
Abstract
A 2023 workshop brought together stakeholders involved in the development and safety assessment of oligonucleotide (ONT) therapeutics. The purpose was to discuss potential strategies and opportunities for enhancing developmental and reproductive toxicity (DART) assessment of ONTs. The workshop was timely, bringing together regulators, industry representatives, consultants, and contract research organization partners interested in the ongoing development of internationally harmonized guidance for nonclinical safety assessment of ONTs. Given DART’s importance in nonclinical safety assessment and the unique attributes of ONTs, the forum discussed case studies, consensus approaches, and areas needing further development to optimize DART strategies. This report covers the workshop proceedings, highlighting methods to achieve a robust DART assessment for ONTs. It includes case studies that described strategies for dose level selection, dosing frequency, species selection, and alternative animal model approaches. Topics also cover surrogate ONT use, exposure of the placenta and embryo/fetal compartment, and weight of evidence approaches. A goal of these workshop proceedings is to describe example approaches to hopefully inform the DART strategy expectations in the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use guidance currently under development for nonclinical safety assessment of ONTs.
Introduction
Background, workshop purpose, and goals
Oligonucleotide (ONT)-based therapeutics are an important and rapidly evolving modality of medicines that use short nucleic acid sequences to modulate gene expression. The first U.S. Food and Drug Administration (FDA) approval of an antisense oligonucleotide (ASO) occurred roughly 25 years ago and the first small interfering RNA (siRNA), gained regulatory approval in 2018. As of 2025, there has been a significant increase in the number of globally approved ONTs available on the market, along with numerous others currently undergoing clinical trials. These compounds hold advantages for patients of rare diseases and are rapidly becoming more prevalent in the development of treatments for more common indications. 1 As ONTs expand in scope and complexity, nonclinical safety assessment continues to evolve, with different strategies being justified by sponsors (eg, companies, nonprofit foundations).
A major component of the nonclinical safety evaluation of a drug or biological product is the developmental and reproductive toxicity (DART) assessments. Due to the importance of this nonclinical focus area, the lack of unified guidelines for ONTs, and the momentum in the pharmaceutical community as it gains experience with designing DART packages specific to ONTs, a workshop was held to exchange information across DART experts and toxicologists working on ONT development. The workshop was organized by the Health and Environmental Sciences Institute (HESI) DART Technical Committee and took place 17–18 October, 2023, in Washington, DC. Speakers and participants included representatives from the pharmaceutical industry, contract research laboratories (CROs), and global regulatory agencies. The workshop served as a forum to determine areas of consensus and areas in which gaps or challenges exist when determining best practices for DART assessment of this drug class. Some of the presentations from the workshop were subsequently presented at the 2024 Society for Toxicology and 2024 Society for Birth Defect Research and Prevention Annual Meetings. While ONTs include a diverse set of chemically synthesized molecules containing short DNA or RNA strands, the scope of compounds covered in the workshop was limited to siRNAs used to leverage the RNA-induced silencing complex for directed RNA degradation and ASOs, used to recruit RNase H for directed RNA degradation sterically block the targeted RNA.
DART studies for pharmaceuticals are typically divided into segments designed to collectively cover assessment of one complete reproductive life cycle from conception in one generation through conception in the following generation 2 (Fig. 1). The full reproductive cycle is defined by separate stages (designated A to F) and is covered in individual studies. The most common approach is to evaluate stages A and B (premating to implantation) in a fertility and early embryo development (FEED) study, stage C (implantation to closure of the hard palate) in an embryo–fetal development (EFD) study, and stages D–F (fetal maturation, birth to weaning, weaning to sexual maturation) in a pre- and postnatal development (PPND) study. EFD studies are generally expected to be conducted in two species (a rodent and a nonrodent, described further in the section “Species Selection”), at least one of which should be pharmacologically responsive. For biological drugs, these studies should only be conducted in two species if both are pharmacologically responsive.
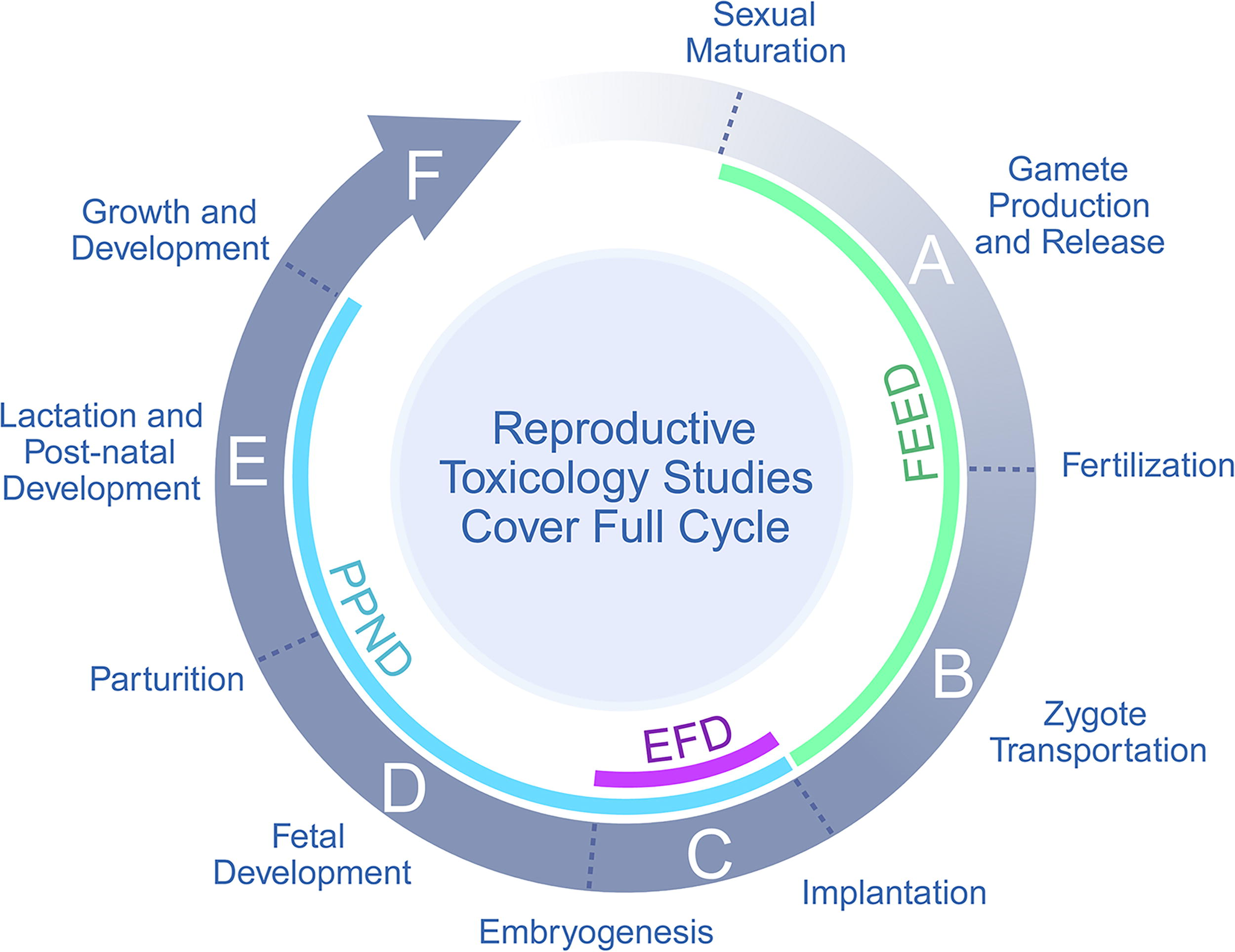
The stages of the reproductive cycle are assessed to support pharmaceutical development. Stages are typically evaluated using three in vivo study types to cover all stages labeled A–F. Semicircles indicate segments of the reproductive cycle during which animals are administered the drug candidate in the fertility and early embryo development (FEED), embryo–fetal development (EFD), and pre- and postnatal development (PPND) studies.
The timing of DART studies relative to the clinical development is described in International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH M3(R2)) and commonly includes the conduct of EFD studies prior to enrollment of women of childbearing potential (WOCBP) or prior to Phase 3 depending on the region. 3 The FEED study is conducted prior to Phase 3, and the PPND is conducted prior to submission of the marketing application. The strategy for DART assessment used for a particular development program differs depending on modality and product attributes, existing information on mode of action (MoA) or repeat-dose toxicity, intended patient population, and indication. Specific principles are applied to the conduct of DART studies to ensure dosing and exposure during critical developmental periods, to select adequate doses, and to fully evaluate the effects of exaggerated pharmacology, as well as effects of chemical structure and, if appropriate, off-target pharmacology. Adherence to these principles could lead to modifications in the standard study designs when applied to ONTs, including which stages of the reproductive cycle are included in a single study, selection of species, use of a surrogate ONT or other alternative model, optimization of dosing paradigms, and collection of supporting data such as absorption, distribution, metabolism, excretion, and pharmacodynamics (PD).
ONT drugs are regulated as small molecules; however, they have some attributes of large molecules. There is currently no internationally harmonized regulatory guidance specific to ONTs, although the Pharmaceuticals and Medical Devices Agency (PMDA) in Japan issued a guideline on nonclinical safety assessment of ONTs in 2020. 4 Existing testing strategies have used a hybrid approach of applying principles for both small molecules (ie, ICH M3 3 ) and biologics (ie, ICH S6 5 ). The DART ICH S5(R3) guidance includes considerations for both small molecules and biologics, and therefore, all principles unique to DART testing for ONTs are covered. Nevertheless, many questions remain regarding how to apply some of these principles to fit with the unique product attributes of ONTs. The workshop was therefore designed to develop specific recommendations for DART assessment for ONTs. In November 2024, the ICH Management Committee endorsed a concept paper to create a new ICH Safety Guideline titled “Nonclinical Safety Evaluation of Oligonucleotide-Based Therapeutics,” which will serve to clarify regulatory expectations in this area. The concept paper indicates that the recommendations for DART assessments will be among the topics to be harmonized. FDA also released a draft guidance document in November 2024 on nonclinical safety assessment of ONTs, 6 including a section describing DART expectations. The basic principles in the draft FDA guidance generally align with the output of the workshop, and it is the author’s hope that the example approaches discussed herein will inform the DART section of the forthcoming ICH guidance document.
