitative cerebral blood flow (CBF) (units of mL/g per min) was measured using MR based pCASL. A whole-body volume coil was used for transmission, and a mouse brain surface coil was placed on top of the head for receiving. Paired control and label images were acquired with a train of Hanning window-shaped radiofrequency pulses of duration and spacing 200/200 μsec and the following parameters: flip angle = 25 degrees, slice-selective gradient = 9mT/m, labeling duration = 2100 msec, echo spacing = 0.33 msec. A two-dimensional multi-slice spin-echo planner imaging sequence was used with a FOV: 18.0 × 18.0mm2, matrix = 128 × 128, slice thickness = 1.0 mm, six slices, TR = 4000 msec, and TE = 20 msec with 1200 repetitions. 19 Manual shimming was applied after automatic shimming with the lowest possible frequency obtained before the image was acquired. Ten measurements were obtained before the usable 120 measurements as a test run to optimize the phase adjustment between two successive tagging radiofrequency pulses.
1 H-MRS was acquired in the first setup experiments using the 2 × 2-surface coil, a single 1 H-voxel of interest was defined as the bilateral dorsal hippocampus with a size of 12.48 mm3 (2.0 mm × 5.23 mm × 1.2 mm). Acquisition parameters for applied PRESS spectroscopy sequence include: TR = 1500 msec, TE = 135 msec, flip angle (FA) = 90 deg, rotation (R = 0 deg), spectral width (SW = 60 Hz), and averages (A = 400). Manual shimming was applied after automatic shimming on a shimming volume 30% larger than the spectroscopy voxel to maximize the suppression of outer-voxel water. A second acquisition with 10 averages and no water suppression was acquired and used to calculate metabolite concentrations relative to water.
1 H-MRS was acquired in the second set of experiments using the cryocoil; acquisition parameters for the LASER spectroscopy sequence 20 for the cryocoil include: TR = 2500 msec, TE = 21 msec, FA = 90, R = 0 deg, SW = 250, and A = 200. The LASER sequence was used for the second set of experiments to maximize the suppression of outer-voxel water and use a short TE. This maximizes signal to noise, allows for the measurement of metabolites not observable at higher TEs, and increases sensitivity to metabolite concentrations. The single 1 H-voxel of interest was defined as the bilateral dorsal hippocampus. Manual shimming was applied after automatic shimming on a shimming volume 30% larger than the spectroscopy voxel to maximize the suppression of outer-voxel water. The lowest possible full width half maximum (FWHM) was obtained, and any spectra with a FWHM greater than 40 Hz as determined by the LCmodel processing software was eliminated. A second acquisition with 10 averages and no water suppression was acquired and used to calculate metabolite concentrations relative to water.
pCASL data analysis
MANGO software (version 3.8, UT Health, San Antonio, TX) was used to quantify the blood perfusion to the brain. pCASL images were overlaid on top of the T2-weighted structural images in MANGO software to visualize the regions of interest (ROI). Labeling efficiency (e.g., >0.85) was used to verify the quality of the image series. Four mice were excluded because of a poorly acquired image. The pCASL images were analyzed using a custom, in-house developed MATLAB (Natick, MA) script, based on published equations. 21 A single image slice was used for the CBF quantification with approximate coordinates of 1.5–1.7 mm anterposterior).
1 H-MRS data analysis
The concentrations of the metabolites were derived from the Linear Combination model (LCModel) on a Linux operation system.
22,23
The concentrations of the metabolites (in μM) were computed using the following equation:
where [m] is the concentration of the specific metabolite, Sm is the metabolite intensity obtained from 1 H-MRS, Swater is the water intensity, [water] is the water concentration (55.14 mM at 310 K), Cn is the correction for the number of equivalent nuclei for each resonance, and Cav is the correction for the number of average. 18,24 For each spectra, a signal to noise ratio was calculated by LCModel; the FWHM estimate of 0.049 was used as a cutoff for the cyrocoil 1 H-MRS results. Nine mice were excluded because of a FWHM greater than 0.049.
Statistical analysis
JMP Software version 12.0 (SAS institute, Cary, NC) was used for statistical analysis. A repeated-measures analysis of variance (ANOVA) was used for RAWM. For all other end points, a one-way ANOVA was used comparing injury groups. If a significant main effect was found, post hoc analysis was used to compare groups. The normality assumption was assessed using the Shapiro-Wilk test, and heterogeneity of variances was assessed with the Levene test. Differences between mean were considered significant at α = 0.05. Graphs were generated using GraphPad Prism version 7.0. Values are expressed as mean ± standard error of the mean (SEM), unless otherwise noted. Scatter plots represent individual mice. No difference was found between the sham group three days post-surgery compared with the sham group at 28 days post-surgery; therefore, these groups were collapsed into a single sham group for all end points. Number of mice used for each end point are indicated in the figure or figure legend.
Results
At least 75% of the 1.7 million TBIs that occur in the United States each year are classified as a mild TBI. 25,26 In the present study, we used a CHI to model mild TBI. As reported previously, 12,13 and confirmed here (Fig. 1), the CHI model does not cause gross structural lesions to the brain. At a one day post-injury time point, we find markers of axonal injury in the neocortex (Fig. 1A, B). At 28 days post-CHI, we find diffuse astrogliosis (Fig. 1C, D), with the highest GFAP staining corresponding to the same region of the brain with the greatest amyloid precursor protein (APP) staining at one day post-CHI.
