
Abstract
BK polyomavirus, a virus that latently resides in tubular epithelial cells of human kidneys, represents a major clinical problem for immunocompromised patients undergoing kidney transplantation. No proven effective specific antiviral therapies other than reduction of immunosuppressants exist. We exploited splice-modulating antisense oligonucleotides (ASOs) to block expression of the viral “gatekeeper protein” T antigen to specifically inhibit viral replication. Candidate oligonucleotides were screened for inhibitory activity in BK virus-infected cells, resulting in up to 97% reduction of viral T antigen mRNA and virus-encapsulating protein. The most promising ASO candidates were validated in BK virus-infected human kidney epithelial cells, showing sequence-specific activity that was exacerbated by chemical modifications. Administration of the candidate oligonucleotide in mice revealed long-lasting uptake in proximal tubules of the kidney, where BK virus resides. A final optimization round yielded an oligonucleotide displaying superior activity. Studies in transformed cells revealed a discernible shift in T antigen splicing not observed with a scrambled control, indicating that targeting of the donor splice site was responsible for the observed effects. Here, we demonstrate proof-of-principle for a direct-acting, splice-modulating, universal ASO that distributes to sites where BK viral replication occurs, warranting further development of this novel therapeutic to combat BK virus replication in kidney transplant patients.
Introduction
Breakthroughs in immunosuppression have profoundly impacted kidney transplantation success and have significantly reduced 1-year graft rejection and improved patient survival. 1 However, the introduction of potent immunosuppressive agents has increased patient susceptibility to opportunistic infections that threaten kidney allograft function.2–4 In particular, post-transplantation reactivation of BK polyomavirus (BKV), a nonenveloped, double-stranded DNA polyomavirus, is considered a growing problem in kidney-transplanted patients among clinicians. BKV infection occurs early in childhood and establishes a lifelong latent infection in the kidneys and urogenital tract with high seroprevalence rates of >90% worldwide.5–7
While rare and generally nonproblematic in immunocompetent individuals, BKV reactivation can be a significant problem for immunocompromised kidney transplant patients. Shedding of BKV particles into the urine (viruria) occurs in 30%–50% of patients. BKV in the blood (viremia) is observed in 10%–20% of individuals in the first-year post-transplantation, 8 whereby levels >103 viral particles per milliliter of blood dramatically increase the risk of polyomavirus-associated nephropathy (PVN), and >104 viral particles per milliliter of blood is the consensus level for clinicians to taper immunosuppression, thereby increasing the risk of graft rejection. PVN occurs in 1%–10% of kidney transplant patients and is characterized by interstitial fibrosis, tubular atrophy, inflammation, and risk of allograft loss. 8
The BKV genome has three functional regions (Fig. 1A), namely: (1) a promoter-containing noncoding control region (NCCR); (2) an early coding region (ECR) that contains sequences for the T antigen proteins; and (3) the late coding region for viral structural capsid protein synthesis. Importantly, all early control and late structural proteins are expressed from an early or a late pre-mRNA by differential splicing, respectively. An intron within the ECR pre-mRNA allows for alternative splicing and production of the protein isoforms: large T antigen (TAg), small T antigen (tAg), and truncated T antigen. Viral infection or reactivation results in ECR transcription and alternative splicing of the ECR pre-mRNA, primarily yielding TAg protein to stimulate cell cycle progression, deter apoptosis, and recruit host cell DNA machinery to the viral promoter to initiate viral DNA replication. Accumulation of viral DNA in the cellular nucleus prompts TAg to bind to the late (NCCR) promoter and activate late coding region transcription9,10 and production of the virus-encapsulating VP1–3 proteins.9–11 Importantly, the pivotal master regulator role for TAg in guiding this replication process underscores TAg as an attractive target in preventing the production of nascent BKV.
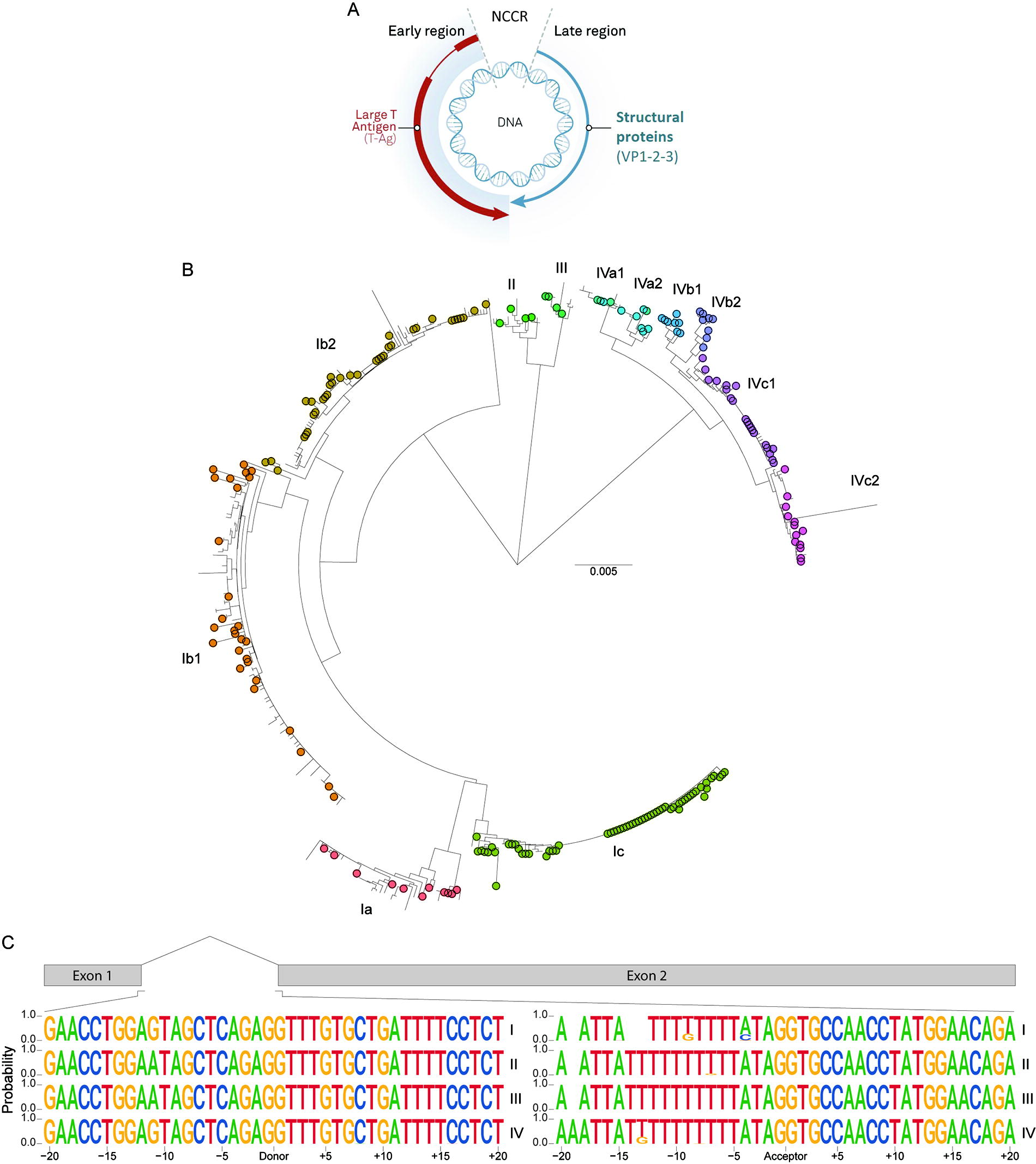
BKV splice site conservation.
Specific treatments for BKV do not exist. Reducing immunosuppression is standard care, prompting the host immune response to clear the infection. However, while generally successful in reducing viral titers to acceptable levels (<104 viral particles per milliliter of blood), this approach is slow and substantially increases the risk of allograft rejection and lowers long-term graft survival. 12 Intravenous immunoglobulins (IVIG), (brin)cidofovir, and leflunomide have been used in conjunction with immunosuppression reduction, but data supporting efficacy in consistently reducing BKV viremia are lacking. 13 More recently, several monoclonal antibody approaches targeting BKV in blood have entered phase 2/3 clinical trials,14,15 however, with mixed results where data have been reported. 16 In addition, allogeneic “off-the-shelf” multi-virus-specific T cells advanced to phase 3 clinical trials but were discontinued. 17
We hypothesized that applying antisense oligonucleotides (ASOs) could be a suitable approach to combat BKV after kidney transplantation as: (1) BKV replication and pathogenesis are restricted to the kidney; (2) chemically modified ASOs are fast-acting and long-lasting splice regulators that are reabsorbed by the tubular epithelium of the kidney, where BKV resides; and (3) BKV replication is heavily reliant on alternative splicing of the ECR (and resultant TAg protein production), a process that can be perfectly addressed by ASOs. 18
Materials and Methods
Retrieval of public sequences
Genomic sequences of BKV isolates were downloaded from the publicly available NCBI database on August 30, 2022, using the “rentrez” package (v.1.2.3) in R with the term: “txid1891762 AND complete genome OR V01108.1.” Only the isolates reporting a unique complete genomic sequence were used for in silico analysis. Isolates FNL-9 (Accession: AB269824.1), MM (Accession: V01109.1), 575 (Accession: MW023596.1), and 717 (Accession: MZ189087.1) were removed manually due to sequence abnormalities in TAg (duplications or deletions).