The workshop opened with an introductory session, followed by multiple case studies addressing different aspects of DART strategies and study designs, and closed with a round table session. The introductory session included presentations by speakers representing the Oligonucleotide Safety Working Group (OSWG) DART subgroup that authored the original white paper 7 on considerations for DART for ONTs (see the section “Example Approaches and Points of Consideration”); regulatory bodies from the United States, European Union, and Japan; and CROs partnering with companies developing ONTs. Case studies were organized into focus topics, including dose selection and dosing schedule, species or model selection, and unique scenarios that warrant consideration outside of a traditional approach for DART assessments. Finally, the round table discussion featured representatives from industry, regulatory bodies, and authors of the OSWG 2014 DART subcommittee article. Key consensus points were highlighted and discussed during this closing session, and areas requiring further data support were identified.
Overall, the workshop identified several approaches to apply DART principles described in ICH S5(R3). These workshop proceedings present common approaches and best practices identified in the workshop. Additionally, this white paper highlights areas in which ongoing generation of data may inform future approaches for designing DART strategies for ONTs, including the use of in silico, in vitro, or alternative in vivo models. Finally, the paper highlights areas where modality-specific guidance on ONT DART safety assessments could supplement the existing DART guidance for drugs and biologics.
Introductory concepts in application of DART principles to ONTs
Several key concepts are introduced and described in this section to serve as background information before summarizing the workshop presentations and example approaches. ONTs have unique product attributes (Fig. 2) that require consideration on a per case basis for applying DART principles. ONTs are chemically synthesized and include chemical modifications that distinguish them from endogenous nucleic acids, which may elicit toxicities related to chemical/physical structure. However, ONTs likely fall somewhere in between small molecules and biologics for target and species specificity, as they are designed to hybridize with specific nucleotide sequences that may be species-specific but also have some potential to hybridize to unintended targets. They are designed to partition rapidly into target cell types where they reside and exert PD for an extended period, resulting in unique kinetic and pharmacological properties that need to be considered when designing the DART study dosing regimens. ONTs often contain a delivery conjugate component, such as N-acetylgalactosamine (GalNAc) or transferrin receptor antibody, which facilitates enhanced uptake into the cells of the target tissues (and avoids other tissues). The same “platform” consisting of conjugate, linker, and modifications can be used across ONTs, raising the question of the ability to utilize data from one ONT program to another program using the same platform. Finally, the use of a weight of evidence (WoE) approach for understanding DART risk can be applied to ONTs but may need to be coupled with experimental or computational data generation to ensure all attributes are accounted for when considering human risk potential.
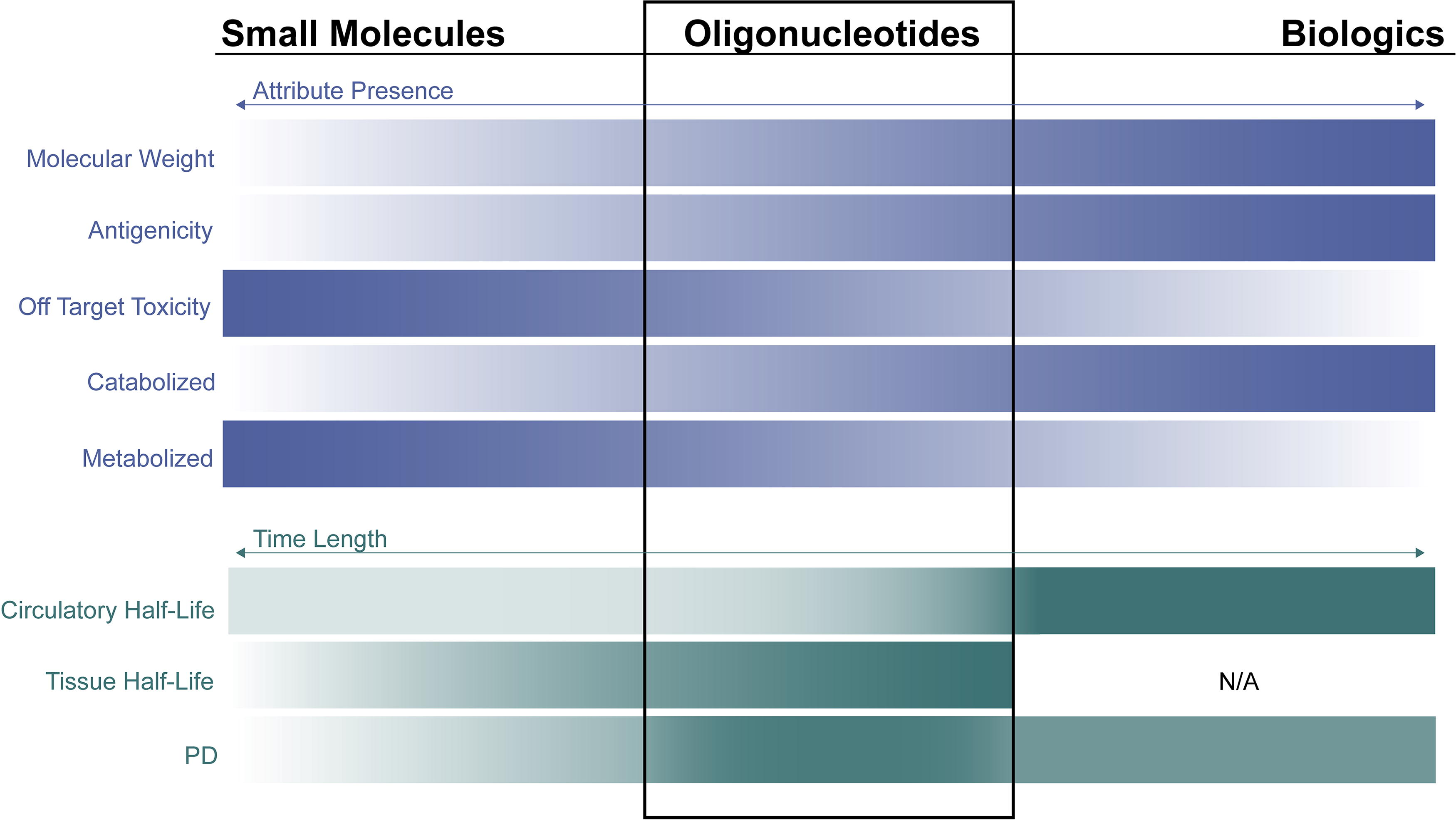
Oligonucleotide attributes compared to small molecules (new chemical entities; NCEs) and biologics (new biological entities; NBEs). Darker shading indicates the modality has a greater degree of the indicated properties.
On-target, off-target, and chemical/physical property-related toxicity potential in DART studies for ONTs
On-target toxicity of an ONT refers to exaggerated and adverse pharmacologic effects due to engagement (hybridization-dependent) with the target RNA of interest in the test system being evaluated. Off-target toxicity refers, in part, to effects caused by modulation of unintended targets (also hybridization-dependent); these may be related biologically or totally unrelated to the target RNA/protein of interest. Off-target toxicity can also refer to adverse effects due to mechanisms beyond ONT hybridization (hybridization-independent), such as effects of chemical or physical structure (Fig. 3). 8
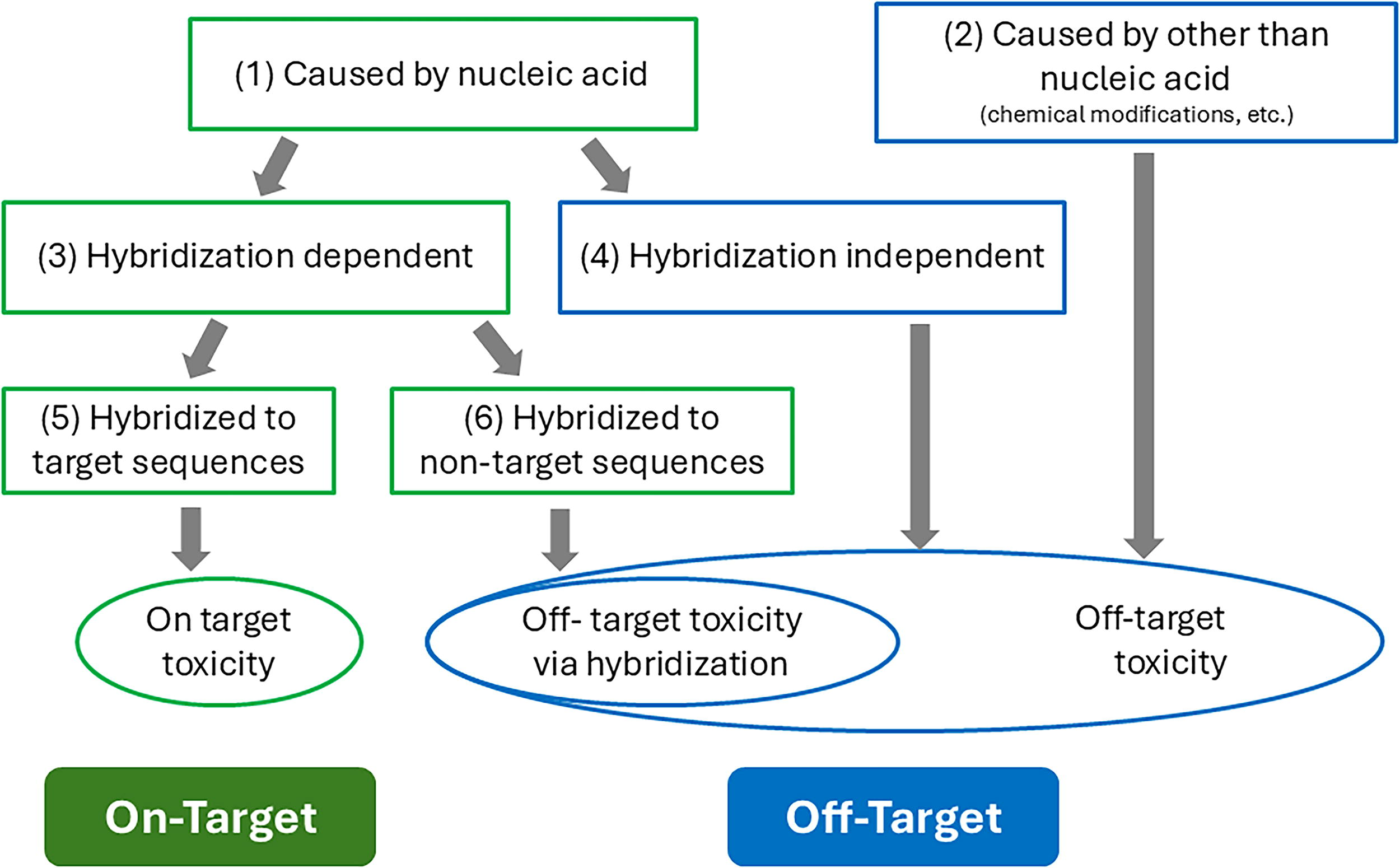
Classification of toxicities induced by ONTs (oligonucleotide therapeutics). This figure illustrates the classification of toxicity mechanisms associated with ONTs. Toxicity is broadly categorized into two types: (1) toxicity caused by nucleic acid and (2) toxicity caused by factors other than nucleic acid, such as chemical modifications that were intended to improve in vivo pharmacokinetics (PK). Toxicity caused by nucleic acid is further subdivided into (3) hybridization-dependent toxicity and (4) hybridization-independent toxicity. Hybridization-dependent toxicity involves engagement with RNA sequences and is further divided into (5) on-target toxicity (due to hybridization with the intended target sequence) and (6) off-target toxicity (due to hybridization with unintended nontarget sequences). In contrast, (4) hybridization-independent toxicity is a result of chemical structures specific to the nucleic acid molecules or physicochemical properties not mediated by hybridization, such as changes in innate immunity mediated by toll-like receptors (TLRs). Notably, off-target toxicity comprises three components: (2) toxicity caused by factors other than nucleic acid, (4) hybridization-independent toxicity, and (6) hybridization-dependent off-target toxicity. Understanding these mechanisms provides critical insights into risk assessment and the development of safer ONTs.
