
Abstract
Duchenne muscular dystrophy (DMD) is a severe X-linked disorder caused by mutations in the DMD gene, resulting in a lack of dystrophin protein. This leads to progressive muscle wasting, cardiac and respiratory dysfunction, and premature death. Antisense oligonucleotide (ASO)-based therapies represent a promising approach to treating DMD, with several already approved by the FDA. However, the levels of dystrophin restoration achieved in clinical trials are often insufficient for meaningful therapeutic impact, highlighting the urgent need to enhance ASO efficacy. One potential strategy is to improve muscle pathophysiology, which is compromised in DMD due to cycles of necrosis and regeneration, chronic inflammation, and fibrotic and adipose tissue replacement. These disease characteristics may limit ASO efficiency. In this study, we evaluated the combination of tricyclo-DNA-ASO targeting the Dmd exon 23 with 20-hydroxyecdysone (20-E), a steroid hormone known to activate the protective arm of the renin-angiotensin-aldosterone system, enhance protein and ATP synthesis, and exhibit anti-inflammatory and antifibrotic properties. Mdx mice were treated with ASO alone or in combination with 20-E for 8 weeks. While both treatments restored similar levels of dystrophin and significantly improved functional outcomes such as the distance run and maximum speed in the treadmill exhaustion test, other improvements like the specific force and the decrease in the force drop after eccentric contraction were observed only with the combination therapy. Importantly, the cotreatment was well tolerated without liver or kidney toxicity. These findings provide proof of concept that combining 20-E with ASO therapy can ameliorate dystrophic pathology and improve muscle function in a DMD mouse model. By targeting both dystrophin restoration and muscle pathophysiology, this combined approach may offer a therapeutic strategy with the potential for meaningful clinical benefits, warranting further investigation and potential translation to patients.
Introduction
Duchenne muscular dystrophy (DMD) is a severe X-linked neuromuscular disorder characterized by progressive muscle wasting that affects one in 5000 male newborns. 1 This disease progresses rapidly to a loss of ambulation, followed by cardiac and respiratory insufficiencies, which ultimately leads to early death. DMD is caused by mutations, mainly deletions (65%), in the DMD gene encoding dystrophin. 2 These mutations result in the generation of an out-of-frame mRNA, which in turn leads to the introduction of a premature stop codon and the subsequent loss of dystrophin expression. 3 Dystrophin is a membrane-associated protein that links the cytoskeleton with the extracellular matrix (ECM). 4 Its primary function is to confer resistance to the contractile forces experienced by the myofibers, thereby maintaining the structural integrity of the skeletal muscle. In the absence of functional dystrophin, the sarcolemma becomes unstable and vulnerable to rupture during muscle fiber contractions.
Currently, exon-skipping therapy represents one of the most promising strategies for the treatment of DMD. Given that this pathology is mainly caused by frameshift or point mutations, 2 the exclusion of one or more exons appears to be an effective method to restore the reading frame and, consequently, the expression of an internally deleted but functional protein. 5 This therapeutic strategy involves the use of short single-stranded nucleic acid sequences, known as antisense oligonucleotides (ASOs), to bind to specific pre-mRNA targets and modulate their maturation. To mediate exon skipping, the use of naked DNA or RNA has been replaced by chemically modified oligonucleotides that are more resilient to nuclease activity and exhibit a superior affinity for RNA substrates. Initially, a 2′O-Me (2′O-methyl RNA) and a phosphorodiamidate morpholino oligomer (PMO) were developed and tested clinically for the treatment of DMD.5,6 Some PMO-based therapies have been approved in the United States and Japan for the skipping of exons 45, 51, and 53.7–10 However, these ASOs exhibit poor distribution to target tissues and dystrophin restoration levels are very low. 11 Alternative chemistries and new generations of ASO are currently being developed to overcome these limitations. 12 Among them, tricyclo-DNA (tcDNA)-ASO conjugated to palmitoyl acid have shown particularly promising potency in the mdx mouse model, mainly due to their properties of biodistribution and RNA affinity.13–15 Although this novel class of ASO appears to offer a promising therapeutic avenue, there is scope for enhancing its efficacy in the treatment of DMD.
Different strategies can be employed to potentiate the efficacy of ASO. In the context of muscular dystrophies, the improvement of the muscle pathophysiology may play an important role. Indeed, muscles affected by DMD undergo cycles of necrosis/regeneration, which result in chronic inflammation and progressive replacement of fibers by connective and adipose tissue. 16 These characteristics of the disease may limit the efficacy of ASO therapy. In this study, we used a steroid hormone, 20-hydroxyecdysone (20-E), which is extracted from invertebrates or plants. This hormone mimics the angiotensin 1–7 (Ang1-7) and activates the protective arm of the renin-angiotensin-aldosterone system via the activation of the Mas receptor. 17 20-E has previously demonstrated anabolic, antifibrotic, and anti-inflammatory effects and has been shown to increase protein synthesis in mdx mice.18,19 20-E has been purified to pharmaceutical grade for use as a drug candidate (BIO101) in several diseases including sarcopenia and COVID-19. 18 In a global multicentric phase 2/3 study, BIO101 showed a significant reduction of 44% of the risk of respiratory failure or death for patients with severe respiratory symptoms due to COVID-19. 20 In the field of neuromuscular diseases, a phase 2 interventional study performed in Europe and the USA demonstrated an increase in the primary endpoint, that is, the 400-m walk test, for patients with sarcopenia (NCT03452488), 21 leading to an imminent phase 3 for this disease. The effects of BIO101 have also been assessed in preclinical models of spinal muscular atrophy (SMA) and DMD and revealed positive outcomes supporting the initiation of phase 1/2 MYODA clinical trial planned in late 2024 in Belgium and the USA in nonambulant children with DMD. 22
Given the therapeutic potential of this molecule, we hypothesized that the amelioration of muscle histopathology using 20-E may not only improve the overall therapeutic benefit but also synergize the effects of ASO aiming at restoring dystrophin expression. For that purpose, we evaluated a combined treatment of tcDNA-ASO targeting the Dmd exon 23 with or without 20-E in mdx mice.
Methods
Antisense oligonucleotides and animal experiments
Animal care and all experimental procedures complied with the national and European legislation, the ARRIVE guidelines, and were approved by the French government (Ministère de l’Enseignement Supérieur et de la Recherche, Autorisation APAFiS #6518). Mdx (C57BL/10ScSc-Dmdmdx/J) mice were bred in our animal facility at the Plateforme 2Care, UFR des Sciences de la santé, Université de Versailles Saint Quentin and were maintained in a standard 12-h light/dark cycle with free access to food and water. Mice were weaned at weeks 4–5 postnatal and 2–5 individuals were housed per cage.