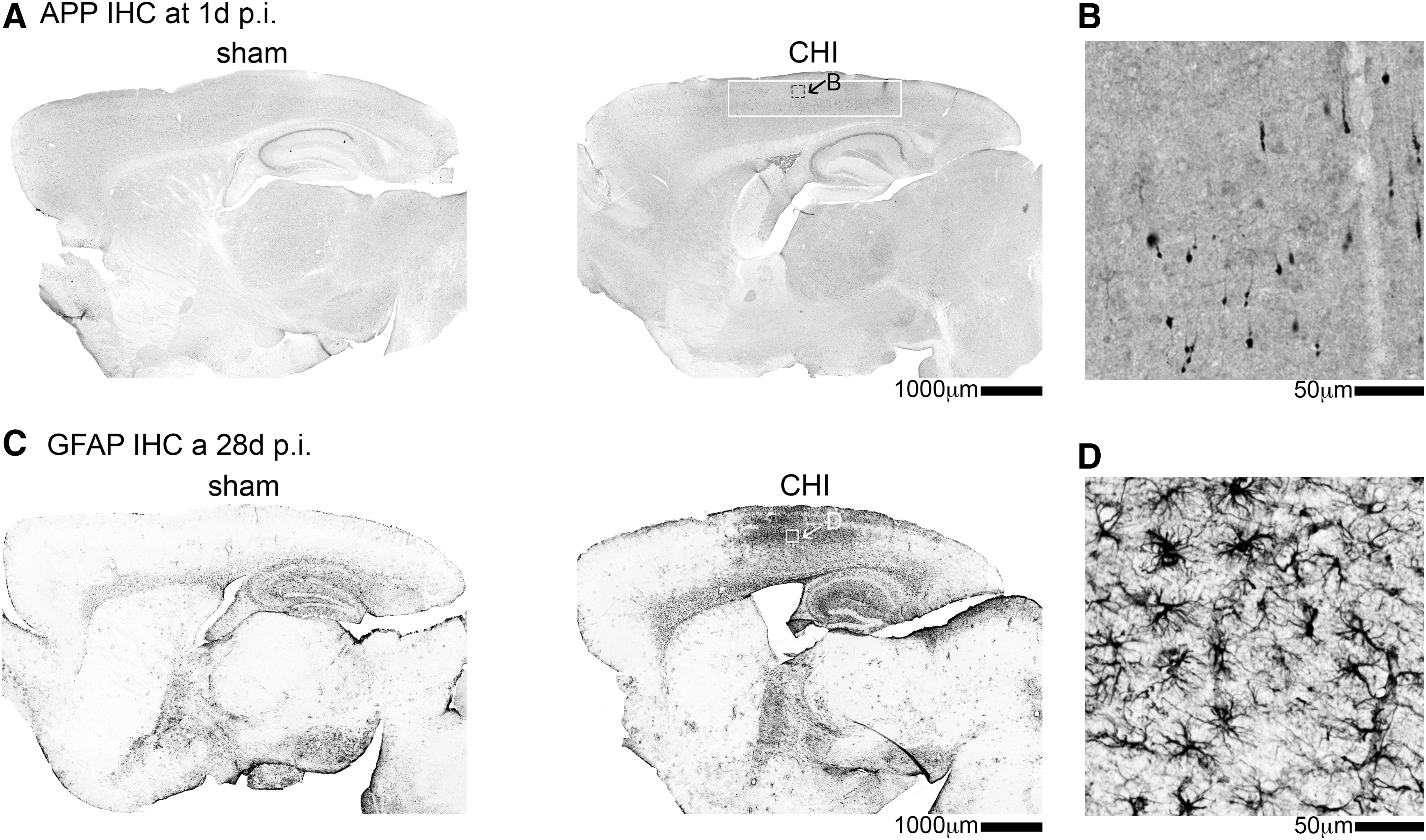
Closed head injury (CHI) causes axonal injury and astrogliosis but no structural lesions. (
C57Bl/6J mice show persistent deficits in mitochondrial bioenergetics after a mild TBI
Total mitochondria were isolated from the cortex and hippocampus, and State-III (adenosine triphosphate [ATP] synthesis capacity), State-VC1 (complex I driven maximal electron transport), and State-VC2 (complex II driven maximal electron transport) were measured using the Seahorse Extracellular Flux Analyzer. In the cortex, we found a stepwise decrease with time post-injury in both State-III and State-VC-I mitochondrial respiration after CHI compared with the sham mice. No statistical significant difference in mitochondrial bioenergetics was found at three day post-CHI compared with sham-injured mice in the cortex (Fig. 2A) or hippocampus (Fig. 2B). By 28 days post-CHI, mice had a significant decrease in State-III and State-VC-I mitochondrial respiration compared with the sham-injured mice (Fig. 2A). In the hippocampus, we found a decrease in State-III, State-VC-I, and State-VC-II mitochondrial respiration at 28 days post-CHI compared with the sham-injured mice (Fig. 2B).
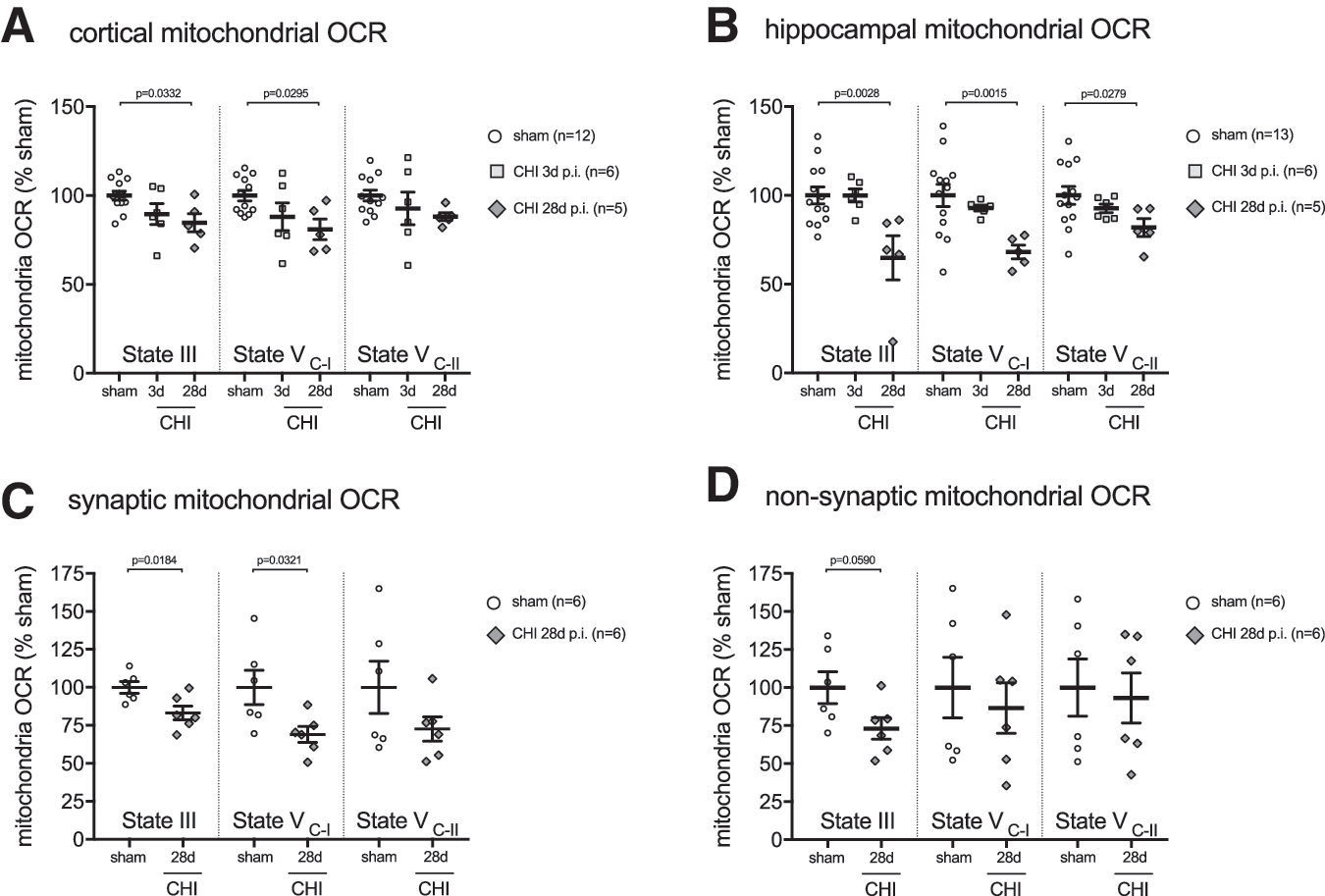
A mild traumatic brain injury (TBI) results in persistent deficits in mitochondrial bioenergetics. (
A brain-region heterogeneity was evident in the CHI-induced mitochondrial bioenergetics dysfunction between the hippocampus and cortex. In the hippocampus at 28 days post-CHI, the deficit in mitochondrial respiration compared with shams was 35% in State-III and 32% State-VC-I. In contrast, the cortex at 28 days post-CHI compared with shams was found to have a 16% deficit in State-III and 20% deficit in State-VC-I. It is unknown whether the regional heterogeneity in mitochondrial bioenergetics is a reflection of different sensitivity of the mitochondria in the brain regions to the effect of the injury, or differences in the degree of the primary injury in the two brain regions.