Both the risk assessment and the development strategies used for xenobiotics, including ONTs, can be influenced by the understanding of the mechanism of toxicity. 9 Information, including in silico analysis using human databases, in vitro assays using human samples, and understanding the biological characterization of the genes involved, can help predict hybridization-dependent toxicity. 10 For off-target (hybridization-independent) toxicity, the toxicologist needs to consider a compound’s structure, its chemical/physical properties (including the impact of a formulation), as well as receptor or protein interactions.8,11
In developmental toxicity studies, maternal administration of an ONT could result in on- and off-target toxicity in two compartments not found in general toxicity studies, the placenta and the developing embryo/fetus. Toxicity in these compartments can be a result of biodistribution of the ONT into these compartments or ONT-induced changes in maternal physiology that are not directly linked to the placental or the embryofetal compartment. The changes that occur over the course of gestation in these two compartments further complicate these evaluations. Translation of any observed toxicity, or lack thereof, needs to be conducted with a knowledge of the basic developmental biology of the test system. For example, in humans, the placenta is the sole organ of nutrient transport and a potential conduit for transfer of xenobiotics to the embryo, while in rats and rabbits, the visceral yolk sac performs these functions early in gestation with the placenta taking over these functions later in gestation. 12 In rats, the visceral yolk sac completely envelopes the embryo during early organogenesis, whereas in rabbits, it remains open, potentially allowing the embryo to be in direct contact with maternal secretions and providing another possible avenue for transfer of xenobiotics to the embryo. 13 Further differences in placenta type and features should be considered when using alternative animal models, such as minipigs, in DART studies. 14 Additional species differences in early development can also influence regulation of embryonic drug exposure, including pH gradient between maternal and embryo/fetal compartments, placental transporters and metabolic enzymes, and protein binding differences within the conceptus and maternal fluid compartments. 13 Therefore, establishing kinetics of ONT exposure in pregnant animals, over the course of organogenesis, could help put the animal study findings into context. However, the presence or absence of an ONT in the fetal compartment during any gestational interval in animals may not predict transfer of the ONT to the human embryo/fetus.
Using a nonbiologically responsive animal model to detect off-target toxicity or toxicity due to chemical/physical properties of an ONT is useful, but any toxicity, or lack thereof, should be translated to human fertility, embryo–fetal development, and postpartum development. If toxicity is observed in the rodent or rabbit, additional investigative work could be necessary to determine if the toxicity is unique to the test system or if it also presents a safety risk in humans. Often the mechanism driving off-target toxicity is unknown and assumed human relevant in the absence of investigative data.
Considerations of pharmacological relevance of test species
Studies in rodent and rabbit are the preferred approach for DART, as these species are defined by ICH S5(R3) as “routine test species” due to being well-characterized and relevant for detecting treatment-related effects. Nevertheless, these routine DART species may not capture on-target assessment as ONTs are sequence dependent and often species-specific, with human and nonhuman primate (NHP) frequently being the only pharmacologically relevant species. When appropriate, a surrogate ONT designed to hybridize with an analogue to the target gene in a routine DART species can provide a means to fully address pharmacology-based concerns for an ONT. This approach has been used with approved ONTs such as patisiran (ONPATTRO®) and nedosiran (RIVFLOZATM) GalNAc siRNAs.15,16 Considerations on characterization and study design for a surrogate molecule are described further in the section “Example Approaches and Points of Consideration.”
A surrogate-based approach may not be feasible based on gene or protein expression differences or absence of the target gene in traditional DART species. Complexity increases with the use of a surrogate ONT conjugated with a species-specific delivery component, such as an antibody. This scenario is discussed further in the section “Species Selection Considerations for Biological-Conjugated ONTs.” The NHP has been the most used species for general toxicity assessment of ONTs based on pharmacological relevance; however, DART assessments in NHP have limited flexibility in terms of design and scope and are less robust as they evaluate a limited number of offspring with single offspring per mother.17,18 This option should be restricted only to cases where on- or off-target concerns cannot be ruled out a priori or routine DART species or alternative models have been thoroughly evaluated and deemed unsuitable.
Consideration of PK/PD (pharmacokinetics/pharmacodynamics) disconnect of ONTs
ONTs have the unique product attribute of being cleared rapidly from systemic circulation, but having longer tissue retention times, leading to prolonged pharmacodynamic effects. Clinical dosing regimens for ONTs, therefore, involve infrequent administration, for example, weekly, monthly, quarterly, or even yearly. As such, general toxicity studies with ONTs are commonly conducted using an intermittent dosing regimen to mimic the intermittent clinical dosing regimen. For DART evaluations, particularly EFD studies, sustained systemic exposure and pharmacologic activity during each critical developmental period are necessary to sufficiently evaluate potential adverse effects on development. For this reason, infrequent, intermittent dosing is not optimal during gestation, and therefore, different dosing regimens, as described further in the section “Dosing Schedule,” have been used for these studies to sustain plasma exposure between doses. Additionally, timing of the onset of target protein modulation should be considered in relation to timing of dosing and degradation of the target RNA. To ensure the intended pharmacological outcome of protein modulation is occurring during the intended exposure periods within DART studies, adjustments to the initiation of dosing may need to be considered, as described further in the section “Dosing Schedule.” Finally, to achieve efficient delivery of ONTs to the site of interest, ONTs may require clinical dosing routes of administration not conducive to replicating in nonclinical DART species, such as intrathecal administration. Consideration should be given to the most appropriate route used in DART studies to mimic relevant clinical exposure.
Use of a weight of evidence approach or a platform approach
A WoE approach as described in ICH S5(R3), ICH S6(R1), and numerous publications 19 can be applied in an ONT DART strategy when sufficient information is available to describe the risk due to on-target pharmacology. A concept more unique to ONTs of using a “platform approach” may be considered and potentially applied to justify the use of a single species for an EFD assessment or obviate the need for a full assessment of off-target DART potential. The platform approach concept was theoretical in nature and had not been applied to approved ONTs at the time of the workshop. In this concept, sufficient data might include EFD data demonstrating no adverse effects for another ONT using the same platform (conjugation, linkers, and chemical modifications) and for which the only difference from the untested candidate is the nucleotide sequence. In this case, the “platform approach” for off-target hybridization-independent toxicity risk could supplement an assessment of the hybridization-dependent DART (on- and off-target) risk to provide sufficient information to inform on overall DART risk. Alternatively, if a DART signal is observed with a specific platform, these data may be useful in a WoE, and potentially no additional DART studies would be warranted. At the present time, these exceptions to a small-molecule EFD strategy are unlikely to apply to most ONTs, and it is recommended that any significant deviation from ICH expectations should be discussed and approved by health authorities.
Workshop Summary
Introductory session summaries
DART for oligonucleotides: Where have we been and where are we now?
The opening presentation of the workshop provided an overview of the efforts undertaken by the OSWG DART subteam. The OSWG was established following the Oligonucleotide-Based Therapeutics Conference in 2007 hosted by the FDA and Drug Information Association. The OSWG consists of subcommittees covering various aspects of safety assessment, with members representing academic and industry researchers. 20 The Reproductive Subcommittee of the OSWG published an article in 2014 describing points to consider in designing science-based reproductive safety evaluations based on the attributes of the specific product.
The points to consider were discussed for specific ONT subtypes, with the caveat that mainly ASOs had been approved for marketing at that time. Following the summary of the state of the field at the time of the OSWG publication, the current questions and status regarding DART approaches for ONTs were briefly introduced to start the workshop, including:
What approaches are currently being used to address the unique attributes of ONTs when designing DART strategies and studies? Have there been any advances in implementation and acceptance of alternatives to using a traditional small-molecule DART strategy approach? Is there enough data on this drug class to list best practices for considerations such as species or model selection; appropriate alternatives to animal models to capture on- and off-target toxicity potential of ONTs; dosing regimen; and study designs for use of surrogate molecules or transgenic animal model approaches? What are some additional considerations that have evolved as the drug class has advanced in terms of complexity of the ONT components, including use of multiple different delivery conjugates? Is there a global regulatory consensus on DART assessment for ONTs?