TcDNA-ASO targeting the donor splice site of exon 23 of the mouse dystrophin pre-mRNA (sequence 5′-CCTCGGCTTACCT-3′, previously described14,23) were synthesized by SQY Therapeutics (Montigny le Bretonneux, France). Palmitic acid was conjugated at the 5′end of a full PS-tcDNA via a C6-amino linker and a phosphorothioate bond as previously described. 23
Groups of 7–10-week-old male mdx mice were treated with 20-E and/or ASO during 8 weeks as follows: 20-E (BOC sciences) dissolved in miliQ H2O was used at a final concentration of 50 mg/kg/day. 20-E was administered by oral gavage 5 days per week. Groups of mice receiving the ASO were injected intravenously once a week with 25 mg/kg/week of ASO. Age-matched mdx groups receiving an equivalent volume of sterile saline were included as controls and C57BL/10 mice were included as wild-type (WT) controls.
One week after the last ASO injection, mice were euthanized by cervical dislocation, muscle and tissues were snap-frozen in liquid nitrogen-cooled isopentane and stored at −80°C.
Functional analysis
Muscle function
Muscle function of mdx mice was evaluated by measuring tibialis anterior (TA) muscle contraction in situ in response to nerve stimulation. Mice were anesthetized under isoflurane throughout the experiment. Body temperature was maintained at 37°C using radiant heat. The knee and foot were fixed with pins and clamps and the distal tendon of the muscle was attached to the lever arm of a servo-motor system using a silk ligature. The sciatic nerve was crushed proximally and stimulated distally by a bipolar silver electrode using supramaximal 0.1 ms-duration square-wave pulses. We measured the absolute maximal isometric tetanic force (P0) generated during isometric contractions in response to electrical stimulation (frequency 75–150 Hz, stimulation train 500 ms). P0 was determined at L0 (length at which maximal tension was obtained during the tetanus). Absolute maximal isometric force was normalized to muscle mass as an estimate of specific maximal force (sP0), that is, specific force.
Fragility was estimated from the force decline resulting from lengthening contraction-induced injury. The sciatic nerve was stimulated 700 ms (150 Hz stimulation frequency). A maximal isometric contraction of the TA muscle was initiated during the first 500 ms. Then, muscle lengthening (10% L0) at a velocity of 5.5 mm/s was imposed during the last 200 ms. All isometric contractions were made at an initial length, L0. Nine lengthening contractions of the TA muscles were performed, each separated by a 60 s rest period. Maximal isometric force was measured 1 min after each lengthening contraction and expressed as a percentage of the initial maximal isometric force. Measurements for both TA muscles per mouse are shown in Fig. 1. The experimenter was blinded to the treatment group when performing the measurements.
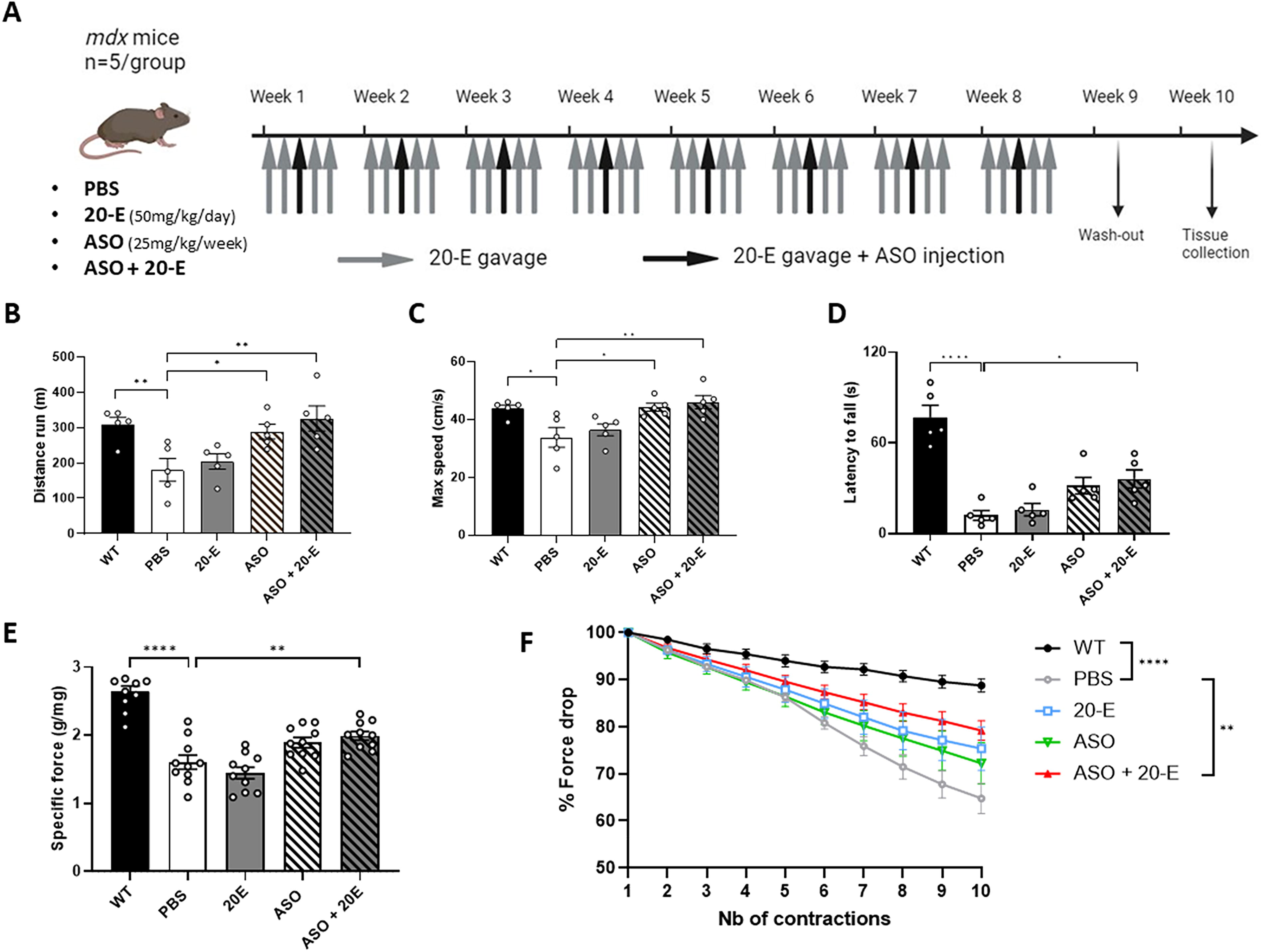
Functional improvement after 20-E and ASO treatment in mdx mice.
Treadmill
Mice were first acclimatized for 30 min in the room of experiment before each session. During 3 days before testing, they were placed on a switched-off treadmill for 30 s followed by a warm-up at 20 cm/s for 5 min. For the test session, mice were placed on the treadmill and the test started at the lowest speed of 5 cm/s to allow a warm-up (3 min). Speed was then increased by 1 cm/s every 30 s until exhaustion. Exhaustion was defined as the moment when the mouse would not continue running on the treadmill for 20 s despite gentle nudges to make it do so. At the end of the running test exercise, the total distance run was measured for each mouse.
Inverted grid
Mice were first acclimatized for 30 min in the room of experiment before each session. They were placed on a grid which was turned over before the timer was started. Each mice is evaluated three times with 300 s of rest between each test. The duration of each test cannot exceed 120 s and the latency to fall was recorded.