Phylogenetic analysis and generation of sequence logos
Whole-gene sequences of TAg were extracted and aligned in five iterations using Prank 19 (v.140110), with gap opening rate and gap extension probability set at 0.025 and 0.75, respectively, followed by manual adjustments to the sequence alignment using sequence landmarks. A neighbor-joining phylogenetic tree was constructed using the Kimura 2-parameter model with uniform rates among sites and 1,000 bootstrap replications in MEGA (v.10.0.5). Manipulation and visualization of the phylogenetic tree were performed in R. Previously reported reference BKV isolates 20 were used to identify genotype clades in the constructed phylogenetic tree. Sequence logos of the donor and acceptor splice sites were generated using the “ggseqlogo” package (v.0.1) in R. Positions were marked as distance from the splice site ranging from −20 to +20.
Oligonucleotides
ASOs targeting TAg splice sites were designed following the guidelines previously described. 21 ASOs were designed to have an overlap of at least one intronic nucleotide and at least one exonic nucleotide for either donor or acceptor splice site. A coding sequence-targeting control ASO was included, as well as a scrambled control. Research-grade ASOs for in vitro experiments consisted of a full phosphorothioate backbone (unless otherwise indicated) and contained either 2′-O-methyl or 2′-O-methoxyethyl modifications (OMe-PS or MOE-PS). For in vitro studies, a 5′ 6-FAM label was included to confirm localization of ASO to the nucleus. All ASOs were synthesized by Eurogentec (Liège, Belgium), Axolabs GmbH (Kulmbach, Germany), or Nitto Denko Avecia Inc. (Milford, MA). ASOs were purified using RP-HPLC, with a second HPLC-IEX ultrafiltration for ASOs intended for in vivo studies. Characteristics that were taken into account when designing included %GC content, predicted basic melting temperature, presence of CpG motifs, and target accessibility using RNAcofold from the ViennaRNA package.
Cells and virus
Human kidney proximal tubular epithelial cells (HK-2; ATCC® CRL-2190™) were maintained in Dulbecco's Modified Eagle medium (DMEM)/F12 (Thermo Fisher Scientific, 31330095) supplemented with 1X Insulin-Transferrin-Selenium, 40 ng/mL triiodothyronine, 10 ng/mL epidermal growth factor, 36 ng/mL hydrocortisone (all purchased from Sigma, Zwijndrecht, The Netherlands), and 100 U/mL penicillin, 100 µg/mL streptomycin solution (Invitrogen, Breda, The Netherlands). Human Renal Proximal Tubular Epithelial Cells (HRPTEpiC; Sciencell, #4100) were maintained in REBM Basal Medium (Lonza, Cat: CC-3191) with REGM™ SingleQuots™ supplements (Lonza, Cat: CC-4127, hereafter referred to as REGM) supplemented with 1% fetal calf serum (FCS). Experiments with HRPTEpiC were performed between passages 4 and 6. The transformed mouse cell line pRPcT1ss1 (a kind gift from Prof. Massimo Negrini at the University of Ferrara, Italy) was maintained in DMEM (Gibco, Cat: 41966-029) containing 1% l-glutamine (Sigma-Aldrich, G7513) and 10% fetal bovine serum (Serana, Cat: S-FBS-AU-015). ASO transfection experiments with pRPcT1ss1 were performed in DMEM with 1% l-glutamine and 2% FCS. All cells were maintained at 37°C and 5% CO2. Fresh vials of the BKV Gardner strain (ATCC, Cat: VR-837™) were freeze-thawed 3× following supplier’s instructions, after which the virus stock was diluted 3.2-fold in REGM and stored as 100–200 µL aliquots in liquid nitrogen.
Transfection of ASO
For testing inhibitory activity in kidney epithelial cells, HK-2 and HRPTEpiC were seeded in 12-well plates (Corning, 3512) at a density of 31,579 cells/cm2 (120,000 cells per well) and 23,684 cells/cm2 (90,000 cells per well), respectively, and grown overnight to a confluency of approximately 80%–90%. Uptake of ASO by pRPcT1ss1 was achieved by seeding cells in 12-well format plates (Corning, Cat: 3513) at a density of 13,158 cells/cm2 (50,000 cells per well) in regular medium containing 2% FCS to achieve 80%–90% confluency the next day. Transfection of ASO was performed using 3 µL Lipofectamine™ 3000 Transfection Reagent (Thermo Fisher Scientific, L3000075) in 100 µL OptiMem (Gibco, 51985-026) per well following manufacturer’s instructions for 5 h in kidney epithelial cells and overnight for pRPcT1ss1. Transfected cells were washed 1× in DMEM/F12 with HEPES (Thermo Fisher Scientific, Cat: 11330032) or DMEM (Gibco, 41966-029) for HK-2/HRPTEpiC or pRPcT1ss1, respectively, after which 1 mL fresh regular culture medium was added.
Infection
Infection of HK-2 and HRPTEpiC was performed by incubating cells in the presence of BKV in REGM (to a final dilution of 1:320 for HK-2 and 1:1,000 for HRPTEpiC) for 2 h at 37°C and 5% CO2. Then, cells were washed 2× in 1 mL DMEM/F12 with HEPES to remove excess virus particles. After final washing, fresh regular culture medium was added to every well. At least 200 µL of original BKV-containing REGM was retained for later quantification of virus DNA (infectious load). Cells were supplemented at 3 and 5 days post-infection (d.p.i) by replacing 20% of the supernatant with fresh medium.
Isolation of RNA and DNA
For the isolation of RNA, cells were lysed in TRIzol™ (Thermo Fisher Scientific), and RNA was isolated using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. DNA removal steps were introduced to remove DNA remnants in the sample during isolation of RNA using the RNase-Free DNase Set (Qiagen, 79254) and after isolation using the TURBO DNA-free™ Kit (Thermo Fisher Scientific, AM1907) in case of BKV-infected cells. Supernatant was collected from BKV-infected cells, and Pierce Universal Nuclease was added for 15 min at room temperature and then inactivated with 5 mM ethylenediaminetetraacetic acid (EDTA) to remove any BKV genomes not incorporated into a viral capsid. BKV genomic DNA was then isolated using the QIAamp DNA Mini Kit (Qiagen, 51306) following the manufacturer’s instructions.
RNA quantification
RNA from cells was used to synthesize cDNA using GoScript™ Reverse Transcriptase (A5003) in Reverse Transcription 10X Buffer (A356A) with Random Primers (C1181) or Oligo(dT) 15 Primer (C110B), dNTP Mix (C114B), 0.1M DTT (P1171), and Recombinant RNasin® Ribonuclease Inhibitor (N251A), all purchased from Promega (The Netherlands). Expression of TAg and VP1 mRNA in BKV-infected HK-2 and HRPTEpiC was quantified using RT-qPCR, with primer sequences shown in Table 1, in SYBR™ Select Master Mix (Thermo Fisher Scientific, 4472908) and run on the CFX384 Touch Real-Time PCR Detection System (Bio-Rad) in Hard-Shell® 384-well PCR plates (Bio-Rad, HSP3805) using the following cycle conditions: 2 min at 50°C, 2 min at 95°C, 40 amplification cycles (10 s at 95°C, 20 s at 61°C, and 45 s at 72°C), and final 10 s at 95°C. Splicing interference in pRPcT1ss1 was studied using RT-qPCR, with primers described in Table 1. The RT-qPCR for pRPcT1ss1 was performed in a Real-Time PCR system (Bio-Rad, CFX384) using the following cycle conditions: 3 min at 95°C followed by 40 amplification cycles (10 s at 95°C, 15 s at 60°C, and 30 s at 72°C), 10 s at 95°C, and a final hold at 4°C. Expression of BKV RNA was analyzed using the delta delta threshold cycle algorithm (ddCT method) and normalized to the expression of glyceraldeyhye-3-phosphate dehydrogenase (GAPDH). In order to further investigate splicing interference in pRPcT1ss1, an agarose gel electrophoresis PCR-based assay was performed on the synthesized cDNA using TAg-specific primers (Table 1). A reaction mixture was prepared containing 5X Green GoTaq Flexi Buffer (Promega, M891A), MgCl2 (25 mM; Promega, A3518), 10 mM PCR nucleotide mix (Promega, U1518), forward and reverse primer (10 µM), GoTaq G2 Flexi DNA Polymerase (5 U/µL; Promega, M780A), and water (Fresenius KABI, B102987). The PCR was executed in a thermocycler with the following cycle conditions: 2 min at 95°C followed by 30 amplification cycles (15 s at 95°C, 15 s at 56°C, and 25 s at 72°C), 5 min at 72°C, and a final hold at 4°C. For the gel electrophoresis, samples were loaded on a 2% agarose gel with Midori Green, including a GeneRuler 1 kb Plus DNA Ladder (Thermo Fisher Scientific, SM1331). Gel electrophoresis was performed at 120 V for 75–120 min, and PCR products were visualized using a ChemiDoc Touch Imaging System (Bio-Rad). DNA bands at approximately 246 bp and 590 bp corresponded to TAg and tAg, respectively. Quantification of band intensity and background subtraction was performed using Image Lab (Bio-Rad). Viral titers in the supernatant of HK-2 and HRPTEpiC, obtained through DNA isolation from supernatant, were determined using Taqman PCR as described previously. 22 In short, isolated DNA was used as input for RT-qPCR analysis using primers described in Table 1 in combination with a 5′-FAM-labeled Taqman probe with sequence FAM-5′-CCAAAAAGCCAAAGGAACCC-3′-BHQ1, to generate a 90-bp fragment within the BKV VP1 gene. Reactions were performed using HotStarTaq Master Mix (Qiagen, Hilden, Germany), primer, probe, and MgCl2, using a CFX96 real-time detection system (Bio-Rad, Hercules, CA) with the following cycle conditions: 15 min at 95°C, followed by 45 amplification cycles (30 s at 95°C, 30 s at 55°C, and 30 s at 72°C). For quantification, a standard of a quantified BKV-positive urine sample was used. The virus load in the supernatant was expressed as viral DNA copies per milliliter.