Regulatory perspective
Pharmaceuticals and Medical Devices Agency, Japan
The PMDA presentation provided a thorough review of the Japanese guideline for evaluating ONTs, 4 primarily focusing on potential toxicities in DART assessments. The guideline classifies ONT-induced toxicities into hybridization-dependent and hybridization-independent types. Concerning on-target toxicity assessment, the presentation emphasized the importance of using pharmacologically relevant animal models and favoring surrogates over NHPs. Exploring hybridization-dependent off-target toxicity, the presentation advocated for tailored evaluation methods such as in silico analysis and RNA sequencing (RNAseq) with human cells. It also highlighted the necessity of comprehensive in vitro gene expression analysis, especially for safety assessment of siRNAs, while ensuring the reliability of in silico algorithms and databases. In assessing hybridization-dependent on- and off-target toxicity, if placental transfer of ONTs indicates fetal exposure below a biologically significant level, it can be used as part of a WoE assessment. Finally, the presentation addressed hybridization-independent off-target toxicity induced by chemical modification of ONTs and highlighted challenges regarding setting dosing frequency in EFD studies plus the acceptance of platform approaches to mitigate toxicity studies with clinical candidates. In particular, the PMDA representative emphasized a cautious stance against platform approaches to evaluate toxicity due to reported sequence-dependent acute toxicity in nonclinical studies with ASOs,21,22 which would not be ruled out with a platform approach.
Dutch Medicines Evaluation Board, Netherlands
The presentation from the Dutch Medicines Evaluation Board (MEB) highlighted that there is no consolidated European Union position on ONTs. Access to data for EU member state regulators is limited since they rely on companies or academia to publish or share their data. When companies choose to apply for optional scientific advice at the national competent authorities or European Medicines Agency (EMA), limited data become available to the regulators in the form of a briefing book and/or investigator’s brochure. When companies submit a market authorization application, full study reports are provided, but at the time of the workshop, the number of EU-approved ONTs was limited. There is no access to data on products that are discontinued early during nonclinical or clinical development, while these data can be highly valuable. This is different compared to the FDA, which is often involved at an earlier stage during development.
A preliminary survey was conducted by the MEB on ∼60 scientific advice letters given by the EMA (committee of human medicinal products) between 2020 and 2023 concerning the nonclinical development of ASOs and siRNAs (article in preparation). Companies mainly asked for advice on the adequacy of the general nonclinical development plan but did not ask specific questions on the design of DART studies (eg, use of surrogates, timing of dosing, choice of species). When evaluating the DART testing strategy as proposed by the companies, it was noted that many different strategies were proposed, following ICH S5(R3) but often without proper scientific justification. This indicated that additional guidance is needed. The MEB representative emphasized wanting to move these strategies away from following a tick-box approach (which could provide a false sense of safety) and steer toward a more science-based approach.
In general, the MEB would like to further explore approaches in which data from similar products can be leveraged to negate the need for conventional DART studies for a new product (ie, WoE approach). It is envisioned that in cases where products have the same chemistry/backbone modifications and they target the same gene, but have a different sequence, a non-DART in vitro or in vivo bridging study could be considered to determine whether this change in sequence would result in any new toxicities. In addition, when DART data are already available for an unconjugated form of the ONT, these data could be used to evaluate the DART risk for a GalNAc-conjugated form with the same sequence.
Food and Drug Administration, United States
The FDA presentation opened with a consideration of the current regulatory landscape for the development of ONTs, and how this has begun to evolve in recent years, noting in particular (1) the availability of the Japanese guidance, (2) publication of a considerations white paper by the EMA, (3) publication of FDA draft guidance to support the development of individualized antisense ONTs for treating severely debilitating or life-threatening diseases, and (4) ICH support for developing harmonized guidance for the nonclinical safety assessment of ONTs. The FDA draft guidance document was issued after the workshop. 6
Specifically, regarding DART safety assessment, it was stressed that ONTs are considered in scope of ICH S5 (R3); however, it was also recognized that specific characteristics of ONTs, in particular their rapid clearance from circulation and prolonged tissue residence time (and consequently prolonged PD), pose unique challenges in designing DART studies, especially EFD studies. Regarding EFD study recommendations, FDA’s position appears to be aligned with that of the PMDA in that EFD assessments are generally expected in two species, and that the intended pharmacological activity should be assessed in at least one species. The FDA was also aligned with the preference for adding a species-specific surrogate arm to the rodent study, rather than defaulting to the NHP, if the NHP is the only species responsive to the clinical candidate. The recommendation for the use of two species is based on the observation that the base sequence has a significant and unpredictable effect on hybridization-independent off-target toxicity, and that different chemical structures can also have unique toxic effects.
DART strategy for oligonucleotides: Perspectives from the CRO
The CRO presentation discussed commonly asked questions and concerns related to the design of DART studies for a nonclinical development program of an ONT. These were important points to highlight as many smaller companies do not have dedicated DART-experienced toxicologists or may be uncertain about when to involve trained DART experts. A first step in outlining any DART plan is to clarify key parameters for the program, including Is the ONT candidate an ASO, a siRNA, or a different construct? Is the indication a rare disease; oncology; a disease involving a large patient population; one in which WOCBP are included early in clinical development and/or at relatively high numbers? Common or uncommon routes of administration and the planned dose schedule have a direct impact. The answers, or working assumptions, influence the final study design(s). Similarly, there are program-specific objectives and concerns. DART studies are almost always conducted post Investigational New Drug and after Phase 1 clinical trials, the general toxicology studies and clinical data provide relevant context. Additional questions include “Are there concerns based on the intended pharmacology (on-target)”; “Is an unintended effect a concern, ie, unintended pharmacology or those due to the ONTs chemical/physical properties?” Furthermore, what is the mechanism of toxicity, a direct effect on the embryo/fetus, on male fertility, female fertility, the ability to maintain pregnancy, or a combination of these? Program specific considerations must be balanced with current regulatory expectations and scientific rationale.
DART study designs are well-established and widely accepted, and working within the expected designs for rodent and nonrodent studies is the most effective means of addressing potential human risk. However, ONTs may not fit neatly into standard designs. This could mean considering a nonstandard dose schedule and modified dose levels. Ultimately, there is flexibility in the approach for a DART program, and with appropriate justification and a clear rationale, alternative designs have been accepted by regulatory agencies.
Case studies and discussion
Case studies were presented by 11 speakers representing both small and large biotechnology or pharmaceutical companies that are actively developing ONTs. The case studies and discussion provided examples of approaches for DART testing and highlighted consensus points and areas where a one-size-fits-all approach was not sufficient.
In the first session, general principles were covered, including dose setting and dose frequency across different phases of the reproductive cycle, considering the unique PK, PD, and toxicity profiles of ONTs. The second session focused on species and model selection for ONTs. Specific topics covered by speakers included determining PD relevance in routine DART species (rodent and rabbit), use of an animal analogue/surrogate ONT to the pharmacologic target in a routine DART species, and use of nonroutine DART species, including NHP and minipig. In the third session, speakers presented case studies in which characteristics of the ONT or the indication required unique approaches in the DART strategy. These included considerations for an antibody-conjugated siRNA, an siRNA indicated during pregnancy with a target gene in the placenta only, and understanding lactation phase data within a PPND study with an siRNA. The information gathered from the case studies is presented in two formats. First, each of the presented case studies is briefly summarized in Table 1 (with a more detailed summary provided in Supplementary Data) to provide information on the specific topics, questions, and experiences. Next, consensus points and example approaches used across the various case studies are described.
Case Study Summaries
ASO, antisense oligonucleotide; DART, developmental and reproductive toxicity; EFD, embryo–fetal development; FEED, fertility and early embryo development; GalNAc, N-acetyle galactosamine; GLP, Good Laboratory Practice; ICH, International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use; LNA, locked nucleic acid; MOE, methoxyethyl; NHP, human and nonhuman primate; PD, pharmacodynamics; PK, pharmacokinetics; PMO, phosphorodiamidate morpholino oligomer; PPND, pre- and postnatal development; siRNA, small interfering RNAs.
Example approaches and points of consideration
The case studies and discussion throughout the workshop highlighted areas where multiple approaches can be applied depending on the specific program, such as dose level selection and species or model selection. For other areas, consensus was reached on approach, such as dosing frequency during different phases of the reproductive cycle and general application of a surrogate approach. Several areas were identified as needing additional data before the approach could be universally accepted, such as use of a platform approach, use of alternative assays including in vitro, ex vivo, or nonmammalian in vivo assays, and understanding of toxicokinetic characteristics of ONTs in reproductive tissues and compartments. These approaches or areas for further investigation are summarized in the sections below.
Dose level selection
Identification of a relevant high dose is essential to a scientifically robust DART evaluation. As stated in ICH S5(R3), dose justification for DART should be based on all available information, including pharmacology, PK, and repeat-dose toxicity. For an ONT, in addition to the general ICH S5(R3) considerations of on-target pharmacology-related PD, the potential for off-target effects and accumulation within organs often directly influence dose selection. The selection of lower dose levels follows the standard practice of evaluating a dose response and establishing a no-observed-adverse-effect level (NOAEL) with consideration of expected or intended clinical doses. Due to the unique kinetic and PD properties of an ONT, target tissue concentration should be considered in dose setting decisions to avoid nonrelevant tissue burden. This step may require additional studies or endpoints, such as exposure assessment in rabbits, as this species is not commonly used for general toxicology. Ultimately, the choice of a high dose and dose range follows established DART and general toxicology practice with ONT-specific considerations. This theme was consistent throughout the workshop; however, discussions also emphasized the need for flexibility and a strong emphasis on determining relevance of a high dose and dose range for each ONT program.