Restraint-induced unconditioned fear
Mice were first acclimatized for 30 min in the room of experiment before the session with a light intensity of ∼60 lux. They were restrained by trained experimenter grasping the scruff and back skin. After 15 s, the mouse was released to a new cage and video-tracked for 5 min using the ANY-maze software (Stoelting, USA).
ASO quantification by fluorescent hybridization assay
Using the Precellys (Bertin Instruments, France), tissues were homogenized in lysis buffer (100 mmol/L Tris–HCl, pH 8.5, 200 mmol/L NaCl, 5 mmol/L EDTA, 0.2% sodium dodecyl sulfate) containing 2 mg/mL of proteinase K (Invitrogen) to a final concentration of 50 mg tissue/mL of buffer. After an overnight incubation at 55°C, lysate was centrifugated at 7000 rpm and the supernatant was collected.
A hybridization assay with a molecular beacon probe was used to perform quantification of ASO, as previously described. 23 Briefly, 10 µL of tissue lysates were incubated with a 5′Cy3-DNA complementary probe conjugated with HBQ quencher at 3′ in a black nonbinding 96-well plates (Fisher Scientific). Phosphate-buffered saline (PBS) was added to a final volume of 100 µL per well and fluorescence was measured on a spectrophotometer (Ex 544 nm/Em 590 nm using FluoStar Omega). The amount of tcDNA in tissues was determined using a standard curve built on the measurement of known tcDNA quantities dissolved in the respective tissue lysates of mock-injected animals.
RNA analysis
Total RNA was isolated from snap-frozen muscle tissues using TRIzol reagent according to the manufacturer’s instructions (ThermoFisher Scientific, USA).
To visualize exon-skipping levels, RNA was used for reverse transcription polymerase chain reaction (RT-PCR) analysis using the Access RT-PCR System (Promega, USA) in a 25 µL reaction using the external primers Ex 20Fo (5′-CAGAATTCTGCCAATTGCTGAG-3′) and Ex 26Ro (5′-TTCTTCAGCTTGTGTCATCC-3′). The cDNA synthesis was carried out at 45°C for 45 min, directly followed by the primary PCR of 29 cycles of 95°C (30 s), 55°C (1 min) and 72°C (2 min). A total of 1.5 μL of these reactions were then re-amplified in nested PCRs by 24 cycles of 95°C (30 s), 55°C (1 min) and 72°C (2 min) using the internal primers Ex 20Fi (5′-CCCAGTCTACCACCCTATCAGAGC-3′) and Ex 26Ri (5′-CCTGCCTTTAAGGCTTCCTT-3′). PCR products were analyzed on 1.5% agarose gels with ethidium bromide.
Exon 23 skipping levels were quantified by ddPCR QX 200 (Biorad). Briefly, 1 µg of RNA was retro-transcribed using Luna Script RT SuperMix kit (New England Biolabs) but only 100 ng of cDNA were used to perform the ddPCR analysis. The Supermix for probes (no dUTP) was added to cDNA with probes designed to recognize the exon 23–24 junction and exon 22–24 junction. Droplets were generated thanks to the QX200 Droplet Generator according to the recommendations of the manufacturer. After sealing the plate and running PCR, the plate was read by the QX200 Droplet Reader.
Quantification of markers of fibrosis, inflammation, and angiogenesis was performed by quantitative PCR (qPCR) on the same cDNA using the iTaq Universal SYBER Green supermix (Biorad). The sequences of all qPCR primers and probes are provided in Supplementary Table S1.
Western blot analysis
Muscle sections were collected during cryosection and then homogenized with the Precellys24 (Bertin Instruments, France) in RIPA buffer with 5% SDS and protease inhibitor. After denaturation and centrifugation, supernatant was collected and total protein concentration was determined with the BCA Protein Assay Kit (ThermoFisher Scientific, USA). For the Western blot, 25 μg of protein were loaded onto NuPAGE 3%–8% Tris-Acetate Protein gels (Invitrogen), following manufacturer instructions. Dystrophin protein was stained using Every Blot Blocking Buffer (Biorad). The membrane was labeled with NCL-DYS1 primary monoclonal antibody (NCL-DYS1; Novocastra, Newcastle, UK, dilution 1/1000) and hVin-1 primary antibody (Sigma, dilution 1/25,000), followed by incubation with a goat antimouse secondary antibody (IRDye 800CW Goat antimouse IgG, Li-Cor, Germany, dilution 1/2000). Bands were visualized using the Odyssey CLx system (Li-Cor, Germany) and quantification was done using the Empiria Studio software (Li-Cor, Germany) based on a standard curve made from pooled lysates from C57BL10 (WT) and mdx control for each tissue.
For myomesin-3 (MYOM3) detection, mouse sera were diluted at 1:20 before loading onto 3%–8% Criterion™ XT Tris-Acetate Protein Gel, following manufacturer’s instructions (Biorad, France). MYOM3 protein was stained using the iBindTM Flax Western Device (Fisher Scientific) and by incubating the nitrocellulose membrane with MYOM3 primary rabbit polyclonal antibody (MYOM3, Proteintech, Manchester, UK dilution 1/1000), followed by incubation with a goat antirabbit secondary antibody (IRDye 800CW Goat Antirabbit IgG, Li-Cor, Germany, dilution 1/2000). Bands were visualized using the Odyssey Imaging System (Biosciences, Lincoln, USA). Signal intensities in treated samples were normalized to total protein staining (Revert 700 Total Protein Stain (Li-Cor Biosciences GmbH, Germany), then quantified and normalized to signals from PBS control mice signals using the Image Studio software (Li-Cor, Germany).
Immunohistochemistry analysis
Sections of 10 µm at 120 µm intervals were cut and examined for dystrophin and laminin expression using the rabbit polyclonal antibody dystrophin (dilution 1:500; cat. number RB-9024-P ThermoScientific), which was then detected by Goat Antirabbit IgG (H + L), F(ab′)2 Fragment (Alexa Fluor® 488 Conjugate, dilution 1/500) or the rabbit polyclonal antibody laminin (dilution 1:200; Sigma ref: L9393-2ML), which was then detected by Goat Antirabbit IgG (H + L), F(ab′)2 Fragment (Alexa Fluor® 488 Conjugate, dilution 1/1000), respectively. Images were taken at equivalent exposure times and analyzed with ImageJ software.
Prior to Picro-sirus Red staining, frozen sections (10 µm) were allowed to air-dry for one h at room temperature and incubated in xylene for 10 min to prevent the overstaining of red fibers. Subsequently, the sections were hydrated in an ethanol gradient (100%, 80%, 40%) for 30 s each, followed by a 1-min incubation in dH2O. Thereafter, the sections were stained with Picro-sirius Red (Abcam AB246832) for 1 h. Following the staining procedure, the sections were rinsed twice for 2 min each with 0.1N HCl, then rapidly rinsed with dH2O. They were subsequently dehydrated in an ethanol gradient (70%, 80%, 100%) for 30 s, 30 s, and 1 min, respectively. The sections were then cleared with xylene for 2 min and cover-slipped with Vectamount® permanent mounting medium.