Primer Sequences
BKV, BK polyomavirus; tAg, small T antigen; TAg, large T antigen.
Retrospective examination of the RT-qPCR primer sequences for TAg revealed a single nucleotide (C > G) mismatch in the HRPTEpiC RT-qPCR forward primer against TAg (sequence GAGGAGGATGTAAAGGTAGCTCA with the mismatch underlined). However, RT-qPCR analysis results from separated experiments utilized a cross-comparison between this primer set and alternative primer combinations directed at TAg and revealed that there were no discrepancies in normalized TAg expression (data not shown), indicating that the single nucleotide mismatch did not negatively impact the mRNA expression results described here.
Protein quantification
Cells were lysed in lysis buffer containing 50 mM Tris-HCl at pH 7.5, 150 mM NaCl, 1% sodium dodecyl sulfate (SDS), 0.5% deoxycholate, 0.5% Triton X-100 and Pierce Protease and Phosphatase Inhibitors Mini Tablets (Thermo Fisher Scientific, Cat: A32959). Sample protein concentrations were determined after sonification using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, Cat: 23225), using an internal standard series ranging 62.5–1,500 µg/mL Bovine Serum Albumin (BSA; Life Technologies, Cat: A23016). VP1 protein expression was quantified using the Wes Simple Western System (Bio-Techne). In short, protein lysate samples were analyzed using a 12–230 kDa Separation Module (ProteinSimple, Cat: SM-W004-1) and Anti-Rabbit Detection Module (ProteinSimple, Cat: DM-001). VP1 protein was detected using 1:20 Anti-SV40 VP1 antibody (Abcam, Cat: Ab53977), and GAPDH was used as loading control using 1:20 rabbit polyclonal GAPDH (Cell Signaling, D16H11). Secondary antibodies were Goat Anti-Rabbit Secondary HRP Antibody (ProteinSimple, Cat: DM-001) or 1:20 Goat Anti-Rabbit Immunoglobulins/HRP (Agilent, Cat: P044801-2). The assay consisted of 30 min separation time at a voltage of 375V, followed by 5 min antibody diluent time, 60 min primary antibody time, and 30 min secondary antibody time. Peak area calculation was performed using Gaussian fit of the High Dynamic Range chemiluminescent signal. Peak find threshold and width were adjusted on a capillary-by-capillary basis to ensure proper fitting of the signal. VP1 expression was normalized against GAPDH and expressed as fold change relative to scrambled control.
Quantification of viral DNA
BKV DNA isolated from BKV-infected HRPTEpiC supernatant was transferred to the Department of Medical Microbiology in the LUMC as a fee-for-service for PCR measurement of BKV virus titers using HotStarTaq DNA polymerase and a Taqman primer/probe mixture. Virus titers were calculated using a dilution standard and internal control and expressed as DNA copies per milliliter.
Animals
Animal studies were conducted according to the national guidelines on animal experiments and were approved by the national government body and local ethics committee of the Leiden University Medical Center (project proposal: AVD1160020185326 appendix 8; research plan: PE.18.084.003). Eight-week-old male C57BL6/J mice (Charles River) were housed in individually ventilated cages in the animal facility of the Leiden University Medical Center with ad libitum access to food and drink. A stock solution of 10 mg/mL ASO#03 was prepared in 0.9% NaCl saline solution (Fresenius Kabi, Cat: 0611251/03NL) for intravenous (i.v.) tail vein injection. Animals were distributed over groups (n = 4–6) receiving either (1) saline on day 0 and 7, (2) 40 mg/kg ASO on day 0 and saline on day 7, or (3) 40 mg/kg ASO on day 0 and 7. All animals were closely monitored for body weight, general appearance, and possible signs of distress. Animals were sacrificed at pre-determined time points up to 90 days after first administration by thoracotomy and exsanguination via cardiac puncture under isoflurane anesthesia and Buprenorphine analgesia (0.1 mg/kg Temgesic via subcutaneous injection).
Tissue processing
Tissue samples were collected after whole-body perfusion with PBS (Fresenius Kabi, Cat: M090001/03NL) via cardiac puncture. While multiple organs were collected per animal (kidney, liver, spleen, bladder, heart, brain, testis, muscle, stomach, small and large intestine), only the left kidneys were used for further processing. Organs were subjected to macroscopic inspection and snap-frozen in liquid nitrogen or fixed for 24 h in 4% buffered formaldehyde (VWR International, Cat: VWRKFOR0022AN69001), followed by 4°C preservation in 70% ethanol (Supelco, Cat: 1.00983.1000). Formaldehyde-fixated tissue samples were processed and paraffin-embedded (VWR International, Cat: VWRK2079-A) using a Leica ASP200 tissue processor. Tissue sections were prepared using an HM 355S Automatic Microtome (Thermo Scientific, Cat: 15370795) at a thickness of 4 µm, mounted on Starfrost/VWR coated slides (Superfrost/VWR, Cat: K 220/631-1168), and allowed to dry overnight at 37°C. Tissue slides were dewaxed in xylene (J.T. Baker, Cat: UN1307) and rehydrated in an ethanol gradient. Antigen retrieval was performed using proteinase K treatment for 10 min at room temperature. Washing was performed in TBS/T and tissue sections were permeabilized in 0.3% Triton X-100 (Merck, Cat: 11869) in phosphate buffered saline (PBS), followed by additional washing in TBS/T. Background signal was reduced using Cyto-Q Blockbuster.
Immunohistology
To study ASO uptake in the kidney using immunohistology, ASO uptake was revealed using a rabbit phosphorothioate-specific antibody (pAb) (developed and kindly provided by the research group of Jonathan Watts), from here on called pAb. In short, tissue slides were incubated overnight at 4°C with pAb or isotype control rabbit IgG (DAKO, Cat: X0936) in 2% BSA/5% NGS in TBS/T. Primary antibodies were detected for 1.5 h at room temperature using Goat Anti-Rabbit-HRP in 2% BSA/5% NGS in TBS/T. Peroxidase activity was revealed using DAB (3,3′-diaminobenzidine, Dako, Cat, Lot) for 12 min at room temperature. After rinsing in dH2O, sections were counterstained with haematoxylin (1:4 dilution), dehydrated, and mounted with Pertex. Slides were imaged using the PANNORAMIC Midi II or PANNORAMIC 250 Flash III digital slide scanner (3DHistech Ltd.) with a 20× objective. Image processing was performed using CaseViewer software (3DHistech Ltd.).
Immunofluorescence
For immunofluorescence analysis of ASO uptake, incubation with primary antibodies in 2% BSA/5% NGS in TBS/T was performed overnight at 4°C for pAb, biotinylated Lotus Tetragonolobus Lectin (LTL; Vector Laboratories, B-1325), or isotype control rabbit IgG (DAKO, Cat: X0936). Primary antibodies were detected for 1.5 h at room temperature using Goat Anti-Rabbit 488 (Life Technologies, Cat: A-11008) and Streptavidin 568 (Invitrogen, Cat: S11226) in 2% BSA/5% NGS in TBS/T. Slides were counterstained using Hoechst 33342 (Invitrogen, Cat: H3569), mounted using ProLong™ Gold Antifade Mountant (Thermo Fisher Scientific, Cat: P36930), sealed with nail polish, and allowed to dry overnight at room temperature. Slides were imaged using the PANNORAMIC Midi II or PANNORAMIC 250 Flash III digital slide scanner (3DHistech Ltd.) with a 20× objective. Image processing was performed using CaseViewer software (3DHistech Ltd.).