Toxicity-based endpoints or exposure-margin-based endpoints were the most common means of dose level selection. Table 2 outlines key concepts and approaches and provides a short list of advantages and disadvantages. Dose level selection based on the maximum tolerated dose (MTD) and plasma exposure or dose margin have been used in ONT DART assessments. One advantage of an MTD approach would be a high likelihood that the evaluation will meet regulatory expectations for dose or exposure multiples equal to and above clinical doses or exposures. Potential disadvantages include the possibility of tissue exposure levels that far exceed those relevant to human risk assessment. When utilizing an MTD approach, pilot studies would be essential to determine exposure in blood [ie, max serum concentration (Cmax) and area under the curve (AUC)] and key tissues and the concentration versus effect relationship. Similarly, while the approach of using an exposure margin of >25× highlights systemic parameters, tissue levels of an ONT and their potential for toxicity in the target tissue should be considered.
High-Dose Level Setting for the Oligonucleotide Clinical Candidate or Surrogate Molecule
AUC, area under the curve; Cmax, max serum concentration; HED, human equivalent dose.
Establishing a high dose based on the calculation of a human equivalent dose has been utilized in ONT programs. Justification should be provided to support a scaling factor. An example is the siRNA inclisiran (LEQVIO®) for which extended PD in patients allows for a 284-mg initial dose, repeated at a 3-month interval, and then once every 6 months thereafter. EFD studies in rat and rabbit involved once daily dosing at up to 150 mg/kg/day. 23 Doses evaluated were based on body surface area scaling, with a safety margin of up to approximately 5× or 10× in rat and rabbit, respectively.
Dose level setting based on a maximum feasible dose (MFD) may be relevant but was regarded as less common for an ONT. As the ONT field expands to include novel constructs and a wider scope of indications, the MFD approach may be appropriate. When relevant, there are clear recommendations for how to address this situation.2,3
Dose level setting for a surrogate ONT should focus on the pharmacological response, since off-target (hybridization-dependent or -independent) toxicity of a surrogate would not likely be relevant to the clinical candidate. The dose level selected for the surrogate should be the lowest dose that produces the intended level of target gene modulation in a relevant tissue. Dosing higher should be avoided to minimize potential for nonrelevant effects.
Dosing schedule
Decisions on dose levels and dosing schedule are often interdependent for an ONT. As emphasized throughout the workshop, the dose levels and dosing schedules utilized in DART studies considered factors such as the planned clinical dosing schedule, the chemical/physical properties, PD, PK, and toxicity profile of the ONT being tested to ensure appropriate systemic exposure during the critical DART periods to best inform human risk assessment (Table 3). Typically, administration of an ONT in the clinic is intermittent and infrequent, as exemplified by the siRNA LEQVIO, which in the maintenance phase is given twice yearly. 24
Dosing Schedule Approaches for Oligonucleotide Clinical Candidate or Surrogate Molecule
MTD, maximum tolerated dose; ONT, oligonucleotide.
Despite infrequent dosing in the clinical setting, a consensus was reached during the workshop that, in EFD studies, it is necessary to maintain both PD and adequate systemic exposure throughout organogenesis to account for all potential critical windows of vulnerability to a compound. This principle is consistent across all modalities; however, for ONTs, this should be done with an awareness of balancing frequent dosing to maintain consistent systemic exposure with limiting tissue accumulation and the potential for greatly exaggerated PD or tissue burden-related toxicity that would not be relevant to the clinical dosing paradigm. For DART studies involving dosing during nongestation periods, such as the FEED and later stages of the PPND, a consensus was reached that intermittent dosing as used in general toxicity studies would be adequate to evaluate safety, provided that the dosing interval was at least as frequent as would be expected in the clinic.
There are multiple ways to approach the dosing schedule to strike the balance between exposure during critical windows of development while reducing tissue accumulation, exaggerated PD, or exceeding tolerability (Table 3). One approach is fractionated dosing, which entails dividing the weekly or monthly dose in general toxicity studies, into smaller doses that can be given either daily or several times per week. Often, the high dose and schedule are designed with the aim of limiting parental toxicity while providing an adequate (ie, 25×) plasma AUC-based exposure margin to that in humans at the maximum recommended human dose when adjusted for the different nonclinical and clinical dosing schedules. 2 However, a disadvantage is that it will likely result in a lower daily maternal plasma Cmax compared to that observed in the clinic and could thus reduce relevant exposure to the conceptus. Another approach to demonstrate adequate parental exposure to ONTs is to benchmark the target tissue (eg, liver) concentrations in rodents after fractionated doses against those obtained in general toxicology studies. In this approach, the tissue concentration from animals given the fractionated dose should be equivalent to the tissue concentration from animals in repeat-dose studies given the same dose but at less frequent intervals.
An example of fractioned dose is evident in the DART assessment for the ASO inotersen (TEGSEDI™). 22 The approved dose and schedule in patients is 284 mg once weekly (∼4 mg/kg/week). In a mouse combined FEED/EFD, weekly doses of inotersen were 10.5, 52.5, and 87.5 mg/kg/week; a mouse-active surrogate ASO was dosed at 52.5 mg/kg/week. 25 In male mice, the weekly schedule was utilized throughout the study period (∼10 weeks). In female mice, weekly dosing occurred prior to mating; however, during organogenesis, the dosing schedule was modified (fractionated) to once every-other-day, or 3.5 doses per week, with doses of 3, 15, and 25 mg/kg/dose. In the rabbit EFD study, the 3.5 doses per week schedule was also utilized, with doses of 2.5, 5, and 15 mg/kg/dose (8.75, 17.5, and 52.5 mg/kg/week, respectively). Given the tissue half-life of inotersen in rodents (∼2 weeks) and nonrodents (∼4 weeks), the every-other-day dosing schedule was deemed appropriate and balanced sufficient exposure multiples for evaluation while limiting the potential for excessive tissue accumulation and potential confounding toxicity.
Other study design considerations include species PD relevance or need to use a surrogate, toleration of frequent dosing based on findings from general toxicity studies, or technical challenges with the dosing via the intended clinical route of exposure. In cases where more frequent dosing cannot be tolerated, different cohorts of animals could be used with dosing staggered across cohorts such that different gestation days are covered, but no individual cohort is dosed too frequently. This approach is not used often, as it generally requires large numbers of pregnant animals.
Timing of dosing initiation relative to reproductive cycle stage is another important consideration for dosing schedule. In some cases, a dose may be needed in advance of the typical dose initiation day in a DART study to allow time for reduction in the level or modification to the target RNA/protein of interest, prior to the reproductive and/or developmental milestones being assessed. Key information includes the level of change in target RNA, and possibly protein, and durability following a dose. It should also be considered whether the change is pharmacologically relevant; for example, a 10% reduction may not be meaningful, and additional doses may be necessary before estrous cycle evaluation, mating, or organogenesis. Consideration of the protein half-life is useful in determining if evaluation of the target RNA may be sufficient or if protein levels need to be evaluated. For example, proteins with shorter half-lives will have concomitant changes in the level of RNA and protein after treatment with an ONT, while proteins with longer half-lives will have RNA levels that decrease quickly after treatment but the decrease in protein levels will lag. Therefore, an assessment of RNA levels alone may not provide an accurate picture of how much functional target protein is present. These factors will inform how early dosing should begin and the schedule required to balance wanted effects while minimizing the potential for excessive accumulation.
DART studies can be combined to achieve the appropriate dose initiation timing. For example, if it takes 2 weeks to modulate the protein of interest, it may be more strategic to conduct a combined female FEED/EFD study (stages A–D) rather than separate FEED and EFD studies, so that full pharmacology can be achieved prior to mating and pregnancy. Depending on the length of time to achieve target PD, an initial dose could also be administered in time-mated animals prior to implantation. A disadvantage to dosing before implantation would be if the clinical or surrogate ONT caused pre-implantation loss. A reduced litter size or total litter loss would impact the ability to extend the FEED study into the EFD portion, and a separate EFD study would be needed. One way to understand this risk prior to starting a combined FEED/EFD would be to conduct a dose range finding EFD study, with dosing initiated prior to implantation.
The dose frequency and timing for a surrogate ONT should be adjusted based on intended pharmacology. If the surrogate molecule has long-lasting PD activity, the dosing frequency should be determined based on the durability of PD in the maternal target tissue. The strategy for this approach is to maintain clinically relevant PD but avoid surrogate off-target effects, which would not be clinically relevant. While most participants agreed with this approach, and some case studies implemented this frequency of dosing for the surrogate, the potential for PD activity to occur directly in the embryo or fetus is not completely covered when setting the frequency to elicit PD in the maternal animal. Any available information on ontogeny of the target during embryogenesis should also be considered.
Toxicokinetic considerations
The circulatory half-life for ONTs is short and usually lasts only a few hours, but half-life in the targeted tissue can be markedly longer. The resulting pharmacologic effect can last weeks to months. Therefore, dosing an ONT may result in the unique situation of having PD with no systemic (eg, plasma) exposure at a given time.
Many companies or sponsors measure systemic exposure in DART studies by performing toxicokinetic assessments on the first and last days of dosing in a study. Alternatively, toxicokinetics may be assessed once a presumed steady state level of exposure is achieved. The standard approach may be adapted by adding additional sampling time points to confirm systemic exposure at critical time points, such as mating, implantation, or organogenesis. These basic principles apply to ONTs, but confirming systemic exposure to the ONT itself may not be the most important consideration. Therefore, PD assessments (messenger RNA and/or protein levels) and bioanalysis of relevant tissue(s) could provide a more complete understanding of ONT exposure.
Systemic exposure and tissue concentration data from a small number of approved ONTs were discussed during the workshop (Table 4). These previous studies have investigated whether ASOs and siRNAs reach the fetal compartment by measuring concentration and assessing pharmacodynamics in various tissues, including placenta and fetal plasma, liver, and kidney. Different methodologies and sample types were used across programs. Even so, based on this small dataset, there was evidence that ASOs and siRNAs can be measured in placenta; ASOs were not generally quantifiable in fetal liver, while siRNAs may be present in fetal tissues and plasma at low levels. At the time of writing, there are no data on phosphorodiamidate morpholino oligomers. The marketed ONT nedosiran (RIVFLOZA®) was detected in amniotic fluid in rabbits, but this was the only data available for amniotic fluid. 15 Overall, discussion on fetal exposure focused on the idea that while exposure information could help put potential developmental toxicity findings in context as described in ICH S5(R3) (“concentration in the embryo or fetus can facilitate interpretation of discordant or equivocal evidence of developmental hazard”), it can also be challenging to select the most appropriate tissue or compartment, day of gestation, and time point following dosing. Therefore, no recommendation was made on the inclusion of exposure assessments in DART studies. Importantly, lack of detectable fetal tissue exposure or low-level exposure on a particular gestation day may not rule out human relevance when an embryo–fetal developmental effect is observed. Additionally, when levels are measured in the fetus, ICH S5(R3) acknowledges “a direct comparison to the potential levels in the human conceptus is not appropriate.”