After a CHI, the cortex but not the hippocampus has profound gliosis (microglia and astrocytes) and immune cells infiltrates (macrophages and neutrophils). 12,13 We hypothesized that the infiltration of immune cells and proliferation of glia in the cortex could mask some deficits in neuronal mitochondrial bioenergetics, compared with that seen in the hippocampus. To test this hypothesis, in a second cohort of mice, we performed differential isolation of neuronal presynaptic mitochondria and nonsynaptic mitochondria (neuronal cell bodies, glia, and infiltrating immune cells) in the cortex at 28 days post-CHI. In the synaptic mitochondria fraction, we found a 17% deficit in State-III and 31% deficit in State-VC-I (Fig 2C). In the nonsynaptic, there was a 27% deficit in State-III, while State-VC-II was unchanged from sham levels (Fig 2D).
Mild TBI causes impaired hippocampal-dependent learning and memory
In light of the persistent suppression of hippocampal and neuronal mitochondrial bioenergetics after a CHI, we hypothesized that injured mice would have impairments in hippocampal-dependent learning and memory. To measure the cognitive status of the injured mice, we used the six-arm RAWM test of spatial learning and memory. The CHI-injured mice were found to have a delayed learning curve over the four days of training compared with the sham-injured mice (Fig. 3A). Importantly, both groups, CHI and sham, were eventually able to find the location of the hidden platform while making less than two errors by the fourth day of training (Fig. 3A).
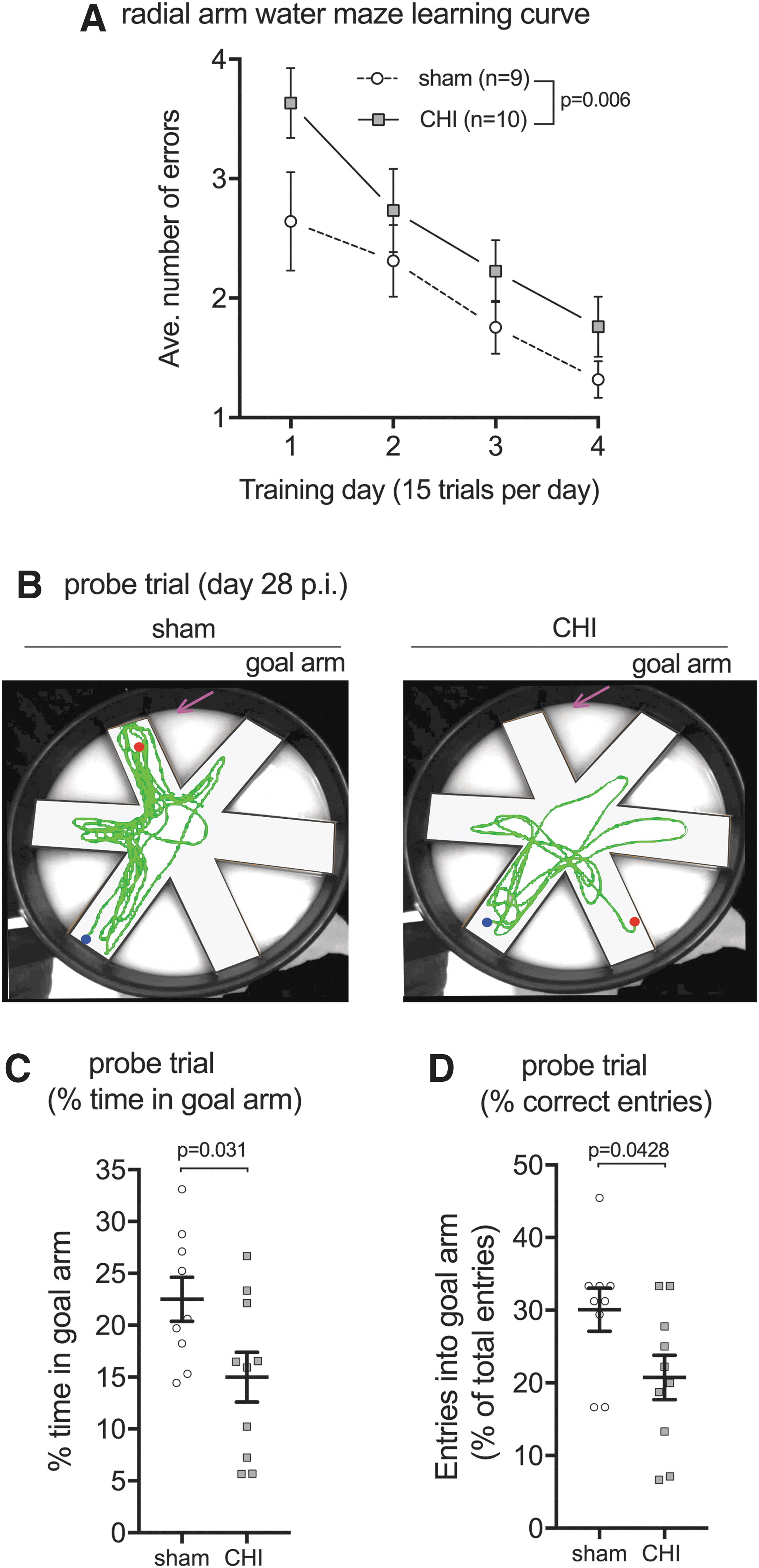
Closed head injury (CHI)-induced deficits in the radial arm water maze. (
Long-term memory consolidation was measured by using a probe trial, where the hidden platform was removed, and the amount of time and number of correct entries into the goal arm were considered an indicator of a memory of the platform location. The probe trial was conducted seven days after the last training session, on day 28 post-injury. As shown in Figure 3B, CHI mice showed no preference for the goal arm, whereas sham-injured mice were found to have a preference for the goal arm. CHI mice spent less time in the target arm (Fig. 3C) and made fewer entries into the target arm (Fig. 3D) compared with sham-injured mice.