Images of the sections were captured using a Leica DFC7000 T camera head attached to a Leica DM IL microscope and analyzed using ImageJ 1.54d software.
Serum and urine analysis
Analysis of serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), bilirubin, creatinine, urea, and albumin levels were performed by the pathology laboratory at Mary Lyon Centre, Medical Research Council, Harwell, Oxfordshire, UK.
Urines were collected using metabolic cages in refrigerated tubes over 24 h. Upon collection, urines were aliquoted and frozen at −80°C for further analysis. Albumin and creatinine were measured using a Mouse Albumin ELISA kit (Euromedex) and Creatinine Assay kit (Biotechne), respectively. Finally, the Pierce BCA assay was used to assess the total protein concentration in urines.
Statistical analysis
All in vivo data were analyzed with the GraphPad Prism8 software (San Diego, California, USA) and expressed as means ± SEM. The “n” refers to the number of mice per group.
Group comparisons were performed using one and two-way analysis of variance (ANOVA) with repeated-measure comparisons when needed. To compare the overall effect of two treatments across the different tissues, the two groups were directly compared using a two-way ANOVA, and the P value of the treatment effect is indicated in the figure legend. The Kruskal–Wallis test was used to compare groups that do not follow a normal distribution (assessed with the Shapiro–Wilk test). Significant levels were set at *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Results
Combined ASO + 20E treatment improves functional outcomes in mdx mice
Adult mdx mice were treated with tcDNA-ASO aimed at skipping the Dmd exon 23 with or without 20-E for 8 weeks (Fig. 1A). At the end of the 8-week treatment, we first evaluated the effect of the combination of 20-E and ASO on the functionality of the restored dystrophin. We assessed the ability of treated mice to sustain continuous exercise over a long period of time using a treadmill exhaustion test. The mice were placed on the treadmill at a speed of 5 cm/s for a 3-min acclimatization period. Thereafter, the speed was increased to 1 cm/s every 30 s until the mice had reached their exhaustion point and could no longer continue. As previously reported, the distance ran by control PBS-treated mdx mice was significantly lower than WT mice (P < 0.01) (Fig. 1B). We observed a significant increase in the distance run in mice treated with the ASO alone (+37%, P < 0.05) compared with PBS but the improvement was even better in mice treated with the combined treatment (+44%, P < 0.01 compared with PBS). This was also the case for the maximum speed achieved by each mouse, where the performance of ASO-treated mice (combined or not) was not different from that of WT mice (Fig. 1C).
To further assess functional improvement, mice were tested on the inverted grid test, which assesses the strength in all four limbs for a maximum period of 120 s. The result of this test demonstrates a clear and statistically significant difference in the average time spent on the inverted grid between WT and mdx mice. WT mice exhibited a mean latency to fall of 76 s, while mdx mice only remained on the grid for an average of 12 s (P < 0.0001) (Fig. 1D). Regarding the treated groups, only the mice treated with both ASO and 20-E demonstrated a significant increase (P < 0.05) in the latency to fall when compared with the PBS group, although no statistical difference was detected between ASO and ASO + 20-E groups. Finally, TA muscles were subjected to a maximum specific force test and resistance to eccentric contractions. Comparing WT and mdx, the maximal specific force is reduced by 40% in mdx mice while the combined therapy significantly recovered 21% of this specific force (P < 0.01) (Fig. 1E). In PBS-treated mice, force decreases to 64.8% after 10 eccentric contractions, whereas WT and ASO + 20-E groups still have 88.8% and 79.2% of their force (P < 0.0001 and P < 0.01, respectively, compared with the PBS group, no statistical difference between ASO and ASO + 20-E groups) (Fig. 1F). These results suggest that combined 20-E and antisense-mediated exon skipping significantly improve functional outcomes in mdx mice.
20-E has no direct impact on exon-skipping and dystrophin restoration levels
To investigate whether 20-E treatment had a direct effect on the ASO treatment, we first evaluated the biodistribution of the ASO by quantifying the amount of ASO in muscle tissues as previously described. 23 No difference was observed in the quantity of ASO between muscles treated with ASO alone and those treated with ASO + 20-E (Fig. 2A). In line with these results, we found no difference in the levels of exon skipping after qPCR quantification (Fig. 2B, C) nor in the levels of dystrophin restoration (Fig. 2D, E). We indeed detected an average of 13% of dystrophin expression in the TA, triceps, diaphragm, and heart of both treatment groups as compared with WT levels. Dystrophin was correctly localized at the plasma membrane, as confirmed by immunostaining performed in the TA, triceps, diaphragm, and heart (Fig. 2F). Quantification of dystrophin immunostaining also confirmed no difference between ASO and ASO + 20-E treatments (Supplementary Fig. S1). These results suggest that 20-E treatment had no direct effect on ASO biodistribution, exon-skipping efficacy and dystrophin restoration. In order to investigate further the reason for the greater functional benefit of the combined ASO + 20-E treatment in comparison to ASO alone, we analyzed various pathophysiological features in the treated muscles.
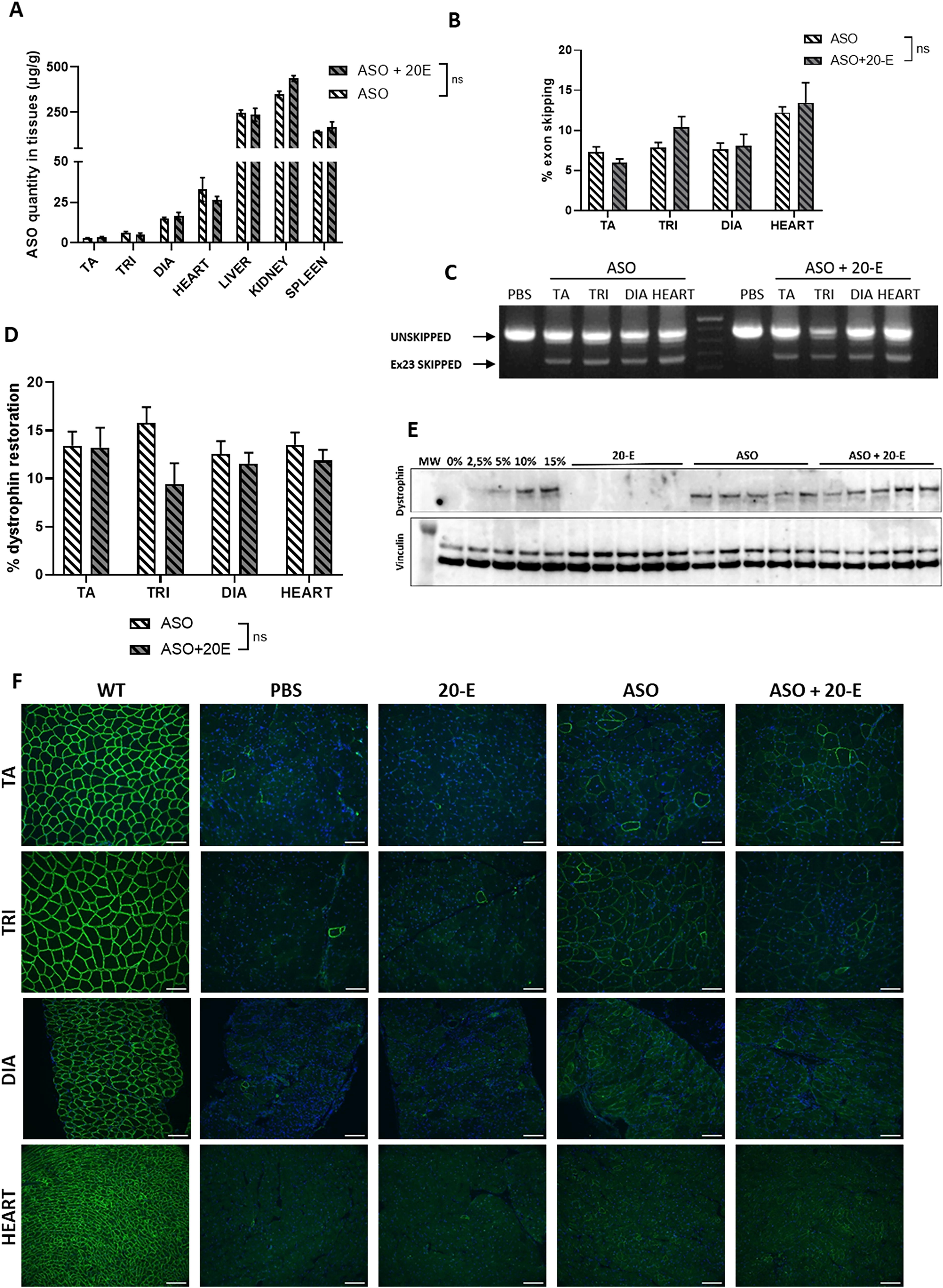
20-E has no impact on exon-skipping and dystrophin restoration levels.