DART Data Summary for Approved Oligonucleotides
Lead molecule was detected, but not surrogate molecule.
Detectable in one nonclinical species, but not in another.
ONT not detected, but lipid components were present in milk.
DART studies were performed using SC administration.
ALS, amyotrophic lateral sclerosis; DMD, Duchenne muscular dystrophy; FCS, familial chylomicronemia syndrome; F, fluoro; hATTR, hereditary transthyretin-mediated amyloidosis; HoFH, homozygous familial hypercholesterolemia; IT, intrathecal; IV, intravenous; MT, maternal toxicity; N/A, not applicable; OMe, O-methyl; PS, phosphorotioate; PO, phosphodiester; Rb, rabbit; Ro, rodent; SC, subcutaneous; 5-10-5 designates a gapmer, with the middle number indicting the number of 2ʹ unmodified subunits; ✓, test article-related effects were identified by this study; X, no test article-related effects were identified by this study; —, not evaluated.
Systemic exposure to an ONT may occur via other routes such as seminal exposure and via milk during lactation. Small molecules have a low transfer to seminal fluid. 38 The maximum transfer of a low molecular weight pharmaceutical via seminal fluid to a female partner is estimated to result in a blood concentration at least three orders of magnitude lower in the female partner as compared to the man. 39 ONTs are generally larger than small molecules, but smaller than biologics. The absorption of ONTs across the vaginal epithelia and then into the systemic circulation has not been well-characterized but is expected to be limited. Should an ONT reach circulation, such as small molecules, the circulatory half-life of ONTs is generally short, lasting only a few hours. Overall, the potential for ONT transfer to a female partner of a male patient through seminal fluid and reach biologically relevant systemic concentrations is likely to be similar to or lower than that expected for a small molecule. To determine if the seminal exposure would present a risk for female partners of male patients or to a fetus, the expected blood concentration in the female should be compared to the NOAEL from an EFD study. However, more follow-up is needed to confirm actual levels of ONTs in seminal fluid. Levels of various subtypes of ONTs should be measured to understand if there are differences between ASOs, siRNAs, or other ONTs. Information on the bioavailability following vaginal delivery of ONTs is also needed.
ASOs and some siRNAs have been quantified in milk, generally at low levels. However, it is largely agreed that ONTs have low oral bioavailability, 40 so lactational exposure would likely result in negligible levels of systemic exposure to a breastfed infant. The concentrations of marketed siRNAs lumasiran (OXLUMO®) and vutrisiran (AMVUTTRA®) were undetectable in pups (Table 4).26,30 In the case study summarizing a PPND study conducted for a GalNAc siRNA, exposure was detected in pups on Post Natal Day (PND) 4 but was decreased by PND 11, indicating there was low-level to no lactation transfer. ONT formulations may be modified to increase oral bioavailability. 41 Therefore, potential impact on lactational exposure risk should be considered if enhancements to oral bioavailability are made to an ONT. Although these examples help to understand the likelihood of lactational transfer of ONTs in animals, it should be noted that FDA does not consider drug concentrations in animal milk as being predictive of human milk concentrations and, therefore, generally does not support the reporting of animal milk concentrations in the United States Prescribing Information.42,43 Sponsors should consider whether measuring milk concentrations of their specific ONT in animal DART studies would be relevant to assess human risk.
For GalNAc-conjugated ONTs, or other delivery modalities, PD should be assessed in the maternal target tissue (eg, liver, kidney) for ONT delivery, and consideration should be given to potentially assessing PD in the corresponding fetal tissue. Other maternal tissues where the target is expected to be expressed may also warrant analysis. As there may be different expression levels and patterns for the gene throughout embryo–fetal development, thought should be given to the time points at which fetal tissues will be assessed, also factoring in when those tissues are likely to be developed, and the technical difficulty of collecting these tissues prior to term.
Species selection
Rat or mouse is the most common rodent species for DART assessment, and rabbit is the most common nonrodent species for EFD assessment, based on availability of historical control data, short and consistent gestation length, large litter sizes, and availability of animals (ICH S5(R3)). ONTs may not be active in rats or rabbits; however, these routine species used in DART evaluations are still considered to be useful for ONT DART testing to fully assess off-target DART potential of the ONT. Indeed, this has been demonstrated in the DART package supporting the recently approved GalNAc-conjugated siRNA nedosiran (RIVFLOZA), in which malformations of the skeletal and cardiovascular system were observed in the rabbit EFD study but were not detected in the rodent model. Maternal body weight loss was noted following the first day of dosing, but treated rabbits recovered to match the control group’s body weights thereafter. The mechanism driving the malformations remains unclear; however, rabbit was not a pharmacologically relevant species, so the potential for different profiles between rodent and rabbit of off-target toxicity of an ONT was demonstrated in this case.
On-target (hybridization-dependent) DART assessment requires a pharmacologically relevant species. ONTs often induce the intended pharmacological response in NHPs, but as noted, DART assessments in NHPs can be challenging for ethical, logistical, and scientific reasons and are, therefore, not the ideal option. Alternatively, a surrogate ONT with an analogous sequence to the human target can be manufactured with identical chemistry to the clinical ONT and can be used in a routine DART species, typically rat or mouse. The use of a surrogate approach can be combined with assessment of off-target hybridization-independent (chemical-induced) or -dependent (off-target sequence hybridization) toxicity due to the clinical candidate by including a separate arm evaluating the surrogate molecule in a DART study with the clinical candidate (Fig. 4). Use of a rodent surrogate and clinical candidate within the same EFD study would allow for a full assessment in a single species, and the second species can be used to evaluate off-target potential only (Table 5). This rodent surrogate approach can be applied to FEED, PPND, and juvenile animal studies, where relevant. Although the addition of a group of animals administered the surrogate would result in a study using more animals than a standard small molecule DART assessment, this approach can obviate the need to use NHP. Additionally, when considering the number of groups included in a study for off-target assessment of the clinical candidate, there may be instances where fewer than three dose levels are sufficient for risk assessment, as stated in ICH S5(R3).
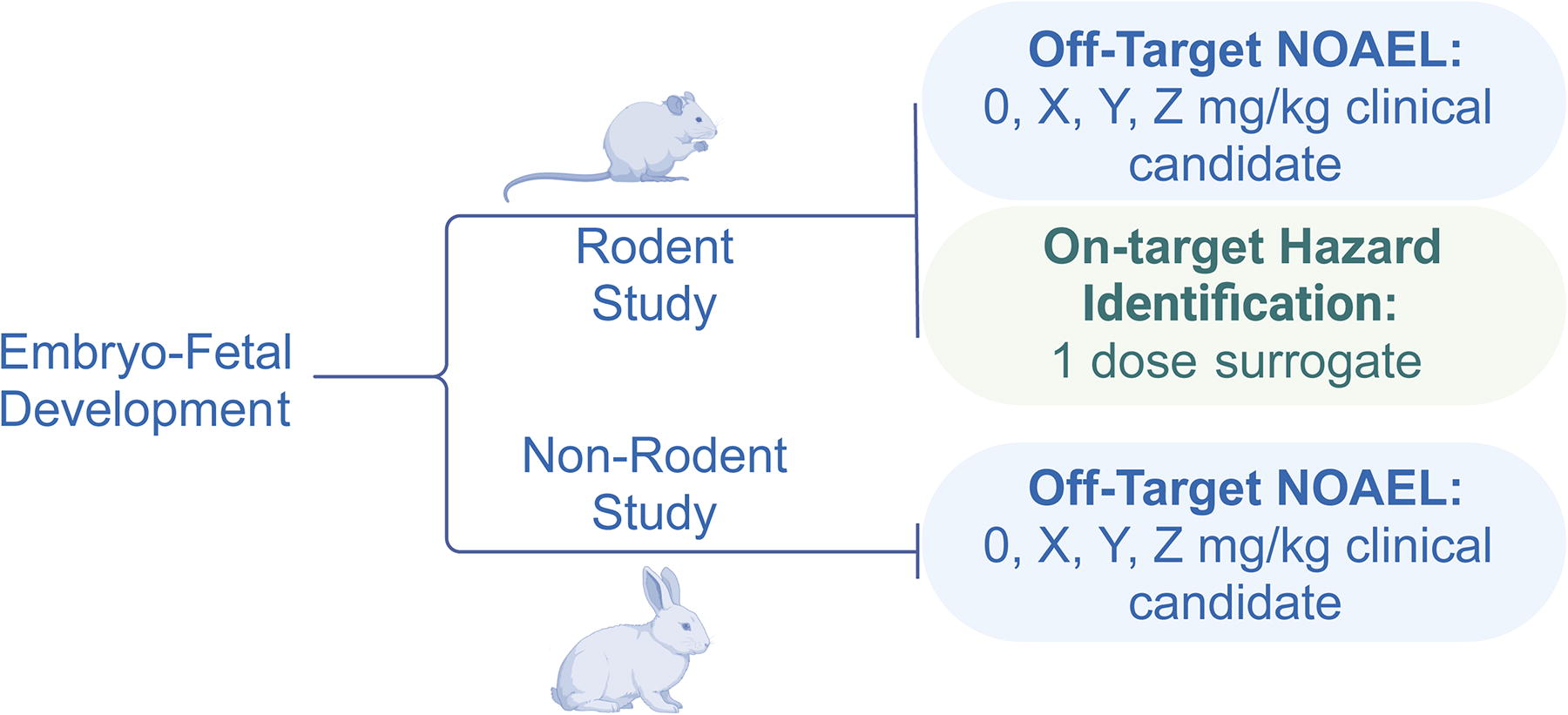
Example EFD assessment for an ONT using a surrogate. In this case, neither rodent nor rabbit is pharmacologically relevant. The rodent EFD study includes a single dose group arm for the surrogate to evaluate potential DART hazard associated with the intended pharmacology, in addition to a dose response with the clinical candidate ONT. The rabbit EFD study will evaluate a dose response of the clinical candidate ONT for off-target DART potential only.