Hybridization ELISA
Quantification of ASO in cells and kidney tissue was performed using hybridization enzyme-linked immunosorbent assay (HELISA) as described by Thayer et al. 23 For the in vitro quantification of ASO#03 and #22-1, standard curves were generated in cellular protein lysate at a concentration of 0.5 mg protein/mL lysis buffer with Pierce protease inhibitors, with ASO ranging from 0.8 to 12,500 ng/mL with a 0 ng/mL control. For the in vivo quantification of ASO#03 in mouse kidney, snap-frozen left kidney samples were weighed on an analytical balance and homogenized in lysis buffer with Pierce protease inhibitors. A standard curve was generated in pooled kidney homogenate from baseline animals (n = 4) at a concentration of 5 mg tissue/mL lysis buffer, with ASO ranging from 0.8 to 12,500 ng/mL with a 0 ng/mL control. Kidney homogenates from saline- and ASO-treated animals were adjusted in lysis buffer to 5 mg tissue/mL. Next, standards and study samples were diluted 1:25 (in vitro) or 1:50 (in vivo) in sample buffer, consisting of 10 mM Tris-HCl (pH 8.0) and 1 mM EDTA. A 5′-biotinylated DNA/LNA capture probe and 3′-digoxigenin DNA/LNA detection probe for ASO#03 were purchased from Qiagen with respective sequences (LNA indicated with +base): /5Biosg/TAG+CT+C+AG+AG and +GT+TTGTGCTG/3Dig_N/. A 5′-biotinylated DNA/LNA capture probe and 3′-digoxigenin DNA/LNA detection probe for ASO#22-1 were purchased from Qiagen with respective sequences (LNA indicated with +base): /5Biosg/CA+GA+GGTT+TG and +TG+C+T+GA+TTT+T/3Dig_N/. A 20 nM probe mixture in hybridization buffer consisting of 60 mM Na2PO4 (pH 7.0, dibasic), 1 M NaCl, 5 mM EDTA, and 0.02% Tween 20 was added in a 1:1 ratio to every sample. Probe hybridization was performed in a thermal cycler at 95°C for 5 min, 40°C for 30 min, with a final hold at 12°C, after which samples were transferred to MSD GOLD 96-well plates and further incubated at room temperature on a shaker (650 rpm) for 30 min. Washing in-between all steps was performed using 1× KPL wash buffer (VWR International). Plates were incubated with 0.5 µg/mL sheep polyclonal Anti-Digoxigenin (200 µg; Roche, Cat: 11333089001), conjugated with the MSD GOLD SULFO-TAG NHS-Ester Conjugation Pack 1 (Meso Scale Diagnostics, Cat: R31AA-1), in 1% MSD Blocker A Kit (Meso Scale Diagnostics, Cat: R93AA-1) in the dark on a shaker at 650 rpm for 60 min. Electrochemiluminescent (ECL) signal detection was performed using MSD GOLD Read Buffer A (Meso Scale Diagnostics, R92TG-1) on the MESO QuickPlex SQ 120 (Meso Scale Diagnostics). Interpretation and quantification of ECL signal intensities was performed using linear or nonlinear regression in R (v4.1.3). ASO concentrations were normalized on protein concentration for in vitro experiments and tissue concentration for in vivo experiments to yield a measure of µg ASO per gram of protein or tissue (µg/g).
Statistics
The following statistical approach was applied, unless otherwise indicated. Experiments were performed in single wells, using biological replicates (n = 3–5), and data are presented as individual replicates and/or geometric means. Individual measurements may have been removed when deemed to act as an outlier or in case of technical inconsistencies. Statistical analysis was performed using one-way analysis of variance or the Kruskal–Wallis and Dunnett test for multiple-to-one comparisons. Conditions were compared with scrambled control and P values below 0.05 were deemed significant.
Results
Renal epithelial cells can be infected by BKV in vitro
To evaluate our BKV-targeting ASOs, we first established cell culture infection models using BKV-permissive immortalized human renal epithelial cells (HK2) or primary human proximal tubular epithelial cells (HRPTEpiC). As TAg protein is poorly detectable using existing antibodies, we initially focused our readouts on TAg and VP1 mRNA and VP1 protein levels. 9 Following infection of HK-2 and HRPTEpiC with BKV, TAg mRNA, VP1 mRNA, and protein were close to undetectable for the first 2 d.p.i. but increased rapidly up to 7 d.p.i. The increased expression of TAg and VP1 mRNA in HRPTEpiC relative to HK-2 cells showed that BKV infection rates were likely higher in primary cells than in the immortalized HK-2 cells. Expression of BKV mRNA and protein was deemed to be sufficiently expressed from 5 d.p.i. onward to allow for the evaluation of ASO inhibitory activity (Supplementary Fig. S1).
Identification of potential ASO target sites highly conserved between BKV subtypes
Four genotypes of BKV have been described based on sequence differences in the viral packaging protein VP1, namely: I, II, III, and IV.24,25 Heterogeneity in genotypes I and IV has prompted the creation of the Ia, Ib1, Ib2, Ic, IVa1, IVa2, IVb1, IVb2, IVc1, and IVc2 subtypes. Genotype I is the most prevalent (60%–80% of the population) and is found worldwide. Genotypes II and III are rare, while genotype IV is predominantly found in East Asia and Europe (estimated 10%–20% of the population). The clinical significance of this BKV diversity is poorly understood.26,27 To develop a universally applicable ASO for the global patient population, we performed phylogenetic analyses using complete ECR sequences to investigate TAg splice site sequence conservation. Retrieval of publicly available BKV genomic sequences at the time of the evaluation yielded 465 unique sequences for alignment, yielding a phylogenetic tree with three major clades consisting of the BKV genotypes: I, II/III, and IV, and corresponding subtypes (Fig. 1B), with 323x genotype I, 17x genotype II, 10x genotype III, and 114x genotype IV. Importantly, two isolates were positioned in different clades than previously reported, namely SHA-43 (IVa1, reported as IVa2) and OKN-19 (IVa2, reported as IVa1). 28
This sequence evaluation enabled us to generate probability sequence logos for the major TAg donor and acceptor splice site (Fig. 1C), with 20-nucleotide flanking regions generated per genotype. The donor splice site showed little inter- and intragroup variation, with an identical 40-nucleotide region in 434 of 465 isolates. In this region, four distinct donor splice site sequence variations were identified with single nucleotide polymorphisms (SNPs) in the less prominent sequences at −11:G>A, +8:C>A, +15:T>G, and +16:C>T, with values indicating the position relative to the donor splice site. Importantly, the −11:G>A mutation was observed in all genotype II and III sequences (28 isolates in total). The remaining sequence variations were unique to single isolates. More sequence diversity between subtypes, in the form of deletions/insertions, was observed for the polypyrimidine tract upstream of the acceptor splice site, as published by others. 29 Very little variation was observed in the first 20 nucleotides of exon 2.
ASO design and activity
Based on these sequence analyses, 20-mers were designed containing full 2′-O-methyl (2′-O-Me) and phosphorothioate (PS) backbone that minimally covered 1 intronic or exonic nucleotide at the TAg 5′-donor or 3′-acceptor site (see Supplementary Table S1). Due to the GC-poor polypyrimidine tract immediately upstream of the acceptor site, only four ASOs for the acceptor site were designed (ASO#04, #05, #12, #13). Eight ASOs targeted the donor site (ASO#01–03, #06–11). A scrambled control ASO, along with a TAg coding region-targeting ASO (ASO#14, exon 1), were designed as controls and predicted not to interfere with TAg splicing.
Candidate ASOs were first tested for activity in BKV-infected HK-2 cells, where carrier-mediated delivery of 50 nM ASO was performed prior to infection. Subsequently, RNA, protein, and culture supernatants were harvested for assessment of ASO activity, revealing most ASOs to mediate BKV parameter reductions (Fig. 2A). Distance matrix computation and hierarchical clustering of log-transformed fold changes of TAg and VP1 mRNA and VP1 protein enabled the clustering of ASOs based on inhibitory profile (Fig. 2B), resulting in a near-complete separation between donor splice site-targeting ASOs (#01–03, #06–11) and acceptor-targeting (#04, #05, #12, #13) or control ASOs (scrambled control and ASO#14). Subsequently, we focused on ASOs targeting the TAg donor splice site. Treatment with ASO#01, #03, or #11 revealed but modest reductions in TAg mRNA expression levels (P > 0.05), while statistically significant attenuation of both VP1 mRNA and protein levels was observed. These data provide the interesting possibility that even subtle reductions in the gatekeeper levels of TAg could have far-reaching consequences for the downstream expression levels of late coding region mRNAs and protein (Fig. 2A–B).