Regional variations in CBF after CHI
Multimodal neuroimaging has the potential to be used as surrogate measures of mitochondrial bioenergetics. We sought to determine in a pilot experiment whether CBF and 1 H-MRS would be sensitive biomarkers of metabolic disturbance after CHI in mice. We chose to measure CBF and 1 H-MRS because these two MR modalities are commonly available clinically and could be completed in one scanning session. To measure CBF, we used the pCASL method. pCASL has higher signal to noise ratio and better labeling efficiency than other arterial spin labeling techniques. Also, pCASL allows for regional analysis to look for heterogeneity in CBF. 21,27 Examples of the CBF perfusion maps are shown in Figure 4A. To quantify the regional heterogeneity in CBF after injury, we chose to analyze four distinct ROIs: (i) proximal cortex (closest to injury); (ii) adjacent cortex; (iii) hippocampus; and (iv) thalamus (Fig. 4B). The ROIs were based on our previous studies, which defined the patterns of neuropathological changes in the CHI model. 12,13
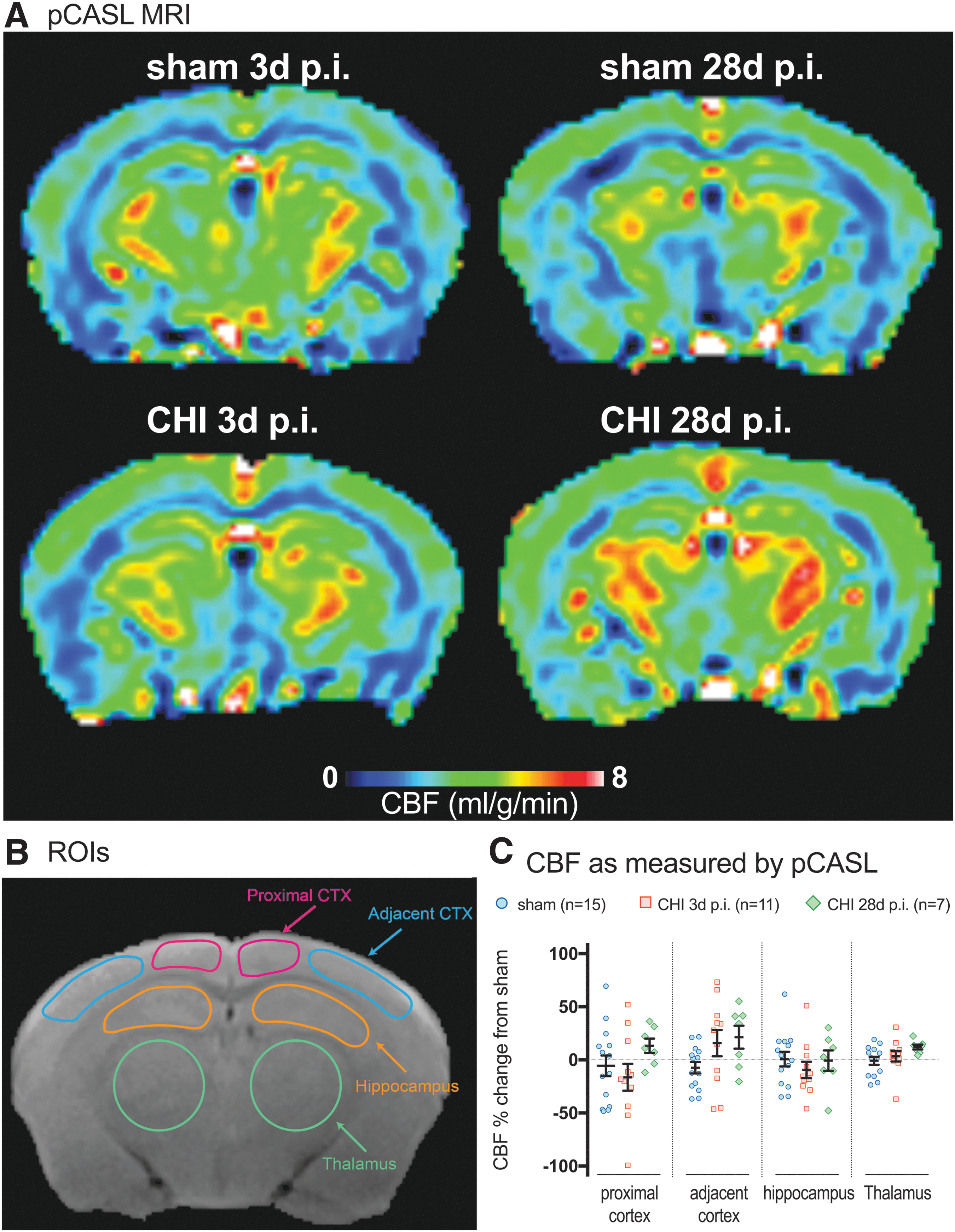
Cerebral blood flow (CBF) as measured by Pseudo-Continuous Arterial Spin Labeling (pCASL). (
We observed subtle regional and temporal changes in the pattern of the CBF. Specifically, a 16.4% decrease in CBF was found in the three days post-CHI group in the region of the cortex most proximal to the site of injury compared with the sham and 28 days post-CHI group (Fig. 4C). In the cortex adjacent to the site of injury, an increase in CBF was found in the three days post-CHI group (15.8%) and 28 days post-CHI group (21.4%) compared with the sham group (Fig. 4C). Similar to the proximal cortex, a small decrease was found in CBF in the hippocampus in the three days post-CHI group (-9.4%) compared with the sham or 28 days post-CHI group (Fig. 4C). The thalamus was found to have a slight increase in CBF in the 28 days post-CHI group (12.1%) compared with the sham or three days post-CHI group (Fig. 4C).
Apparent from the scatter plots in Figure 4, a sizable amount of variability in CBF was seen within the groups. While patterns of CBF changes after a CHI are evident, none of the changes were found to be statistically significant. The amount of variability between the brain regions was relatively proportional to the size of the ROI, indicating that the smaller ROIs were pushing the limits of the scanner. A goal of the study was to determine the feasibility of using pCASL to detect CHI-induced changes in CBF. Results of post hoc power analysis showed that to detect an effect size of 10–20% change in CBF in the neocortex or hippocampus caused by a mild TBI would require an unrealistic number of mice per group (n = 144–56, respectively). The number of mice needed to detect a similar magnitude change in the thalamus was reasonable (n = 21). The results suggest that further improvements in the MR methods to increase the sensitivity of CBF in small ROIs may allow for MR to be used to detect changes in CBF after a mild brain injury in mice.
A mild TBI decreases hippocampal metabolites associated with energy metabolism (PCr) and neuron function (NAA and tCho)
The second multi-modal neuroimaging sequence acquired in the same scanning session with pCASL was 1 H-MRS in the bilateral dorsal hippocampus using a Bruker brain surface coil. Previous studies have demonstrated reliable detection of metabolites with a voxel of interest covering the bilateral dorsal hippocampus. 18 While we anticipated a larger change in metabolites in the cortex immediately adjacent to the site of injury, this region was not selected as the ROI because the size and shape of the region would result in a voxel of interest that we believed would be too small to measure metabolites accurately. Figure 5A shows a representative spectrum of the metabolites acquired by 1 H-MRS in the dorsal hippocampus of a mouse.
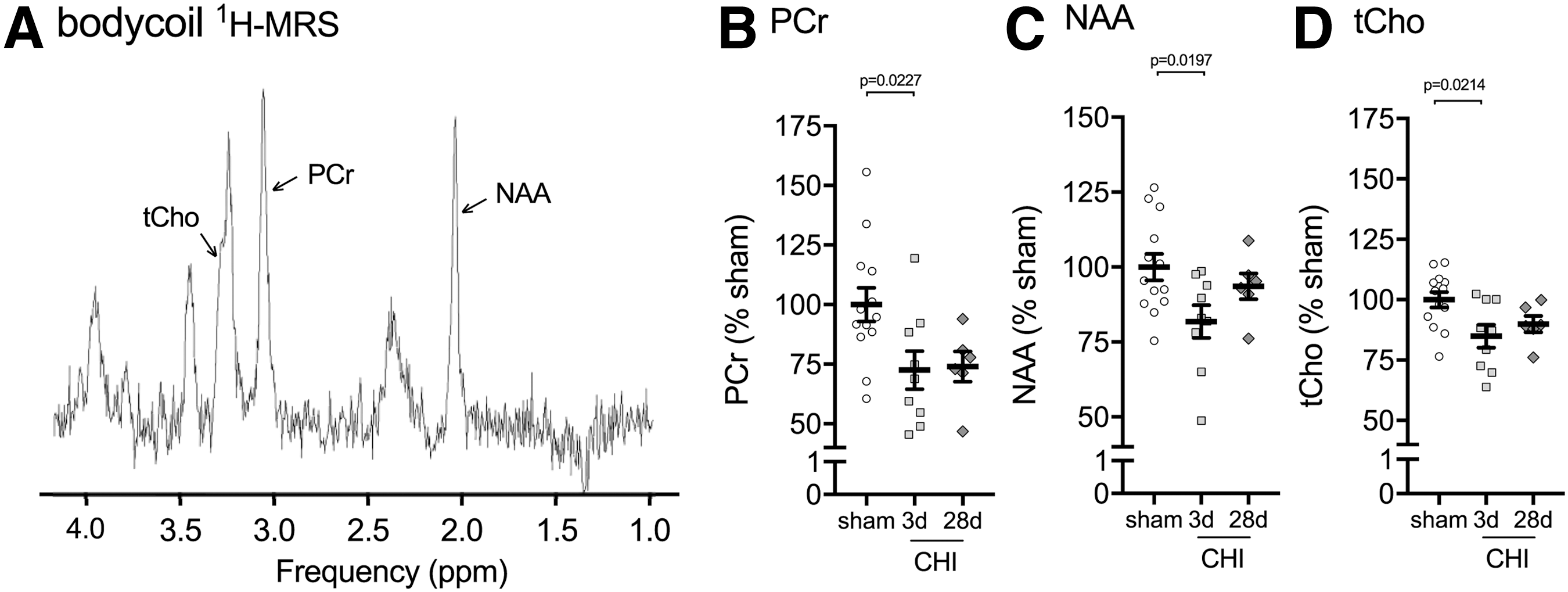
Closed head injury (CHI)-induced changes in hippocampal metabolites measured by brain surface coil
1
H-MRS. (
A total of five metabolites were acquired by 1 H-MRS: phosphocreatine (PCr), N-acetylaspartate (NAA), glutamate (Glu) taurine (Tau) and total choline (tCho). Of the five metabolites, three metabolites were found to be significantly decreased after the injury. PCr was found to be reduced in the hippocampus at three days post-CHI (-26%) and 28 days post-CHI (-27.5%) compared with the sham-injured group (Fig. 5B). NAA was reduced at three days post-CHI (-18.2%) compared with the sham-injured group (Fig. 5C). tCho was found to be reduced in the hippocampus at three days post-CHI (-15.1%) and 28 days post-CHI (-10.1%) compared with the sham-injured group (Fig. 5D). In comparison with the pCASL, power analysis on the pilot study of 1 H-MRS determined that a reasonable sample size would be needed in future studies to detect CHI-induced changes in PCr, NAA, and tCHo by 1 H-MRS in the dorsal hippocampus (n = 15, 7, 4; per group for PCr, NAA, tCho, respectively, for 20% effect size).