Typical Developmental and Reproductive Toxicity (DART) Study Package for Oligonucleotides Based on Pharmacological Relevance of Routine DART Species (Rodent and Rabbit)
EFD in second species is warranted to assess effects related to chemistry and potential off-target hybridization. Less than two species may be justified, see text for examples.
To patient population or based on target (ie, exogenous viral/bacterial target).
Surrogate molecule characterization considerations
Selection of a surrogate ONT that would be appropriate to assess on-target toxicity in the DART nonclinical safety assessment for an ONT would need to meet the following criteria: pharmacologically active in the test species, knockdown of the target gene equal or greater in magnitude than what is expected for the clinical candidate ONT in human, and similar sequence length and chemical modifications of the surrogate to the clinical candidate. The approach of using a surrogate ONT does not replace the assessment of the clinical candidate, and both would be assessed in the DART studies, as discussed above (the section “Species Selection”).
Surrogate ONT manufacture and characterization were also discussed. Many case studies using a surrogate approach cited that “research grade” material (not manufactured under Good Manufacturing Practices conditions) was sufficient because the primary purpose of the surrogate is to demonstrate the target PD and evaluate potential DART hazard associated with the PD effect only. Surrogate material is not used in the clinic and not appropriate to use for setting safety margins for human risk assessment.
Consensus was reached that the surrogate approach was a preferred tool to assess toxicity related to target knockdown rather than using NHPs. Both a surrogate approach and NHP DART approach provide a hazard identification assessment; however, the advantage of using the surrogate model is that a more robust DART assessment can be conducted in a routine species (typically rodent). This recommendation contrasts with what is stated in ICH S6(R1) for biopharmaceuticals regarding preference for using the clinical candidate in NHP instead of the use of a rodent-active surrogate. Nevertheless, this preference is promoted in PMDA guidance and draft FDA guidance as an acceptable approach in applicable cases.
Transgenic or disease-state animal model characterization considerations
The final topic related to alternative models for assessing on-target toxicity was using transgenic animal models or inducing disease-state pharmacology when it is not present in a normal, healthy animal model. Characterization of these alternative models should be performed prior to moving forward into the DART studies. The characterization of a transgenic model needs to be sufficient to show that the reproductive and developmental characteristics of the strain are suitable and stable enough for use in DART testing. Interactions with regulatory authorities were encouraged to ensure alignment with the scientific justification supporting the use of these alternative models.
Species selection considerations for biological-conjugated ONTs
Tremendous progress has been made to enhance the metabolic stability, pharmacokinetics, and potency of ONTs, including their efficient delivery to target tissues outside of the liver. More recently, delivery of ONTs to skeletal muscle, heart, and central nervous system, among other extrahepatic tissues has been demonstrated by conjugation of ONTs to ligands including monoclonal antibodies, antigen binding fragments, cell-penetrating peptides, lipids, and others. 44 Thus, the number of different types of conjugated ONTs in development is expanding to apply RNA targeting therapies to a broader range of tissues and diseases.
There are additional considerations for DART assessment of protein-conjugated ONT therapeutics relative to unconjugated ONTs (eg, linker-ONT; free ASO) or chemically synthesized conjugates such as GalNAc. A comprehensive understanding of the safety profile should be based on pharmacological and nonspecific properties of all components of the conjugated ONT. In the case of a biological moiety (eg, mAb or fab) conjugated to an ONT, potential for exaggerated pharmacology of the biological delivery component needs to be considered in addition to that of the ONT. The cross-reactivity of the conjugated ONT to preclinical species is an important factor when forming the safety assessment strategy. Reproductive risk assessment may include the use of rodent-active surrogates with comparable chemical modifications and pharmacological properties to the clinical candidate ONT. Transgenic mouse models that have the human transport receptor knocked in should also be considered for conjugated ONTs that only bind to a human receptor, allowing the clinical candidate to be assessed in an alternative mouse model. In cases where nonhuman primates are the only pharmacologically relevant species, an alternative species (e.g., rodent) in which there is an appropriately qualified rodent-active surrogate ONT conjugated to an appropriately cross-reactive delivery component should be considered.
In cases where an alternative to NHP DART evaluation is an option for complex ONT conjugates, testing reproductive toxicity for each pharmacological component (delivery protein conjugate and the ONT) separately in a routine DART species such as a rodent may be the most prudent and practical approach while being mindful of the potential for additive effects and differences in tissue distribution of the separated components compared to the clinical candidate. Alternatively, a rodent surrogate protein–ONT conjugate or “double surrogate” containing both a protein and ONT that are active in rodents can be tested. A WoE integrative assessment can be considered in lieu of an animal study for protein-conjugated ONTs, if class-specific effects are well-established or when sufficient data are available to inform on reproductive risk of the delivery conjugate receptor binding and the target knockdown. As for all modalities and ONTs, the use of alternative approaches will require scientific justification and may include risks that should be considered and discussed with health authorities to gain alignment.
Nonroutine DART species, NHP, and Minipig
There may be programs in which routine DART species (rodent and rabbit) cannot be justified. Furthermore, in some cases, a surrogate ONT may not be feasible based on expression differences or an absence of the target RNA between traditional DART species and human. In the workshop, there were no examples of DART studies using nonroutine species. However, two presentations were provided for the use of nonroutine species to conduct juvenile animal studies in support of pediatric indications. The first example was an ONT treatment being developed for Angelman’s disease, which was designed to activate the silenced parental ubiquitin protein ligase E3A (UBE3A) allele in neurons and compensate for loss of the maternal UBE3A allele. Safety studies in juvenile monkeys (with active maternal UBE3A allele) were used to investigate the toxicity of activation of the paternal allele in addition to the active maternal allele to specifically study neuronal development and behavioral development. 45 The intended clinical delivery route for this ONT was intrathecal, which is considered an unrealistic route in rodent DART and juvenile animal studies. The juvenile NHP was considered the most relevant species for toxicity studies to support the pediatric clinical population as the ONT is pharmacologically active in NHP and because of the feasibility of intrathecal administration.
The second example of a nontraditional species was the use of the juvenile minipig to support drugs in development for pediatric indications. 46 The minipig has been a nonrodent species for general toxicity of ONTs and shows a similar profile of ONT-specific toxicity as that seen in NHPs. As evidence, an 8-week once weekly ASO dosing (subcutaneous route) toxicity study to evaluate similarities and differences between juvenile (from 1 to 50 days of age) and adult minipigs, 47 juvenile minipigs were considered to represent a viable model. Advances in this model, such as neurobehavioral tests and techniques for specialty routes (eg, intrathecal), could lead to the wider use of the minipig as a nonrodent model for ONTs. A minipig model is available for EFD assessment 48 ; however, to date, no minipig EFD studies have been published with an ONT.
Additional data available to support off-target DART assessment of oligonucleotides
During ONT development, other in silico, in vitro, and in vivo data are generated that could potentially be applied to the DART assessment. This information is important when determining whether a WoE or platform approach could be used to assess DART risks for a particular ONT, as further discussed in the section “Overall Workshop Takeaways.” The types of assessments that would typically be performed during ONT development were outside the scope of the workshop and were only discussed in general terms but were presented at the subsequent Society of Toxicology and Birth Defects Research and Prevention sessions and have been reviewed by Kornbrust, Cavagnaro. 49 The information briefly summarized below and in Table 6 includes techniques for identifying potential hybridization-dependent and hybridization-independent modes of toxicity. Additional recommendations by the OSWG were also recently published. 8
Alternative Approaches to Evaluate Potential Off-Target Developmental and Reproductive Toxicity Effects
RNASeq, RNA sequencing; WEC, wound epidermal cell.
During the design and optimization of ONTs, in silico assessments are performed to determine the potential for hybridization-dependent actions, and ONT sequence selection is designed to limit the number of potential unintended hits to the human genome. When genes are identified that could hybridize with the ONT, additional follow-up is often necessary to determine the probability that the ONT could alter expression of these genes, including determining the number of hybridization mismatches, the position within the off-target gene where hybridization takes place, and the tissues where the gene is expressed. The function of off-target genes is also important in assessing the potential for safety concerns, and this is especially true for EFD studies because genes can have different functions in developing embryos and in the placenta than in adult organs.
Once a candidate ONT is selected, the total set of potential off-target genes identified in silico can be assessed in experimental expression profile analysis to study potential downregulation related to off-target hybridization. Often, the ONT is transfected into either cell lines or primary cells in 2D cultures, and changes in expression of potential off-target genes are determined by quantitative PCR analysis. If significant changes in gene expression (eg, 10-fold of comparable on-target potency) are identified at relevant concentrations, further examination of those genes is warranted, and if appropriate, perhaps using in vivo techniques. In cases where in silico assessment highlights potential hybridization for an unintended target that would be expressed only in reproductive, placental, or embryo/fetal tissues, appropriate cell types capturing expression should be utilized.
The methods described above are relevant for predicting off-target hybridization-dependent effects; however, ONTs have also the potential for causing toxicity via hybridization-independent mechanisms. One approach to evaluating hybridization-independent off-target effects in vitro is to assess gene expression changes in cells in an unbiased manner by using RNAseq or other similar methodologies. Such methods do not require knowledge of specific gene sequences and can detect changes in gene expression irrespective of the MoA. In these methods, human cell lines or primary cells can either be transfected or treated with the ONT of interest to identify changes in gene expression. One caveat is that these methods are relatively new, and considerable work is often needed to optimize the cell systems, determine next steps, and understand the translation of the in vitro results to potential in vivo effects.