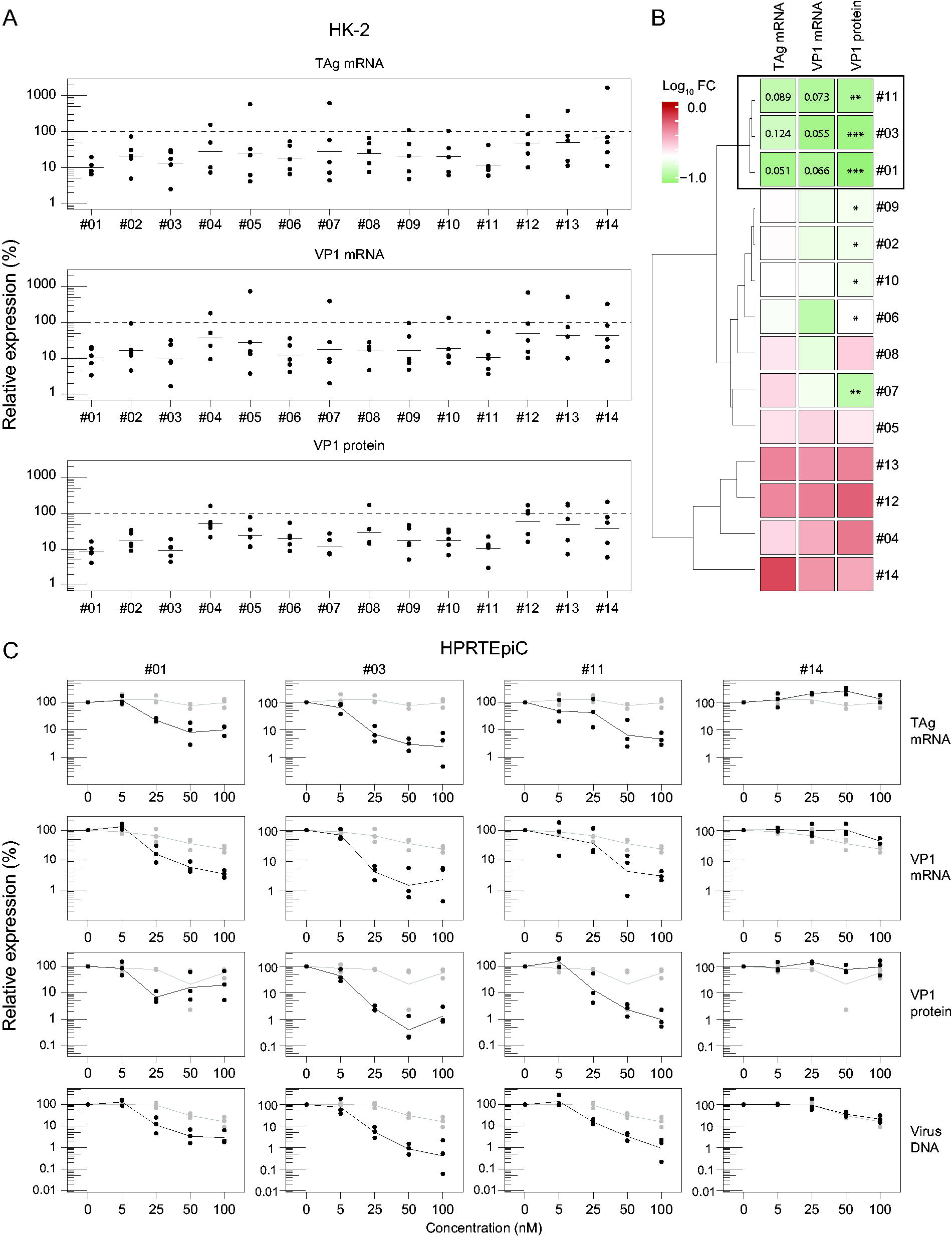
Screening and validation of candidate ASOs in HK-2 cells and HRPTEpiC.
Based on this first screening, we selected ASO#01, #03, and #11 (Fig. 2B) for further testing and validated their inhibitory activity in HRPTEpiC, which display a higher rate of infection allowing for more robust assessment of ASO activity. As shown in Supplementary Fig. S1, HRPTEpiC demonstrated greater susceptibility to BKV infection and more closely represented kidney infection and the reactivation process in humans. Therefore, HRPTEpiC were subsequently employed as the primary cell culture model for ASO activity testing. Here, we performed dose-response studies with carrier-mediated delivery of 0, 5, 25, 50, or 100 nM ASO, after which cells were infected with BKV and RNA, protein, and viral supernatant harvested 7 d.p.i. Cell viability was not compromised by the ASO treatments (data not shown). As shown in Fig. 2C, at 25 nM scrambled control or ASO#14, TAg and VP1 mRNA were markedly reduced, and VP1 protein levels attenuated by 22.7 ± 2.0% (mean ± standard error of the mean (SEM)), while virus production was only suppressed by 6%–7% in control-treated conditions as compared with 94.7 ± 1.8% upon treatment with 25 nM ASO#03. Maximum suppression of the BKV parameters was achieved in the 50–100 nM range for ASO#03 and ASO#11 (Fig. 2C). Highest administered concentrations of scrambled control or ASO#14 also reduced BKV parameters (Fig. 2C, right panel), likely indicative of a combination of hybridization-dependent and hybridization-independent off-target effects.30,31
ASO activity is dependent on phosphorothioate chemistry and dose
Unmodified ASOs are rapidly degraded by nucleases and therefore require chemical modifications to prolong their stability and improve cellular uptake. 32 While RNA-based drugs that employ PS backbone modifications have been observed to concentrate in the renal proximal tubules (PT), they are also associated with PS chemistry side effects. 33 Given that ASO#01, #03, and #11 are fully phosphorothioated, we sought to determine the effect of reducing PS chemistry on BKV inhibitory activity in HRPTEpiC. For this, we designed ASOs with 19, 12, 9, or 6 modifications, whereby the PS residues were maintained at the 5′ and 3′ ends to maximize exonuclease resistance (see Fig. 3A). As compared with scrambled control-treated cells, uptake of full PS-modified ASOs resulted in potent suppression of BKV mRNA, protein, and DNA. An approximate 50% loss of antiviral activity was observed upon reduction to 12 PS modifications, with minimal suppression of BKV with 9/6 PS backbone modifications (Fig. 3B). ASO#03 activity, in particular, was strongly affected by a reduction of PS modifications in the middle region of the ASO, pointing toward an increased sensitivity to endonucleases 34 or alteration of ASO structure 35 that could impact hybridization to the ECR pre-mRNA. Collectively, these studies support the need for PS modifications in ASOs for effective BKV targeting and indicate that ASO#03 is the most promising antiviral ASO.
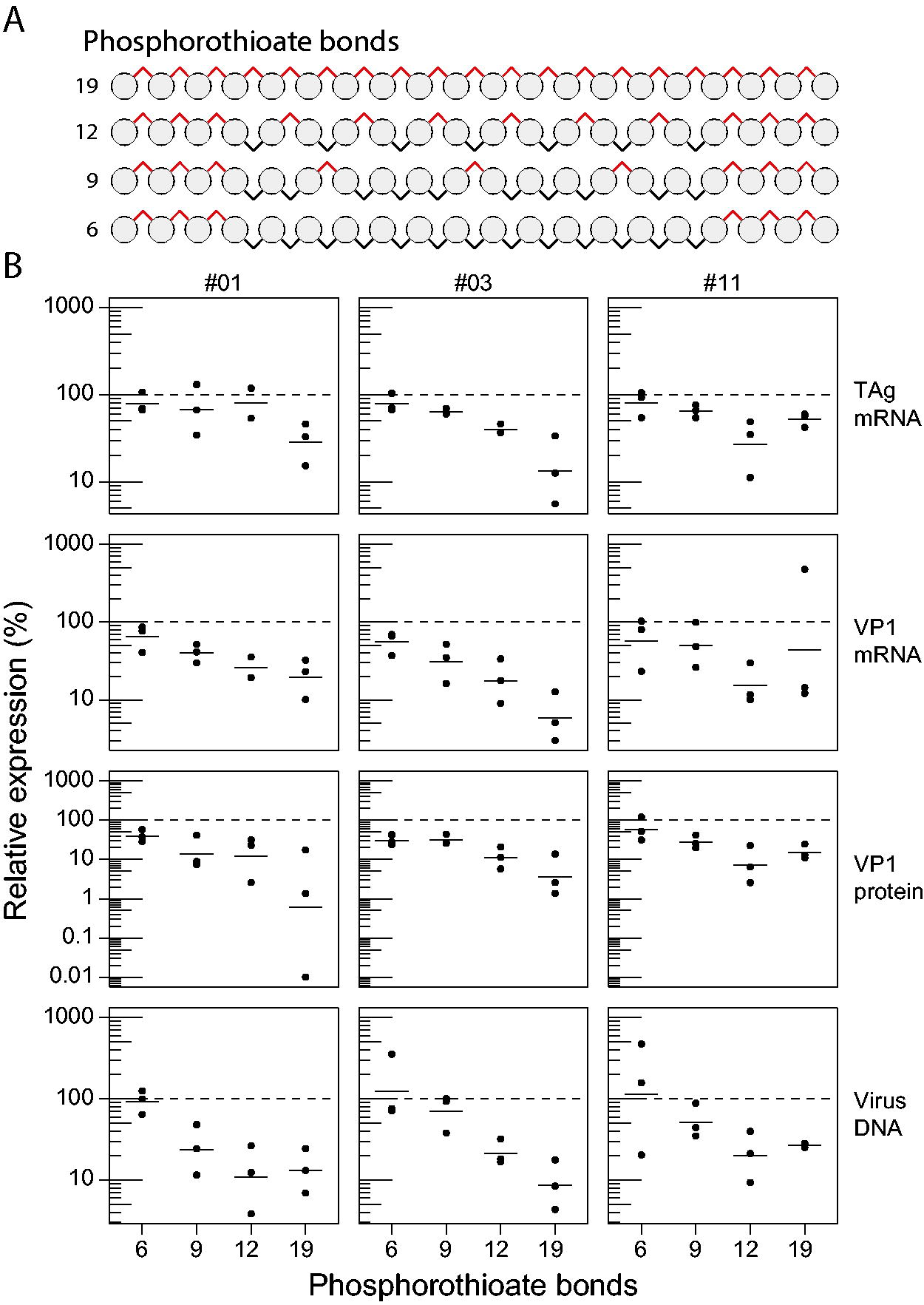
Inhibitory activity is linked to phosphorothioate chemistry.