To confirm the changes in hippocampal metabolites found in the pilot study, we generated a second cohort of sham and CHI mice to repeat the 1 H-MRS. To increase the signal to noise, we used a Bruker cryocoil. The cryocoil provided a 2.5-fold increase in signal to noise, which is a 6.25-fold improvement in imaging time. In addition, we used an improved water suppression sequence called LASER, 20 which allowed us to reduce the echo time while still suppressing the outer voxel water (Fig. 6A). These improvements in cryocoil and LASER sequence increased the total number of metabolites detected from five to 18 (Table 1).
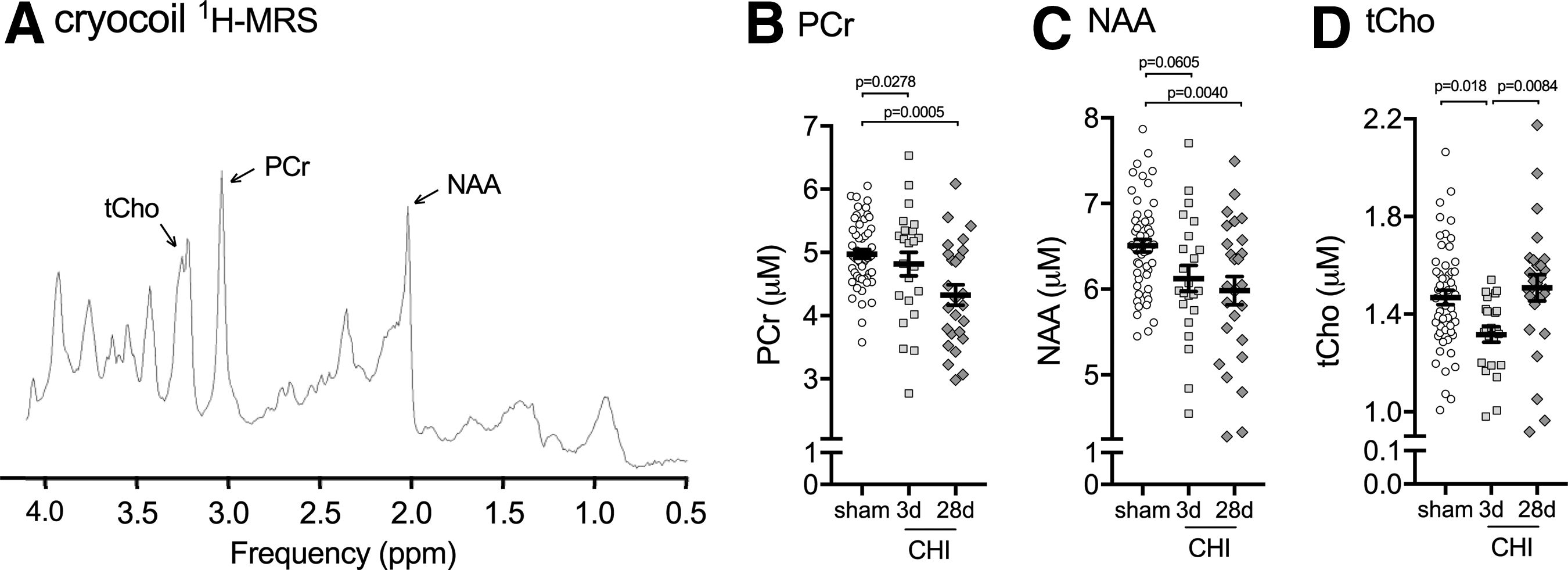
Closed head injury (CHI)-induced changes in hippocampal metabolites measured by cryocoil
1
H-MRS. (
CHI, closed head injury; SD, standard deviation; Ala, alanine; Cr, creatine; PCr, phosphocreatine; GABA, gamma-aminobutyric acid; Glu, glutamate; Gln, glutamine; GSH, glutathione; Oms, myoinositol; Lac, lactate; NAA, N-acetylaspartate; NAAG, N-acetylaspartylglutamate; tau, taurine; –CrCH2, creatine methylene group; tCHo, total choline; Lip, mobile lipids; and MM, macromolecules.
The pattern of hippocampal metabolite changes seen with the cryocoil 1 H-MRS was in agreement with the changes seen using the brain surface coil; however, the magnitude of the changes in metabolites was smaller, which likely reflects a decrease in variability associated with greater sample size and reduced variance from the 2.5 times more signal-to-noise using the cryocoil compared with the brain surface coil. We found that PCr was decreased 3% at three days post-CHI and 13% by 28 days post-CHI compared with sham levels (Fig. 6B). NAA was decreased by 6% at three days post-CHI compared with sham levels (Fig. 6C). In contrast to the brain surface coil where NAA levels had normalized by 28 days post-CHI, we found an 8% decrease in NAA at 28 days post-CHI compared with sham levels (Fig. 6C). The most consistent changes between the brain surface coil and cryocoil 1 H-MRS were found with tCho. A 10% decrease in tCho was found at three days post-CHI compared with sham levels (Fig. 6D), with levels of tCho returning to sham values by 28 days post-CHI.
Cryocoil 1 H-MRS as a radiological biomarker of TBI-induced deficits in brain metabolism and neuronal function
We next sought to determine the usefulness of cryocoil 1 H-MRS as a surrogate indicator of deficits in brain metabolism and neuronal function. We chose to use two threshold values with varying degrees of stringency. We wanted to assess how accurately we could separate injured mice from sham mice while maintaining the robustness to identify injured mice with deficits in surrogate indicators of brain metabolism or neuronal function. One standard deviation below mean sham levels of the metabolites was used as our low stringency threshold. For high stringency, we used a threshold just below the lowest sham value.