As described in ICH S5(R3), in vitro and ex vivo methods have been evaluated as alternatives to animal studies to predict potential effects on embryo–fetal development. Some examples include the use of zebrafish, whole embryo culture of rat or rabbit embryos, and in vitro models (e.g., embryonic stem cells). Such methods could also potentially detect off-target effects related to hybridization-dependent and -independent mechanisms. The use of these models as part of the DART evaluation is potentially acceptable to regulators, but they must be rigorously qualified for use as a DART screen, which can be an extensive process. Additionally, these models have generally been developed for the assessment of small-molecule pharmaceuticals or chemicals; therefore, the applicability of alternative DART models to assessment of ONT remains unknown.
Overall Workshop Takeaways
Current questions and status of DART considerations for ONTs since the 2014 OSWG publication were briefly introduced to start the workshop. Several best practices, new approaches, additional questions, and opportunities were identified in the workshop, based on the examples shared, the discussion, and the round table wrap-up. In response to the questions posed to set the stage for the workshop, key points for effectively evaluating DART for ONTs are summarized below.
What approaches are currently being used to address the unique attributes of ONTs when designing DART strategies and studies?
Consistent with the previous recommendations from the OSWG DART subgroup and regulatory expectations for a small molecule evaluation, many of the examined case studies used routine species in standard DART studies, that is, rodent and rabbit in EFD studies, and rodent in FEED and PPND studies, with the goal of capturing potential on- and off-target DART effects. For ONTs, including ASO and siRNA in development or approved, strategies around dose level setting and frequency included a hybrid of small- and large-molecule approaches based on product attributes. Although dosing paradigms differed, most strategies included a dosing frequency that maintained systemic exposure to evaluate off-target toxicity or maintain PD response during gestation periods, when the embryo is rapidly developing. Overall approaches varied depending on the specific strategy employed, including type of model, attributes of the specific ONT or conjugate component, and whether a WoE was being used.
Approaches to dose level setting, dose frequency during nongestation phases of the reproductive cycle, surrogate molecule or alternative model study design, species or model selection, and exposure or PD measurements varied based on product attributes across case studies and sponsors (Tables 1–4).
Have there been any advances in implementation and acceptance of alternatives to a traditional small-molecule DART strategy approach?
A key discussion point at the workshop was whether a platform approach could be used to predict effects on DART endpoints for a series of chemically identical ONTs, without having to conduct a full DART program for each ONT. Specifically, if a particular ONT was tested in a comprehensive DART package, could the hazards or lack of hazards identified in those studies be applied to another ONT with the identical chemical structure, if there were no specific concerns related to the pharmacologic activity of the second ONT? The platform approach for leveraging data from another ONT using the same platform was briefly mentioned in the original white paper 7 ; however, there were no examples presented in the workshop or existing in publicly available information using this approach.
Regulators from different regions were not completely aligned on the use of the platform approach, with concern that even small differences in ONT sequence could lead to differences not only in potential on-target effects but also in off-target toxicity (both hybridization-dependent and -independent). Regulators from all regions agreed that there were important caveats to applying this approach. First, the “platform” should be defined with respect to the conjugation structure, chemical linker, and chemical modifications, and only ONTs that were identical in all these aspects should be considered for the platform approach. Thereafter, additional information should be collected for any new ONTs within the platform, including the use of robust in silico, in vitro, and even in vivo tests to thoroughly evaluate the potential for off-target hybridization-dependent or sequence-related toxicity. New generations of ONTs are being created rapidly, so there could be difficulty in finding a completed DART assessment for a sufficiently similar platform to read across if there are differences in chemical/physical properties of related ONTs.
Additional areas for further development were identified that could alter the need to apply a strictly small-molecule and two-species EFD approach, including the use of novel in vitro or ex vivo methods if validated for use with ONTs.
For highly species-specific ONT conjugates, strategies for assessing the toxicity of both aspects of the ONT conjugate would need to be developed. In developing novel strategies for DART assessments, including the platform approach, it was considered important to have early and frequent dialogue with regulators to ensure that the proposed approach would be acceptable.
Is there enough data on this drug class to list best practices for considerations such as species or model selection; appropriate alternatives to animal models to capture on- and off-target toxicity potential of oligonucleotides; dosing regimen; and study designs for surrogate or transgenic animal model approaches?
Consensus was reached on the following study design approaches, which accommodate attributes of many ONTs.
Dose frequency during gestation:
Off-target assessment of the clinical candidate should use a study design or dosing paradigm such as daily or every-other-day dosing where consistent systemic exposure is maintained during gestation. On-target assessment of the clinical candidate or surrogate should use a dosing paradigm where consistent target knockdown of a similar magnitude to that expected in the clinic is maintained during gestation. Dose frequency during nongestation periods of the reproductive cycle:
Systemic exposure does not need to be maintained continuously throughout nongestation periods of the reproductive cycle (premating, mating, lactation). This would likely lead to tissue burden-related toxicity that would not be reflective of the clinical dosing scenario where infrequent (monthly to yearly) dosing is sufficient for long-acting target modulation. A cadence of weekly dosing in the nongestation period of the FEED or PPND studies could more closely mirror the clinical frequency and provide sufficient coverage of key phases of the reproductive cycle. Dose level setting
Standard high-dose selection principles described in ICH S5(R3) can be applied while accounting for dose frequency and target knockdown differences between clinical and nonclinical studies. One common approach to account for a sufficiently high-dose and different dosing frequency in an EFD study is to use a fractionated dosing strategy. For example, a weekly dose level from a general toxicity study could be fractionated one-seventh or one-third of the dose and administered every day or every other day, respectively, to maintain the same weekly dose level while maintaining consistent exposure during gestation. Species selection
The routine DART species (rodent and rabbit) were considered appropriate for DART studies, with at least one species showing pharmacological activity. Even in the absence of PD, the use of these species was justified to thoroughly evaluate off-target hybridization-dependent and -independent effects. The use of a surrogate molecule that could be used to test the effects of PD was preferred to use of NHPs for DART. Surrogate molecule study design
A surrogate ONT will provide a hazard identification assessment of DART potential due to target knockdown but does not replace off-target evaluation of the clinical candidate or provide dose or exposure margin information for use in risk assessment. All study design considerations for the surrogate should be driven by the target modulation. Dose selection, frequency, and timing should be selected in a manner that minimizes potential for off-target effects of the surrogate, which would not be relevant to clinical dosing. Alternative in vivo model study design
Alternative to routine DART models, such as disease-state models, should be well-characterized for background DART anomalies, and ensuring expected pharmacology is produced when dosed with the clinical candidate. Exposure and PD assessment
Limited data were available regarding exposure potential of the placenta, fetal compartment, and milk. Generating placenta and fetal exposure or gene modulation data for an ONT or ONT class could support study design decisions or potentially be included as a component of a WoE approach; however, species differences in placental transfer dynamics and overall value of this data for identifying human risk should be considered. Effects on embryo–fetal development can be produced even when the ONT does not accumulate in the placenta or cross to the fetal compartment. Therefore, a lack of exposure to the fetus does not preclude the need to conduct EFD or PPND studies. What are some additional considerations that have evolved as the drug class has advanced in terms of complexity of the ONTs components, including the use of multiple different delivery conjugates? Complexity is inevitable for the ONT DART testing strategy as the range of conjugation components and chemical modifications expands to target new tissue compartments and broadens the indication pool. As discussed in the example of antibody-conjugated siRNAs, species relevance of DART models and understanding of specific pharmacokinetic properties will drive many of the strategy decisions, but a case-by-case scenario described in ICH S6(R1) and Cavagnaro, Berman
7
will continue to be necessary as ONT product attributes evolve. Is there a global regulatory consensus on DART assessment for ONT? Regulators representing seven different global health authorities participated or delivered presentations in the workshop, indicating increasing collaboration between industry and regulators on the common goal of achieving the most appropriate and scientifically justified DART strategies for ONTs. There was consensus from health authority representatives that DART assessments on ONTs are needed, which should generally follow the principles for small molecules outlined in ICH S5(R3). However, since the practices may differ based on product attributes, the proposed approach should be discussed with regulators on a case-by-case basis. There was some difference of opinion regarding a “platform approach”; however, there was agreement on the need to have a well-characterized platform and generate appropriate data with ONTs within the platform to support this approach. It was noted that consensus will be reached during the drafting of the ICH guidance that is currently ongoing at the time of this writing.
Conclusion
Considerations for DART assessments of ONTs as part of nonclinical safety assessment have progressed since last reviewed in terms of number of available examples, acceptance of DART data or strategies, and thinking toward alternative approaches. The key considerations in this area and the approaches laid out have continued to evolve. DART remains a complex area within nonclinical safety testing and will continue to require case-by-case consideration on applying the best strategy for ONTs based on specific product attributes. Early and frequent dialogue with regulators is encouraged when forging a novel testing approach. Nevertheless, the field is maturing, and there is evidence of increased regulatory acceptance of scientifically justified approaches for ONTs described herein.
Footnotes
Acknowledgments
The authors would like to thank Rudiger Cordts (BI) for his role during the workshop as an additional workshop scribe, Alexandra Taraboletti (HESI) for her graphic design support, and those who provided scientific review of the article as part of internal reviews.
Disclaimer
This article reflects the view of the authors and should not be construed to represent or quoted as being made on behalf of or reflecting the position of their organizations or policies. GSK presented research and contributed to one of the case studies used in this article. All studies related to the case study were conducted according to GSK’s Policy on the Care, Welfare, and Treatment of Lab Animals and reviewed by the Institutional Animal Care and Use Committee at GSK or by the ethical review process at the institution where the work was performed.
Author Disclosure Statement
S.M.B., E.B., B.R.H., L.M., S.R., J.S., T.-W.K., D.L.M., and C.S. are employed by companies that develop ONT therapies.
Funding Information
This HESI scientific initiative is primarily supported by in-kind contributions (from public and private sector participants) of time, expertise, and experimental effort. These contributions are supplemented by direct funding (that largely supports program infrastructure and management) that was provided by HESI’s corporate sponsors. A list of supporting organizations (public and private) is available at ![]() ¼3314.
¼3314.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.