Impact of 20-E on mdx pathophysiological features
One of the characteristic hallmarks of fibrotic changes in mdx skeletal muscles is the excess deposition of collagens and other ECM components. 24 We thus performed Sirius red staining (Fig. 3A, B) and quantified collagen accumulation in TA and diaphragm muscles from the different treatment groups. A comparison of WT and mdx mice revealed a 52.8% increase in collagen staining in the TA of mdx mice (Fig. 3C) and a 85% increase in the diaphragm (Fig. 3D). The administration of ASO and ASO + 20-E resulted in a significant reduction of collagen in the TA compared with PBS controls, with a decrease of 26.2% and 32.3% respectively, and a similar trend was observed in the diaphragm, with 19.6% and 18.4% reductions (Fig. 3C, D). Similarly, collagen staining was performed on the triceps and heart, yielding comparable results (Supplementary Fig. S2A–D). We further quantified the expression of fibrosis markers such as collagen I and connective tissue growth factor (CTGF), and of the inflammation marker tumor necrosis factor (TNF) by qPCR, and demonstrated a significant treatment effect for the ASO and ASO + 20-E groups in the TA and diaphragm (Fig. 3E, F).
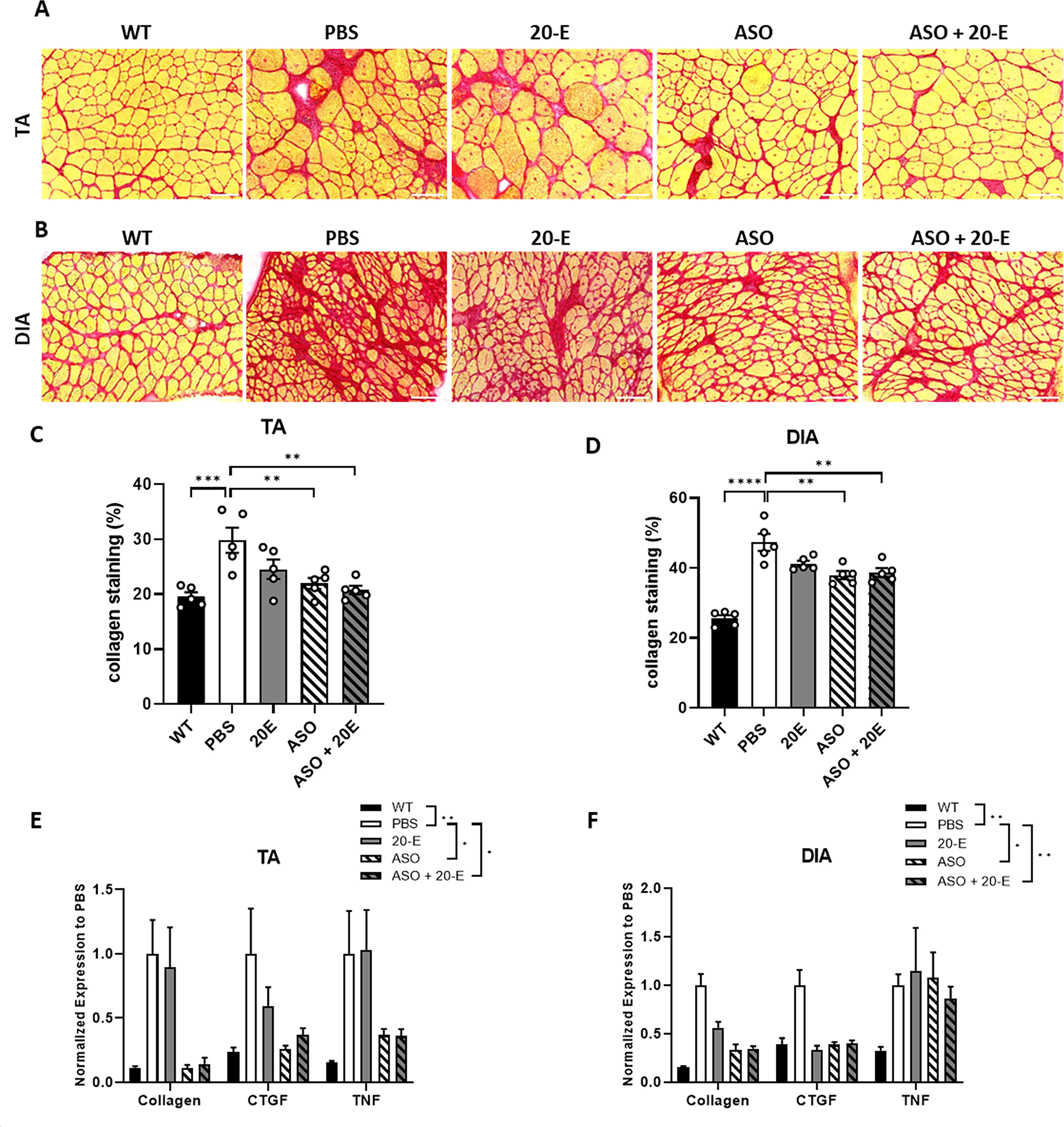
Effect of combined treatment on fibrosis and inflammation. Sirius red staining of TA
Given the previously described anabolic effect of 20-E on dystrophic muscles, we next examined the impact of the various treatments on the size of muscle fibers in the TA muscles of all groups of mice. The fiber size distribution profile in mdx muscles is markedly different from WT muscles as shown in Fig. 4A and B. TA muscles from mdx mice indeed exhibit a higher number of small fibers (<500 µm2) and a reduced number of large fibers compared with WT mice, indicative of strong regeneration (Fig. 4B). Interestingly, the distribution profile of muscles treated with ASO + 20-E appeared the closest to WT profile. ASO + 20E treatment significantly decreased the number of small fibers (<500 µm2) from 35% in control mdx mice to 17.6% in mice treated with the combined ASO + 20-E therapy in TA muscle (P < 0.05) (Fig. 4C). Conversely, the number of larger fibers with a diameter between 1500 and 2000 µm2 increased, exhibiting a distribution pattern analogous to that observed in WT mice (Fig. 4D). A comparable trend was observed in muscles treated with the ASO alone; however, the difference was more statistically significant for the combined therapy. The overall mean area of muscle fibers which is typically reduced in mdx muscles (1445 µm2 in mdx compared with 1911 µm2 in WT muscles), was also significantly increased only with the combined treatment (Fig. 4E). This was also observed in the diaphragm (Fig. 4F–H), highlighting the positive impact of the ASO + 20-E treatment on dystrophic histopathology.