For PCr, the one standard deviation threshold was able identify 35–58% of the three days post-CHI and 28 days post-CHI mice, respectively; while only falsely identifying 13% of the sham-injured mice as having a deficit in PCr (Fig. 7A). With the high stringency threshold, we eliminated false detection of PCr deficits, and still maintained 13–19% of three days post-CHI and 28 days post-CHI mice, respectively, identified with deficits in PCr (Fig. 7B). NAA was able to identify 42–43% of the 28 days and three days injured mice, with a false positive rate of 19% in the sham-injured mice when a one standard deviation threshold was used (Fig. 7C). The detection rate of brain-injured mice with deficits in NAA was reduced to 13–27% of three days and 28 days post-CHI mice, respectively, when all false positives were eliminated (Fig. 7D). The discrimination potential of tCho was low, only identifying 27% of three days post-CHI mice while having a false positive rate of 14% in the sham-injured mice (Fig. 7E). The discrimination potential of tCHO to identify brain-injured mice was less than 10% when all false positives were eliminated (Fig. 7F).
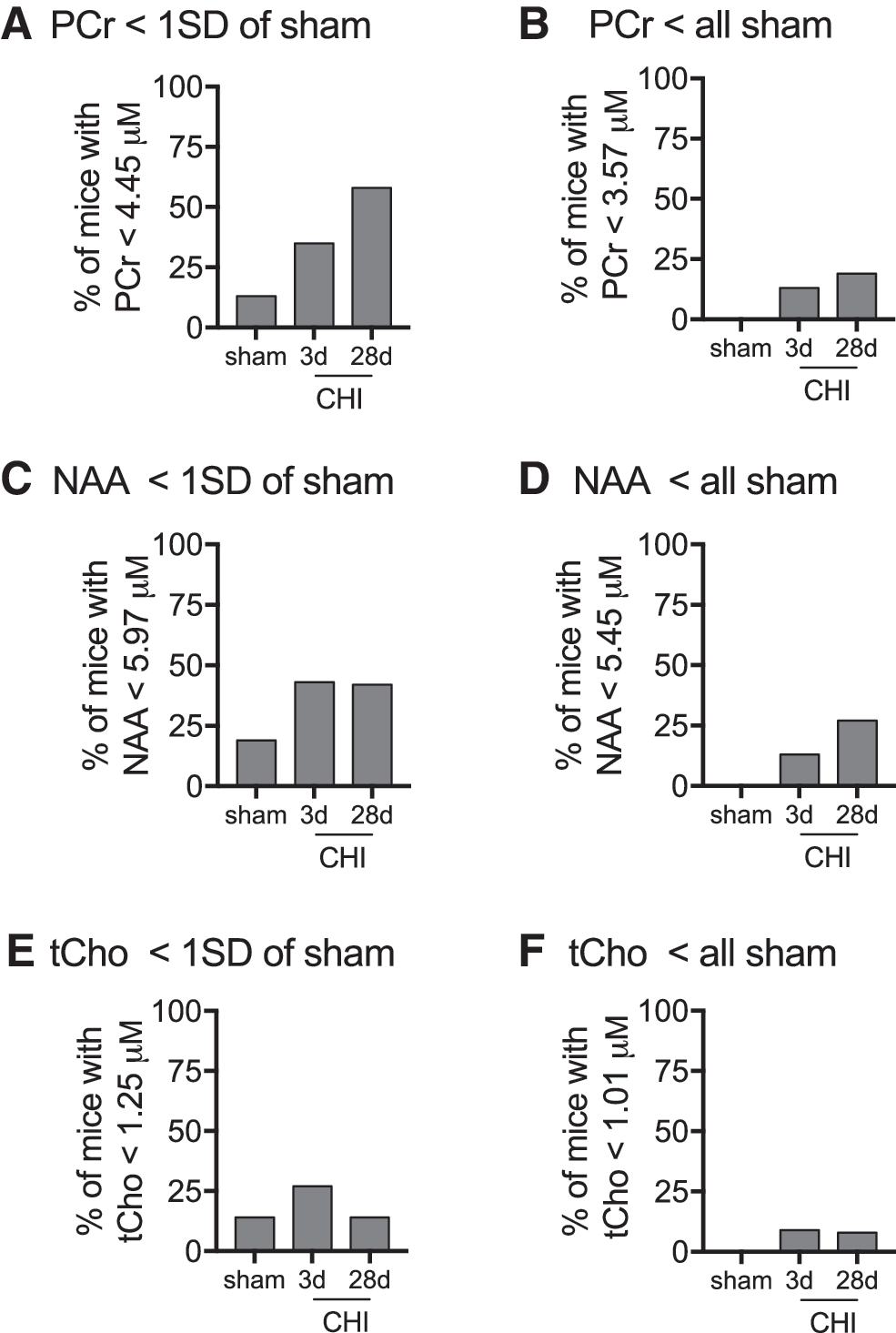
Discriminative potential of
1
H-MRS as a surrogate indicator of deficits in brain metabolism and neuronal function. The percentage of mice with a PCr (phosphocreatine) value less than 4.44 μM (i.e., one standard deviation (SD) of sham mean levels (
Female mice show increased vulnerability to CHI-induced changes in hippocampal metabolites
To assess sources of variability that contribute to changes in hippocampal metabolites, we stratified the data by sex (Table 2). In female mice, we found six metabolites that were showing a significant effect of CHI; however, in male mice, no statistically significant changes in hippocampal metabolites were observed. The metabolites that were found to be significantly affected by the CHI included PCr (Fig. 8A), NAA (Fig. 8B), and tCho (Fig. 8C), which showed a similar injury-induced pattern of changes in both the female and male mice. In addition, three metabolites were found to be changed by the injury only in female mice, including gamma-aminobutyric acid (GABA) (Fig. 8D), lactate (Fig. 8E), and myoinositol (Fig. 8F).