In vivo analysis of ASO distribution
In order to directly treat BKV, it is essential that the therapeutic distributes to tubular epithelial cells of the kidney, where the virus resides and replicates upon activation.3,9,36 As PS-modified ASOs have previously been described to distribute to the kidney, and in particular to tubular epithelial cells, 18 we assessed ASO#03 distribution in mice to validate this. For this, C57BL6 mice received an i.v. administration of 0.9% NaCl (saline) or 40 mg/kg ASO#03 and were sacrificed at 1, 3, 7, 14, 30, and 90 days post-administration. No signs of distress, injection site reactions, or behavioral abnormalities were observed in mice that received saline or ASO#03.
ASO was visualized by immunohistochemistry and immunofluorescence in mouse kidneys using a pAb (kindly provided by Dr. Jonathan Watts). Hematoxylin-DAB pAb staining of mouse kidneys resulted in a faint background signal in kidney cortex tubules and outer stripe of the outer medulla (OSOM), but far less in the inner stripe of the outer medulla (ISOM) in both ASO- and saline-treated animals (Fig. 4A, large panel). This background signal was not observed in isotype controls, indicating a dependence on inherent characteristics of pAb and not related to ASO accumulation. ASO was predominantly detected in convoluted PT, with low levels in cortical straight PT and minimal detection in straight PT of the OSOM (Fig. 4A–B, ASO-treated panels). ASO-treated mice displayed an endolysosomal staining pattern that was restricted to tubular structures of the kidney cortex, a pattern that was not observed in the outer medulla (Fig. 4A, small panels). Immunofluorescence staining for pAb and the PT-specific marker LTL revealed that endolysosomal uptake of ASO was virtually exclusive to PT structures (Fig. 4B, bottom panel).
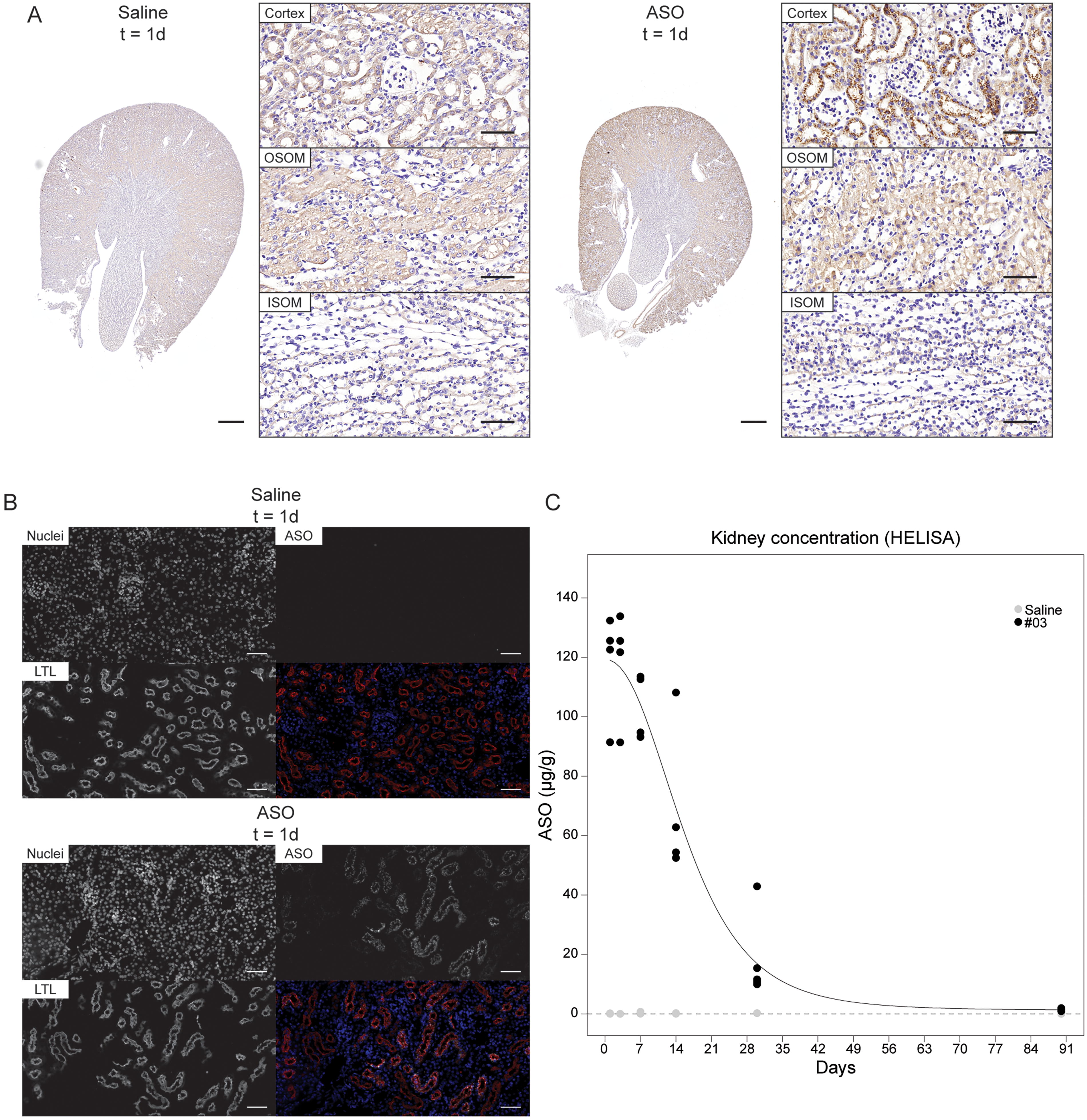
ASO#03 is taken up by proximal tubular epithelial cells in the kidney. C57BL6 mice (n = 4 per group) received intravenous administration of saline or 40 mg/kg ASO#03, and renal uptake of ASO was studied at multiple time points.
We also quantified kidney ASO content by exploratory probe-based HELISA. 23 Administration of 40 mg/kg ASO revealed 116.8 ± 9.1 µg ASO per gram of kidney tissue 3d post-administration, with approximately 50% of ASO remaining at day 14 (Fig. 4C). As shown in Supplementary Table S2, multiple dosing of ASO#03 yielded higher levels of ASO in mice sacrificed on day 14 (172.6 ± 28.0 µg/g based on day 0 and day 7 dosing vs. 66.4 ± 13.1 µg/g with single dose on day 0) and was still minimally detectable after 90d in mouse kidneys (4.78 ± 0.31 µg/g vs. 1.38 ± 0.17 µg/g, respectively).
Screening and optimization of new ASO candidates
Having identified ASO#03 as a highly active BKV-targeting RNA therapeutic, we initiated a final cycle of ASO screening and optimization with 9 novel 20-mer candidate sequences (ASO#15–24) in HRPTEpiC (Fig. 5A), in efforts to identify the most potent 5′-donor splice site of TAg-targeting ASO. ASO#15–24 were administered by carrier-mediated transfection of HRPTEpiC followed by an infection with BKV. ASOs #03, #14, and scrambled control were included as reference ASOs. However, ASO#15–24 were generally characterized by marked suppression of BKV mRNA expression and viral DNA production (minimally >90%), while protein expression was reduced by up to 98% for ASO#19–22 versus scrambled control (Fig. 5A). Clustering of ASO activity using distance matrix computation and hierarchical clustering confirmed ASO#19–22 as the most potent BKV-targeting ASOs, prompting their selection for chemistry optimization studies (Fig. 5B).