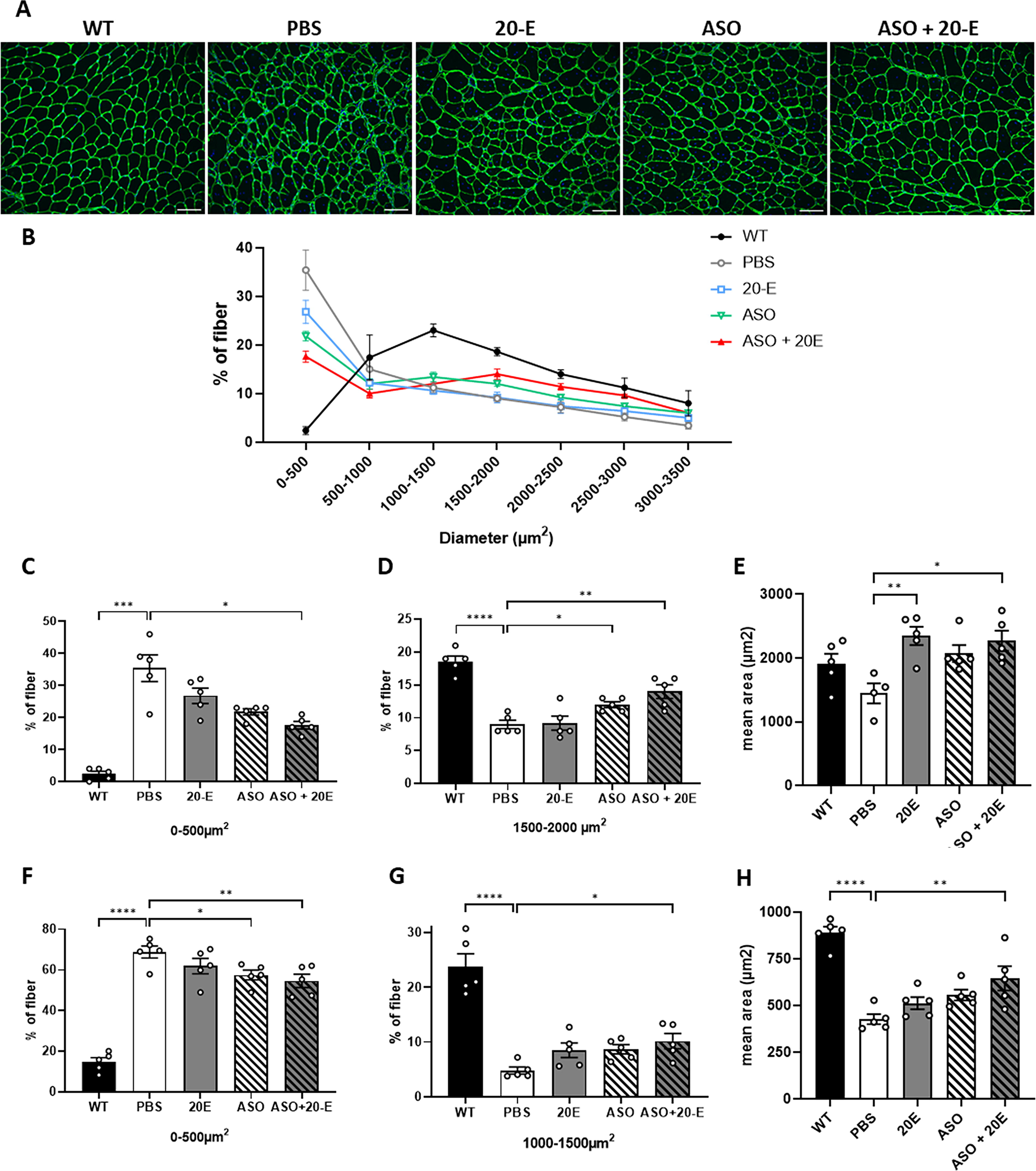
Impact of 20-hydroecdysone on histopathology.
Combined ASO + 20E treatment improves dystrophic biomarkers and is well tolerated
We next evaluated the levels of various known dystrophic biomarkers such as creatine kinase (CK) and MYOM3 in the serum of treated mice. The concentration of CK and MYOM3 in mdx mice is 8.2- and 83.3-fold higher, respectively, than in WT mice (P < 0.05, P < 0.0001) (Fig. 5A, B). The administration of ASO and ASO + 20-E resulted in a significant reduction in serum CK concentration (Fig. 5A). A comparable outcome was observed with regard to the concentration of MYOM3 (Fig. 5B). Given that these biomarkers are indicative of muscle damage, a reduction in their levels suggests that muscles are less impaired after both ASO and ASO + 20-E treatments.