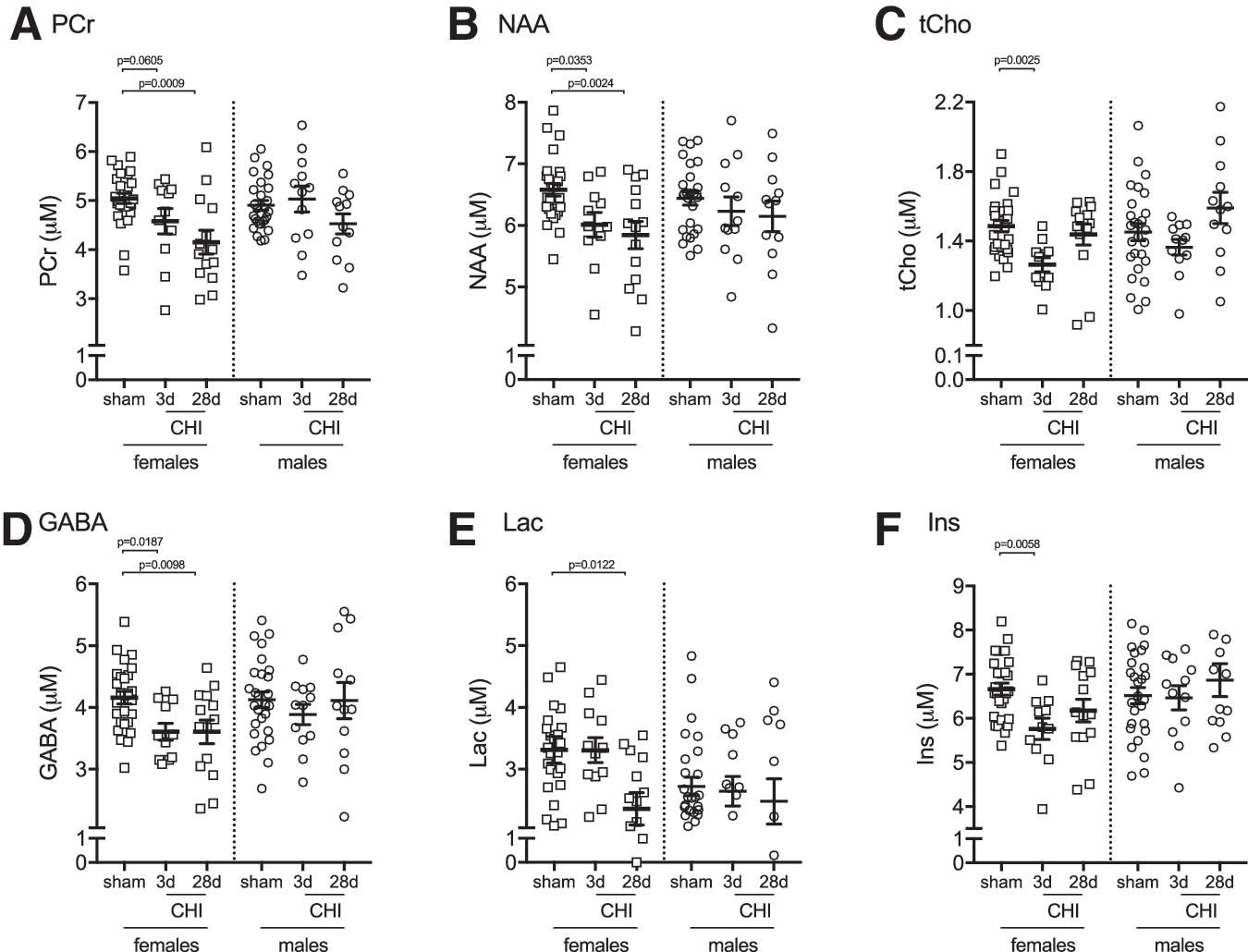
Stratification by sex identifies closed head injury (CHI)-induced changes in six hippocampal metabolites measured by cryocoil
1
H-MRS. A significant change was found in (
CHI, closed head injury; SD, standard deviation; Ala, alanine; Cr, creatine; PCr, phosphocreatine; GABA, gamma-aminobutyric acid; Glu, glutamate; Gln, glutamine; GSH, glutathione; Oms, myoinositol; Lac, lactate; NAA, N-acetylaspartate; NAAG, N-acetylaspartylglutamate; tau, taurine; –CrCH2, creatine methylene group; tCHo, total choline; Lip, mobile lipids; and MM, macromolecules.
Discussion
Two key findings from the studies are reported here. First, lasting deficits in mitochondrial-driven energy metabolism were found after a single, comparatively mild, diffuse brain injury. The injury-induced changes in mitochondrial metabolism were strongest in the hippocampus and synaptic nerve terminals. Unexpectedly, the deficits in mitochondrial bioenergetics worsened with time. TBI is one of the most well-described risk factors for the development of dementia. 4,5,28 The chronic effects of TBI on mitochondrial bioenergetics could be part of the mechanism contributing to the enhance risk of dementia developing, potentially associated with a premature aging of mitochondria.
The second relevant finding was the ability of MR spectroscopy to provide a way to monitor energy metabolism and neuronal health after a TBI noninvasively. Our results expand the utility of 1 H-MRS beyond humans and large animals toward the use of 1 H-MRS in mouse models of mild TBI. The ability to use the same efficacy biomarker in pre-clinical and clinical studies can provide a translational bridge in the important experiential medicine stage of drug development. 29
A mild TBI induces changes to mitochondrial bioenergetics similar to those seen with aging
The mechanisms by which a TBI hastens the onset of dementia are poorly defined. Aging is known to cause global decreases in brain metabolism, including both oxidative phosphorylation 30 and aerobic glycolysis. 31 Mitochondria are the primary source of oxygen radicals, and this underlies the mitochondrial free radical theory of aging. 32 Mitochondrial enzymes are at risk of damage when the antioxidant buffering capacity does not match demand. Increases in mitochondrial oxidative damage are known to occur with age. 33 Similarly, after TBI, elevated levels of intracellular Ca2+ are buffered by mitochondria, protecting the cell but potentially damaging mitochondria by the generation of oxidative stress. 34 Mitochondrial oxidative damage after TBI preferentially targets pyruvate dehydrogenase, the gate-keeper of mitochondrial respiration and ATP production. 35 Once pyruvate dehydrogenase is damaged, pyruvate cannot be converted into acetyl CoA, stopping the flow of electrons into the electron transport system by nicotinamide adenine dinucleotide and flavin adenine dinucleotide.
Prior work has shown that TBI suppresses mitochondrial respiration up to 72 h after the brain injury. 36 It has been assumed that with time after TBI, the damaged mitochondria would be eliminated by mitophagy, or the damaged mitochondria would lead to the death of the host cell. Our results contradict this assumption and suggest that in the case of a mild TBI, the deficits in mitochondrial bioenergetics propagate across time. Our finding suggests that TBI leads to changes in mitochondria akin to those seen with aging. Future studies are warranted to experimentally test whether long-term mitochondrial dysfunction after a mild TBI contributes to TBI-induced deficits in cognitive function and enhanced risk for dementia, and to determine whether therapeutic interventions targeting chronic mitochondrial dysfunction could reduce that risk.
Hippocampal mitochondria are selectively vulnerable to the effects of a mild TBI
Selective neuronal vulnerability is a hallmark of neurodegenerative diseases. Evidence suggests that neurons in general, and particularly neurons in the CA1 region of the hippocampus, have increased vulnerability to oxidative stress 37 and have decreased ability to buffer Ca2+, which contributes to the greater neuronal mitochondria dysfunction after a TBI. 3,38,39 In agreement with neuronal selective vulnerability, we found that a CHI resulted in a statistically significant suppression of mitochondrial respiration in the neuronal pre-synaptic mitochondria. Our previous results had suggested that the neocortex, most proximal to the direct forces of the CHI, was associated with the most pathology as determined by reactive gliosis. 12,13 The larger decrease in mitochondrial bioenergetics in the hippocampus compared with the neocortex, therefore, was unexpected. The decrease in the hippocampal energetic capacity did at least temporally correlate with hippocampal dependent learning and memory impairments in the brain-injured mice in agreement with previous studies from our group assessing mitochondrial function in the basal ganglia and its correlation to motor function both in TBI and aging. 40,41 We tested the possibility that gliosis in the cortex was masking the neuronal decrease in bioenergetics. However, our results do not support this argument because both synaptic and nonsynaptic mitochondria were found to have decreased respiration.
There are two additional explanations for the selective vulnerability of the hippocampal mitochondria to TBI. The first possibility is that neurons in the hippocampus are more sensitive to the damaging effects of oxidative stress, and this resulted in increased damage to the mitochondria. The second possibility is that biomechanical forces of the CHI affected the hippocampus more than the cortex. Studies are ongoing to test these two possibilities.
Head injuries are strongly correlated with cognitive deterioration and neurodegenerative dementia, including Alzheimer disease. 42 It is assumed widely that the effects of a mild TBI, such as dizziness and headaches, are temporary. Evidence suggests that this is not the case, and even a mild TBI can result in progressive brain atrophy. 43 All blows to the head do not lead to neurodegeneration; however, they do increase the likelihood that dementia may develop in the person's lifetime. Our work highlights a potential mechanism by which a mild TBI could lead to cognitive decline through a process of premature mitochondrial aging. Therefore, we sought to test translatable biomarkers that could be used to identify patients with mild TBI who are showing signs of TBI-induced mitochondrial aging.