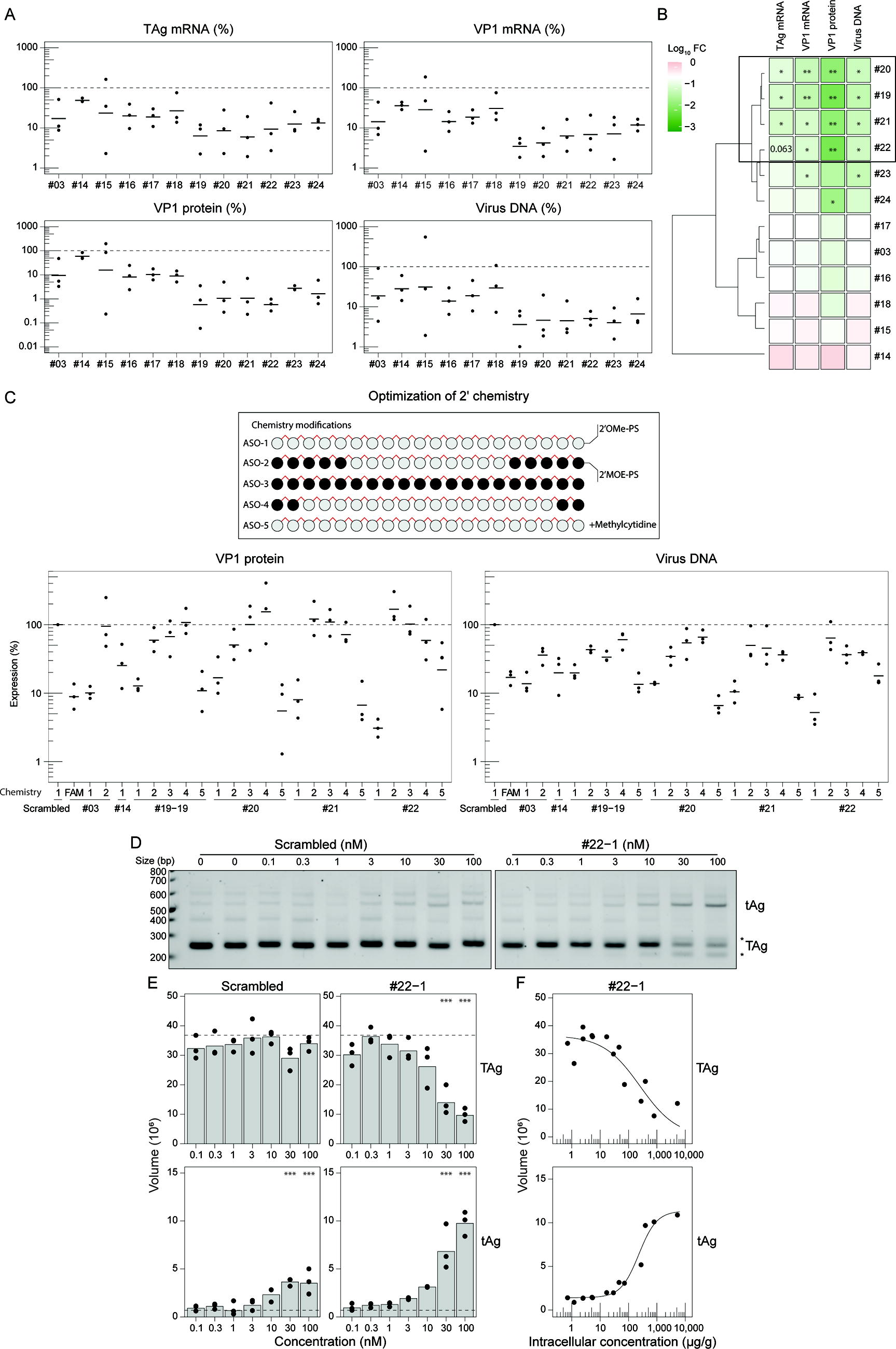
Identification and optimization of the BKV lead candidate: ASO#22
In efforts to enhance ASO activity by optimizing the affinity and stability of our BKV-targeting ASOs, we evaluated the effect of substituting 2′-O-methoxyethyl modifications (MOE) at the 2′-position of the pentofuranose moiety. Furthermore, we also introduced 5′-methylcytosine (5mdC) to our existing full 2′-O-Me and PS ASOs to decrease potential immunostimulatory effects.37,38 Importantly, ASO#19 was adjusted to a 19-mer sequence (denoted #19-19 in Fig. 5C) due to a G>A mutation observed in BKV genotypes II and III that could impact inhibitory activity in these BKV genotypes (Fig. 1C). The location and degree of chemistry modifications introduced to ASO#19–22 are depicted in Fig. 5C and annotated as follows: (1) ASO-1 (100% O-Me-PS); (2) ASO-2 (50/50% O-Me/MOE-PS); (3) ASO-3 (100% MOE-PS); (4) ASO-4 (80/20% O-Me/MOE-PS); and (5) ASO-5 (100% O-Me-PS with 5mdC). For these studies, carrier-mediated transfection of candidate ASOs #19–22 in HRPTEpiC was followed by infection with BKV. FAM-labeled scrambled control, #03 (with 5′-FAM) and ASO#14, as well as unlabeled ASO#03-1 (without 5′-FAM) were taken along as reference or control (n = 3 biological replicates; Fig. 5C). Here, ASO activity was assessed by reductions in VP1 protein and viral DNA reductions 5 d.p.i. FAM-labeled scrambled control, ASO#03 (with 5′-FAM), and ASO#14, as well as unlabeled ASO#03 (without 5′-FAM), were taken along as reference or control (n = 3 biological replicates; Fig. 5C). These studies strongly suggest that 2′-O-Me chemistry is best suited for TAg splice site-targeting ASOs and the corresponding attenuation of VP1 protein production (Fig. 5C, left graph) and viral DNA replication (Fig. 5C, right graph), as the introduction of 2′-O-MOE modifications resulted in loss of activity for all tested ASOs. Interestingly, 5mdC modifications were well tolerated and improved the activity of ASO#20 (ASO#20-5) and ASO#21 (ASO#21-5) relative to their demethylated counterparts (Fig. 5C) but were not superior to the activity observed for ASO#22-1 (Fig. 5C, far right). While ASO#03 remained highly active, ASO#22-1 with full 2′-O-Me and PS clearly represented the most potent ASO designed and was selected as our lead candidate for BKV targeting.
Demonstration of splice interference in pRPcT1ss1 cells
We next sought to confirm that the mechanism of action for ASO#22-1 was through hybridization-dependent splice modulation of TAg. For this, a transformed mouse fibroblast cell line (pRPcT1ss1) stably overexpressing the BKV ECR 39 was employed to study ASO-induced splicing interference. Carrier-mediated uptake of increasing concentrations of scrambled control or ASO#22-1 in pRPcT1ss1c was performed, with RNA and protein lysates being collected 24 h post-transfection. TAg splice variants were quantified and compared with intracellular concentrations of ASO.
As shown in Fig. 5D, TAg-specific primers yielded PCR products of 590 base pairs (bp; unspliced), 523 bp (tAg), and 246 bp (TAg) with ASO#22-1 treatment resulting in dose-dependent TAg splicing interference. ASO#22-1 treatment induced a shift from TAg to tAg at 3–10 nM, with perturbation of TAg splicing being accompanied by the appearance of additional PCR products in the 200–300 bp fragment size (Fig. 5D, annotated with asterisks). As compared with untreated cells, 100 nM ASO#22-1 reduced TAg expression to 26.3 ± 3.3%, as compared with 92.3 ± 12.1% with scrambled control ASO (Fig. 5E, top panel). HELISA of cellular lysates revealed that the TAg-to-tAg splice variant shift (Fig. 5F with log-logistic fit) required 100–300 µg ASO per gram of protein to initiate splice modulation. Furthermore, alternative splice site prediction 40 identified a potential cryptic donor splice site at the 201-nucleotide position in the TAg transcript, 42 nucleotides upstream of the constitutive donor splice site, providing a potential explanation for the 200 bp splice variant. No reduction in TAg expression or TAg/tAg splice ratio was observed in scrambled control-treated cells, indicating that ASO#22-1 complementarity to the TAg donor splice site perturbs BKV ECR splicing and attenuates TAg mRNA generation (Fig. 5D–F).
Collectively, the described herein activities have demonstrated that it is possible to design an antiviral ASO with (1) potent anti-BKV activity in cell culture, (2) a broad BKV spectrum, and (3) a biodistribution profile that is clinically relevant (uptake in tubular epithelial cells). ASO#22-1 demonstrated the best overall profile and has therefore been nominated as a clinical development candidate to undergo further preclinical studies.