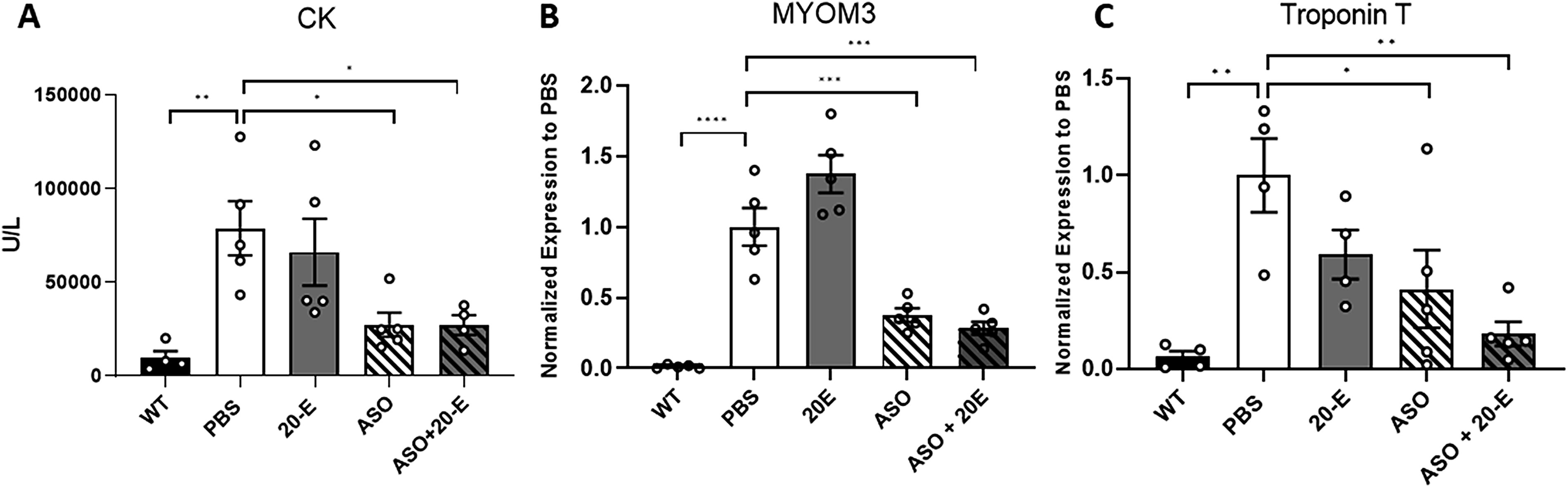
Impact of 20
Other markers of the dystrophic phenotype can be measured such as troponin T (TnT), which is a marker of regeneration in striated skeletal muscle. 25 In mdx mice, this marker is expressed 16.6-fold more than in WT mice (P < 0.01) (Fig. 5C). ASO treatment resulted in a notable reduction in TnT expression by 59% (P < 0.05) and the combined treatment of ASO + 20-E demonstrated an even more pronounced effect, leading to a significant decrease in the amount of TnT mRNA by 82% compared with PBS-treated mice (P < 0.01), although not statistically different from ASO treatment (Fig. 5C).
Serum and urine albumin are biomarkers of kidney damage or acute kidney injury that are elevated in mdx mice in comparison with WT mice. 26 As illustrated in Fig. 6A and B, albumin levels are significantly elevated in both serum and urine of mdx mice compared with WT mice. The ASO treatment, as well as the combined treatment, resulted in an improvement in the quantity of albumin present in the serum, demonstrating a reduction of 10.4% and 9.4%, respectively (Fig. 6A). Urinary albumin levels on the other hand were only significantly improved in the presence of 20-E (alone or with ASO) (Fig. 6B), demonstrating the ability of 20-E to improve renal function in mdx mice.
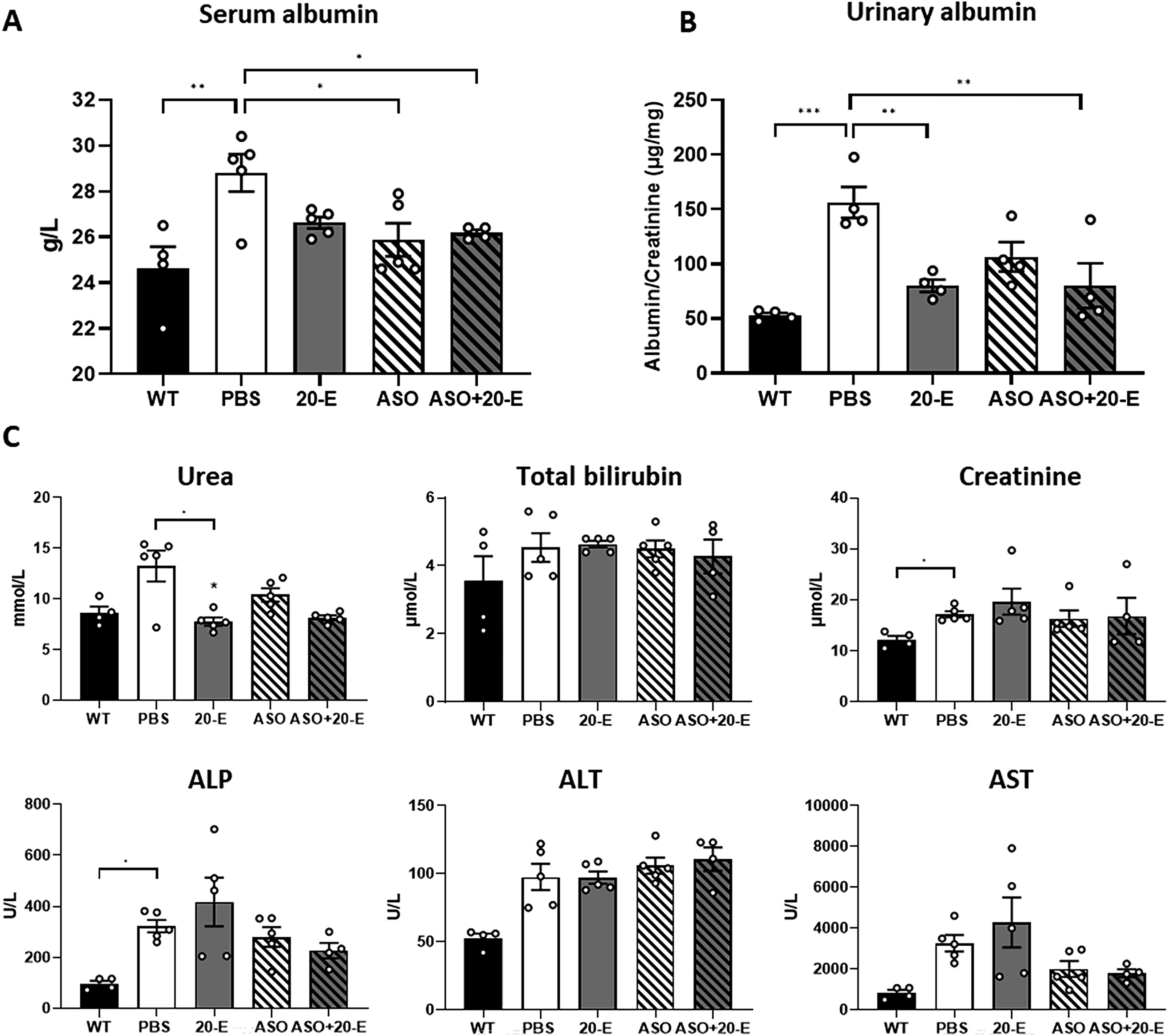
Serum and urine biochemistry. Quantification of albumin in serum
Finally, to ensure that the combination of 20-E with ASO did not induce any specific toxicity, we analyzed the serum levels of various general biomarkers in mdx mice following the different treatments. Quantification of serum urea, albumin, ALP, bilirubin, ALT, and AST revealed no significant changes both in ASO and ASO + 20-E treated mice compared with PBS mice (Fig. 6C).