Advanced neuroimaging is useful at detecting changes in surrogate markers brain metabolism and neuronal health that are altered after a mild TBI
Recent positive acute stroke intervention clinical trials highlight the importance of using advanced neuroimaging-based selection criteria in the successful outcomes of the trials. 44 The Stroke Imaging Research (STIR) group has identified that: (i) Neuroimaging can be used as an efficacy/biomarker or as an outcome assessment in clinical trials; (ii) no one single imaging approach will address all clinical requirements; (iii) use of imaging outcomes as a restrictive selection criterion may reduce the heterogeneity in the study population allowing for studies with smaller sample size requirements. 45 The guidelines outline by the STIR group are exceedingly relevant for mild TBI, because the number of cases of mild TBI are numerous, but the percentage of cases that will have clinical difficulties associated with the mild TBI represent a small proportion of the total number of cases (15%). 46,47
A primary goal of our study was to reverse translate two advanced neuroimaging surrogate markers of brain metabolism, which have shown utility clinically in mild TBI 7 –9 to a mouse model of mild TBI. We chose to use MR methods instead of positron emission tomography (PET) to measure brain metabolism, because of the wider availability of MR scanners in nonacademic clinical centers, and the advantage of not exposing persons to potentially toxic PET ligands. Our two primary MR methods used were pCASL to measure CBF and 1 H-MRS to measure hippocampal metabolites.
In agreement with previous human studies, 48 –51 we were able to detect changes in CBF in mice after a mild TBI. The pCASL sequence used in this study has greater specificity in analyzing CBF than other common methods. 21,27 We found substantial amounts of variability in all groups, however. Because we found that a TBI caused regionally based changes in CBF, with some areas showing hypoperfusion and adjacent areas showing hyperperfusion, it was necessary to use small ROIs. Despite the increased sensitivity of the 7-Tesla scanner and the pCASL sequence, we believe that the small ROIs used in the study were at least in part associated with the substantial variability in CBF seen between animals. In support of our assumption that the size of the ROI was at least partially associated with the increased variability, we found the lowest within group variance in the thalamus and hippocampus, which had the largest ROIs.
Improved MR sequences and scanners hold the potential to overcome the spatial limitations in the ability to define regional variations in CBF in mice after a mild CHI. This is only a limitation for pre-clinical studies, however. While we only see a trend in our mouse models after TBI, the results are nevertheless promising that pCASL could be useful in humans or larger animal models of TBI to identify metabolic disturbances caused by mild TBI.
The second neuroradiological method used to measure metabolic disturbances was 1 H-MRS. In our pilot experiment, we identified three hippocampal metabolites that were significantly decreased from sham levels, namely PCr, NAA, and tCHo. In a second large cohort of mice, we used a more sensitive 1 H-MRS method to validate these three metabolite changes and determine the potential of 1 H-MRS to be a surrogate indicator of brain metabolism and neuronal health. PCr is directly associated with mitochondrial energy metabolism and is used as a source of energy to maintain ATP when levels of ATP are limited. 52 In agreement with the decreases in mitochondrial respiration found in the hippocampus at 28 days after the brain injury, we also found decreases in PCr in the hippocampus at 28 days after injury by 1 H-MRS.
Our data are in accordance with a recent study in athletes who were exposed to a mild brain injury. 53 We found that a significant proportion of the head-injured mice decreased PCr below all sham-injured mice, suggesting that PCr levels accurately define injured versus uninjured animals with metabolic disturbances. In addition, we found that NAA, a marker for neuronal viability, which has been observed to decrease in patients in the acute phases after mild TBI, 8, 54 was also able to accurately define a subset of injured versus uninjured animals.
In contrast, while we found a statistically significant decrease in tCho levels at three days post-injury, tCho values were not able to accurately separate injured from uninjured animals. In agreement with our findings, the peak decrease in tCho after a moderate TBI in rats occurred three days post-injury, after which point the levels of tCho began to rise. 55 Changes in tCho have been reported in persons after a mild to moderate head injury; however, the levels of tCho were found to be increased after injury. 54,56 Similarly, we found that tCho were slightly elevated over sham values at 28 days post-injury. The results suggest a dynamic pattern of tCho values depending on the time after injury when measurements are made. The temporal changes and limited diagnostic potential limits the usefulness of tCho as an efficacy biomarker of intervention studies. The changes in hippocampal metabolites reported in the current study are in contrast to those of a recent study in a rat model of mild TBI, which failed to find differences in cortical metabolites after the injury. 57 Differences between the two studies are numerous and include species, TBI model, brain region, sample size, and spectroscopy method. We believe that the higher field strength scanner, improved water suppression sequence, 20 and the larger sample size used in the current study were all important for our ability to detect changes in brain metabolites after the mild TBI.
Limitations of the current study
Our studies included an equal proportion of female and male mice. Most of our end points were not sufficiently statistically powered to be disaggregated and reported by sex. The sample size available for the 1 H-MRS study was adequate to stratify the data by sex for exploratory analysis. For PCr, NAA, and tCho, we found a similar pattern of CHI-induced changes between the female and male mice; however, the CHI-induced changes in the metabolites did not reach statistical significance in the male mice, likely because of decreased statistical power with the smaller sample size. Interestingly, the exploratory analysis found that GABA, lactate, and myoinositol were decreased by the CHI in the female mice only. Future studies will be needed to determine how sexually dimorphic characteristics, chromosomal differences, and sex hormone levels may contribute to the sex-specific effects on GABA, lactate, and myoinositol.
Multi-modal MR is noninvasive, but in mice, it does require extended periods of anesthesia, with a range of 30–90 min, depending whether one or two sequences are acquired. Using isoflurane, we were able to regulate the depth of the anesthesia to maintain respiration. In addition, each imaging session required the mice to be under anesthesia. Studies have shown that isoflurane can be protective after injury. 58 Other studies have also demonstrated that prolonged isoflurane can impact CBF and mitochondrial function. 59 There is an advantage to repetitive scans in the same mouse, including establishing a pre-injury baseline. We do not know the effect that multiple rounds of extended anesthesia have on the recovery of TBI and brain metabolite levels. Therefore, we did not attempt to establish the time course of MR changes in the same mouse. For these same reasons, we used separate cohorts of mice for our mitochondrial bioenergetics measurements.
Conclusion
In this report, we find that a mild TBI results in a mitochondrial bioenergetics response that is akin to accelerated aging. We found a regional heterogeneity in mitochondrial bioenergetics that suggests selective vulnerability of neurons in different regions of the brain to the effect of TBI. Finally, we validated 1 H-MRS as a surrogate indicator of bioenergetic and neuronal health after a mild TBI.
Footnotes
Acknowledgments
The corresponding author, Adam Bachstetter, PhD, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. DNL was supported in part by a Kentucky Spinal and Head Injury Trust trainee fellowship. Research reported in this publication was supported by National Institutes of Health under award numbers R00 AG044445 (ADB) and P30 GM110787 (ADB), and pilot grant funding from the University of Kentucky (ADB). This work was supported in part by a Merit Review Award # I01BX003405 to (PGS) from the United States Department of Veterans Affairs Biomedical Laboratory Research and Development Program. The content is solely the responsibility of the authors and does not represent the official views of the funding organizations.
Author Disclosure Statement
No competing financial interests exist.