Discussion
In the absence of BKV-targeting therapeutic modalities, immunosuppressive tapering is currently the sole approach clinicians can employ to treat kidney transplant patients suffering from problematic BKV viremia. Existing interventions, including IVIG, (brin)cidofovir, and leflunomide, have demonstrated variable efficacy, 41 and convincing clinical data to support monoclonal antibody approaches against BKV, including MAU868, 16 are currently still lacking. An important caveat for these approaches is that their mechanism of action is either indirect, modulating the activity of host factors, or direct but not specifically targeting BKV replication within productively infected kidney cells. Our universal splice-modulating ASO represents a direct-acting antiviral strategy, limiting BKV replication in kidney epithelial cells and thereby preventing the production and release of nascent viral particles to the surroundings (both neighboring cells and the circulation).
Of paramount importance in designing an ASO targeting BKV is that it is highly active and can be applied as a “one-size-fits-all” therapeutic given the four BKV genotypes observed in the human population.8,9,13 This prompted us to design ASOs targeting the donor and acceptor splice sites in the BKV ECR. However, the proximity of the polypyrimidine tract to the acceptor splice site of the BKV ECR, along with the lesser-conserved sequence in the intronic region flanking exon 2, likely impacted the observed activity of donor- versus acceptor-targeting ASOs. Acceptor-targeting ASOs worked poorly (Fig. 2A and B), while donor-targeting ASOs generally had a clear impact on relevant BKV parameters. Furthermore, the lack of effect by acceptor site-targeting ASOs, which also possess full 2′-O-Me and PS modifications, strongly suggests that the observed antiviral effects by donor-targeting ASOs are not (merely) mediated by ASO uptake and/or indirect immunostimulatory effects. Multiple donor splice-site-targeting ASOs, with varying exonic and intronic content, point toward ECR splicing interference as a surprisingly potent means of inhibiting BKV replication. The minimal antiviral effect observed for scrambled control ASO at high concentrations is not uncommon, as previous studies with the hepatitis B virus-targeting bepirovirsen similarly yielded viral reductions at the highest concentrations of control ASO in nonclinical studies. 42
Optimization of the 2′ sugar moiety chemistry, whereby 2′-O-Me nucleobases were replaced with 2′-O-MOE-modified nucleobases, resulted in a clear inverse relationship between 2′-O-MOE content and BKV-inhibiting capacity. While an explanation for this observation is purely speculative, the bulkier alkyl group of 2′-O-MOE on the pentofuranose moiety could result in steric hindrance for interaction with the ECR pre-mRNA based on the folding properties of the exon–intron junction.43–45 In contrast, the introduction of 5mdC nucleobases substantially improved the activity of ASO#20-5 and ASO#21-5, potentially reducing the immunostimulatory effects of tubular epithelial cell ASO uptake. However, the potent antiviral activity of ASO#22-1 strongly supports its selection as the clinical candidate. Furthermore, as PS protection of the two to three flanking nucleobases is crucial to minimizing 3′-exonuclease-mediated “trimming” of the ASO (resulting in N-1 and N-2 oligonucleotides), we did not evaluate the consequences of PS removal at these locations as the effects on ASO stability and activity are relatively predictable. However, we did evaluate the effect of PS internucleotide linkage removal in the middle region of the ASO, as alternating PS modifications have previously been demonstrated to confer substantial protection and maintain ASO activity. The antiviral activity of the ASOs generally dropped substantially upon introduction of alternating PS modifications, while the activity of ASO#03 was strongly affected (upon reduction of PS-modified linkages from 19 to 12). These data suggest that susceptibility to endonucleases and loss of chirality at key sites can profoundly impact ASO activity and support our selection of fully PS-modified ASOs.
Extensive data exist supporting the primary distribution of chemically modified ASOs to well-perfused tissues such as the kidney and liver, with 20% of the administered dose localizing to the kidneys.18,46 Interspecies differences between kidney cortex and medullary ASO uptake are known, with high cortical and poor medullary ASO uptake observed in rodent kidneys, while a more even distribution between these kidney compartments is observed in non-human primates (NHPs).47,48 Regardless of species, PTECs of the kidney cortex are the major site of ASO accumulation, which clinically could be highly relevant given that single-cell transcriptomic analyses of BKV reactivation in human kidneys revealed a preference for propagation in PTECs, likely due to the high glycolytic/gluconeogenic energy profile in this nephronic segment. 49 Primary accumulation of ASO occurs in the S1 and S2 segments of the PT as a result of high cell surface expression of megalin and cubilin, proteins known to mediate both ASO internalization and transport to the endolysosomal compartment. 50 These receptors are likely responsible for the distribution pattern observed for our fully PS- and 2′-O-Me-modified ASOs in mice (Fig. 4A and B; Supplementary Table S2). Ideally, this distribution data would be complemented with in vivo efficacy studies. However, animal models for BKV infection do not exist or are not feasible due to the narrow range of host species that can support an infection that suitably mimics the human condition. Naturally, alternative approaches would include developing a transgenic model, humanized mice, xenografts, or the use of surrogate viruses, 51 in combination with additional disposition experiments in animal models suitable to study the biodistribution and PK properties of nucleic acid drugs (eg, NHPs or minipigs). The overarching goal of these preclinical and clinical studies is to lay the groundwork for efficacy testing in kidney transplantation patients. Albeit speculative, the ability to halt or ideally clear BK virus replication should be protective to the kidney and potentially sufficient to protect the transplanted kidney from BK virus-associated nephropathy.8,13 Naturally, this antiviral effect relies on a combination of the direct antiviral activity of the drug (our BK virus-targeting ASO) and the “indirect” effects mediated by the residual immune system (∼80% of immunosuppressed kidney transplant patients are able to control BKV reactivation/replication without the need for immunosuppression reduction). Clinical evaluation of this drug will ultimately provide evidence as to whether an ASO-based BK virus-targeting therapy is sufficiently potent to control the virus.
In conclusion, we have identified a direct-acting and highly potent ASO that interferes with the splicing of the BKV ECR. This novel antiviral approach limits production of TAg, the gatekeeper viral protein responsible for driving nascent viral particle production. Importantly, the unassisted uptake of this ASO in the kidney, in particular by PTs, provides hope that BKV reactivation and spread in kidney transplant patients can be limited, reducing the risk of (premature) graft loss.
Footnotes
Acknowledgments
The authors kindly acknowledge Prof. Massimo Negrini (University of Ferrara, Ferrara, Italy) for providing us with the pRPcT1ss1c cells for mechanism-of-action studies; Dr. Jonathan Watts (University of Massachusetts Chan Medical School, Worcester, MA) for providing a polyclonal antibody that detects phosphorothioate-modified backbones of ASOs; and Prof. Joris Rotmans and Dr. Herman Wunderink for discussions regarding BK virus infection-related complications in kidney transplantation patients.
Data Sharing Statement
Access to the raw data detailed in this article (materials and methods, results, and figures) is freely available to noncommercial entities and academic institutions. Commercial entities must contact AiCuris Anti-Infectives GmbH to request access to data and/or supporting documents.
Author Disclosure Statement
None as outlined by the journal, but for full disclosure A.A.-R. declares being employed by LUMC, which holds patents on exon-skipping technology, some of which have been licensed to BioMarin and subsequently sublicensed to Sarepta. As co-inventor of some of these patents, A.A.-R. is entitled to a share of royalties. A.A.-R. further discloses being ad hoc consultant for PTC Therapeutics, Sarepta Therapeutics, Regenxbio, Dyne Therapeutics, Lilly, BioMarin Pharmaceuticals Inc., Eisai, Entrada, Takeda, Splicesense, Galapagos, Sapreme, and AstraZeneca. A.A.-R. also reports being a member of the scientific advisory boards of Eisai, Hybridize Therapeutics, Silence Therapeutics, Sarepta Therapeutics, and Mitorx. Remuneration for these activities is paid to LUMC. In the past 5 years, LUMC also received speaker honoraria from PTC Therapeutics, Alnylam Netherlands, Italpharmaco, and Pfizer and funding for contract research from Sapreme, Eisai, Galapagos, Synaffix, and Alpha Anomeric. Project funding is received from Sarepta Therapeutics and Entrada. J.P., A.J.v.Z., and E.v.d.V. have patents on splice modulation of BKV that are outlicensed to AiCuris Anti-Infectives GmbH (Wuppertal, Germany). Hybridize Therapeutics B.V. and AiCuris Anti-Infectives GmbH provided funding for the research. E.v.d.V. is a shareholder of Hybridize Therapeutics B.V.
Funding Information
This work was funded by the Dutch Kidney Foundation (PPS06: BK-Away) together with Dutch governmental public–private research grant(s) and Starfish Innovations, the Leiden University Medical Center and Department of Nephrology (including NephroSearch), and AiCuris Anti-Infectives GmbH.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.