Discussion
In this study, we used the oral administration of 20-E at a dose of 50 mg/kg/day in combination with an exon-skipping approach using tcDNA-based ASO, aiming at skipping the exon 23 of dystrophin pre-mRNA. The combined treatment was found to ameliorate dystrophic pathology in mdx mice, which was particularly highlighted using functional tests such as the treadmill and inverted grid tests, but also when assessing direct muscle electrophysiology. Our initial hypothesis was that 20-E could potentially enhance the delivery of oligonucleotides to the muscles. Indeed, 20-E has been demonstrated to elicit effects comparable to those of angiotensin 1–7 via the Mas receptor.17,27 Angiotensin 1–7 is known to induce vasodilation, 28 thereby increasing blood flow to muscles. We thus hypothesized that like ang1-7, 20-E may influence vasodilation, thereby increasing the amount of tcDNA-ASO reaching the target tissues. However, quantification of ASO in ASO + 20-E-treated muscles showed no increase compared with ASO-treated muscles. We further confirmed that 20-E had no impact on vascularization by showing no increase in CD31 expression in 20-E treated muscles (Supplementary Fig. S3) in contrast to results previously shown in SMA mouse models. 22 In line with these findings, the percentage of exon skipping and levels of dystrophin restoration were not impacted by 20-E treatment. Therefore, the improvement in muscle function observed in the double-treated mice is not attributable to an increase in dystrophin expression, but rather to the effects of 20-E on the general muscle histopathology. 20-E previously demonstrated a reduction in fibronectin deposition in a model of proximal tubule epithelial cells, suggesting a potential impact on fibrosis, a prevalent phenomenon in mdx mice. Yet, following Sirius red staining and quantification of collagen, as well as CTGF and TNF expression, it appears that 20-E did not significantly enhance the effect of ASOs on fibrosis or inflammation in the TA and diaphragm as both single ASO and ASO + 20-E treatments had a similar positive effect on fibrosis and inflammation. One of the most notable effects of 20-E on the dystrophic histopathology is its capacity to normalize fiber size. We indeed observed that only the mice that received ASO + 20-E exhibited a notable reduction in the number of small fibers (with a diameter of <500 µm2) and an increase in the number of large fibers (with a diameter of between 1500 and 2000 µm2) in the TA. This anabolic feature correlates with the findings of a clinical trial on metabolic syndrome, whereby 20-E was shown to increase muscle mass by 2.5%.29,30 Given the well-documented effects of this steroid hormone, which may account for the functional benefit observed in mice treated with the combined treatment, 20-E may also improve the respiratory function of mdx mice. Previous work evaluating the effect of 20-E has indeed demonstrated an improvement in the airway structure and responsiveness in treated mdx mice. 22 Furthermore, a phase 2 clinical trial on patients with SARS-CoV-2 infection showed a reduction in the risk of respiratory failure and death in patients treated with 20-E. 20 It would therefore be interesting to investigate the potential of 20-E to synergize the effect of ASO on respiratory function in mdx mice in further studies.
20-E exhibited no signs of toxicity when assessing the various serum and urinary biomarkers. In fact, 20-E even demonstrated an improvement in renal function of mdx mice as evidenced by a reduction in albumin excretion. These findings are in line with the good safety profile of BIO101 previously demonstrated in preclinical regulatory studies revealing no genotoxic effects and No Observed Adverse Effect Levels at 1000 mg/kg in repeated dose toxicity studies in rats (26 weeks) and dogs (39 weeks). 18 The safety of BIO101 was further confirmed in a phase I single ascending dose study that led to the selection of doses for the subsequent clinical trials for sarcopenia and COVID-19. 31 Our study in mdx mice further suggests the good tolerability of 20-E when used combination with ASOs.
The therapeutic potential of ASO for DMD has been established many years ago and several ASOs have now even been approved in the USA and Japan.7–10 Yet the approved ASOs only restore low amount of dystrophin expression in DMD patient biopsies and the functional benefit remains marginal in patients, leaving much space for improvement. While ASO-based exon skipping remains one of the most promising therapy for DMD, many limitations still prevent ASO to reach their full potential in the treatment of DMD. Improving ASO delivery to muscles is among one of the most studied challenge and is being addressed both with alternative chemistries and novel conjugations. 12 One of this promising chemistry is the tcDNA, which has previously demonstrated unprecedented uptake in preclinical models of DMD13,23 and which is currently evaluated in a phase 1/2a clinical trial sponsored by SQY Therapeutics, in ambulant and nonambulant DMD boys (Avance1 trial NCT05753462). 15
Several other new generations of ASOs aiming at improving delivery to muscle tissues and facilitating endosomal escape have been developed in the recent years and are currently being evaluated in clinical trials for DMD, 12 including alternative chemistries developed by Wave Life Sciences (NCT04906460) and BioMarin (NCT06280209). Peptide-PMO conjugates such as Sarepta’s vesleteplirsen (NCT04004065) also showed improved exon skipping and dystrophin restoration but were recently discontinued due to safety concerns. Alternatives like PepGen’s PGN-EDO51 (NCT06079736) and Entrada’s cyclic peptide-based approaches are under clinical evaluation, with ongoing efforts to balance efficacy and toxicity. Additionally, transferrin receptor-targeting ASO conjugates from Avidity Biosciences and Dyne Therapeutics have demonstrated enhanced potential for systemic delivery to muscle, offering promise for achieving higher exon-skipping levels.32,33
Besides these recent advances, it is becoming increasingly evident that restoring dystrophin alone may not be sufficient to fully alleviate the dystrophic pathology and reach clinically meaningful outcomes in patients with DMD. It is thus crucial to consider combined therapies aiming at improving muscle histopathology alongside dystrophin restoration. The functional benefit observed in this study in mdx mice treated with ASO + 20-E provides evidence that a combined therapy of 20-E and antisense-mediated exon skipping may offer therapeutic benefits in DMD. This enhancement could enable ASO therapies, such as PMO, which are already in clinical use for DMD, to achieve more clinically meaningful outcomes, potentially leading to significant improvements in the quality of life for patients.
BIO101, the pharmaceutic grade formulation of 20-E, has already received orphan drug designation in both Europe and the United States for DMD and has previously demonstrated favorable pharmacokinetic and safety profiles, supporting its upcoming evaluation in a phase 1/2 MYODA clinical trial. 22 This forthcoming trial will assess the potential of BIO101 in patients with DMD, providing data on its effects as a standalone treatment. However, given that many patients are already receiving ASOs or are enrolled in trials testing new ASOs, our study demonstrating improved functional benefits following ASO + 20-E treatment highlights the potential of a future combined therapy that could be rapidly translated to the clinic.
Footnotes
Acknowledgment
The authors would like to thank the personnel of the platform 2CARE for taking care of the animals used in this work.
Availability of Data and Materials
The primary data for this study are available from the authors upon request.
Author Disclosure Statement
L.G. is cofounder of SQY Therapeutics, which produces tricyclo-DNA oligomers. T.T. is an employee of SQY Therapeutics.
Funding Information
This work was supported by the Institut National de la santé et la recherche médicale, the Association Monegasque contre les myopathies, the Paris Ile-de-France Region and the Fondation UVSQ. M.B. is the recipient of a MESRI thesis fellowship.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.