
Abstract
The Oligonucleotide Nonclinical Working Group (WG) of the European Federation of Pharmaceutical Industries and Associations conducted an industry survey to understand current practices and regulatory expectations for genotoxicity and carcinogenicity assessment of oligonucleotide therapeutics (ONTs), along with historical genotoxicity testing results. The survey, involving 29 pharmaceutical and biotechnology companies, revealed a consistent absence of genotoxicity across a diverse range of oligonucleotide classes and chemistries, consistent with previous observations. Despite the lack of genotoxicity, companies continue to follow standard testing guidelines, with only limited divergence. The survey data support the view that well-established ONT modifications can be considered “precedented,” in terms of negligible genotoxic risk. As such, further testing of new ONT candidates containing only precedented modifications is unwarranted, when defined criteria are met. Further, we propose a pathway for novel ONT chemical modifications to achieve precedented status. The survey results also indicate that alternative strategies for carcinogenicity assessment (e.g., single-species testing) can be accepted by regulatory agencies under certain circumstances. Overall, the survey findings underscore the need for a more tailored approach to the nonclinical safety assessment of ONTs, and the WG proposes development of supplementary questions for International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use S2(R1) guidance to reflect this broad industry experience.
Introduction
Oligonucleotide therapeutics (ONTs) represent a diverse group of drugs, both in terms of chemical modifications, delivery and targeting approaches, as well as the mechanism of action, or “class”.1,2 As a modality, they are synthesized chemically and very often contain modifications to the backbone and ribose sugars that improve their potency, drug metabolism and pharmacokinetics (DMPK), and safety properties.1,3 Although they differ from small molecules (e.g., in size, chemical composition, tissue half-life, and durability of pharmacodynamic effect), ONTs are currently regulated as small molecule “new chemical entities” in the absence of specific harmonized regulatory guidance for drug development. As such, companies often ensure that their nonclinical safety packages are aligned with the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) M3(R2), 4 including a full assessment of genotoxicity and carcinogenicity, in accordance with ICH S2(R1) and ICH S1B(R1), respectively.5,6
In 2017, the European Federation of Pharmaceutical Industries and Associations (EFPIA) established an Oligonucleotide Nonclinical Safety Working Group (WG), which is sponsored by the Preclinical Development Expert Group (PDEG). The published conclusions of an industry survey conducted by the WG on “nonclinical practices and regulatory expectations for ONT safety assessment” 4 indicate some variation in the perceived relevance of current safety-related ICH guidelines to the nonclinical safety assessment of ONTs. Although ICH M3(R2) (nonclinical safety studies for the conduct of human clinical trials for pharmaceuticals) was viewed favorably in its applicability, there was a consensus regarding the “lack of perceived relevance” of ICH S2(R1) (genotoxicity testing and data interpretation for pharmaceuticals intended for human use). Free-text responses within the survey questioned the need to run the standard battery of Good Laboratory Practice (GLP) genotoxicity tests on classes of ONTs for which nongenotoxic readouts (i.e., no DNA damaging potential) had been observed previously for multiple exemplars using established chemistries. This observation is in line with the principles outlined in the European Medicines Agency (EMA) Safety Working Party reflection article on “The assessment of genotoxic potential of antisense oligonucleotides”. 7 It also aligns very well with the findings of the Oligonucleotide Safety Working Group (OSWG) Genotoxicity subcommittee, who published an extensive cross-industry analysis in 2016. 8 They reported that of 32 development programs, no ONT drug candidate in any established class or chemistry was positive in the standard battery of genotoxicity tests. Since this assessment was published, additional ONTs using alternative chemistries, including locked nucleic acid (LNA) antisense oligonucleotides (ASOs), and N-acetyl galactosamine (GalNAc)-targeted small interfering (si)RNAs, have been reported to be nongenotoxic in the ICH S2(R1) battery.9,10
The body of peer-reviewed published data8–11 and industry opinion 4 support the notion that continued testing of ONT candidates of established classes (e.g., ASOs and siRNAs) that contain well-characterized modifications is highly unlikely to yield positive (i.e., DNA damaging) results in GLP genotoxicity tests. To our knowledge and based on published literature, there is no evidence of direct DNA interaction and consequent DNA damage with any ONT. However, genotoxicity could potentially be driven by the primary or secondary pharmacology of an ONT. A theoretical mechanism of genotoxicity could be envisioned if the therapeutic target, or a confirmed hybridization-dependent off-target mRNA for example, had a significant role in DNA repair or cell cycle control, but this possibility is usually excluded by an adequate assessment of the target biology and selectivity evaluation (i.e., no biologically meaningful off-target interactions), without conducting formal genotoxicity testing. Another theoretical genotoxicity risk identified for ONTs is that of triplex DNA formation, 7 although it is relatively straight forward to design out/discharge this risk without the need for experimental approaches. 8 Thus, where a company is developing an ONT candidate within a well-established class, containing only “precedented” modifications from a genotoxicity hazard perspective, and other relevant theoretical risks are discharged adequately, this WG advocates for waiving the requirement for genotoxicity testing in the nonclinical safety package used to enable clinical development based on the data and the negligible genotoxic risk to patients.
Despite the consistent absence of genotoxicity findings outlined above, current ICH regulatory guidance does not outline a scenario to support companies for diverging from standard genotoxicity assessment of ONTs.6,12 Although the EMA has previously questioned the need for continued genotoxicity testing of phosphorothioate (PS) ASOs, the Japanese Pharmaceuticals and Medical Devices Agency (PMDA) more recently published guidance on the nonclinical safety evaluation of ONTs, 13 which states that the genotoxicity evaluation of “chemically modified nucleic acid drugs” should “refer to ICH S2(R1)”. There was some evidence of a more tailored approach being acceptable to the Food and Drug Administration (FDA), given what is detailed in their guidance on Nonclinical Testing of Individualized Antisense Oligonucleotide Drug Products for Severely Debilitating or Life-Threatening Diseases. This draft guidance clearly states that “genotoxicity assessment is generally not warranted” for ASOs containing well-characterized chemical modifications with substantial nonclinical information and clinical experience. 14 However, the WG recognizes that the risk:benefit assessment is quite different for ultrarare, severe, and life-threatening indications (where the anticipated risk to humans is low given that the medicine would be administered to only a few patients) versus more prevalent diseases with larger patient populations and potentially different risk tolerance. The FDA has very recently (November 2024) posted (for public consultation) a new “draft guidance for industry on the nonclinical safety assessment of ONTs.” 15 This guidance indicates that the FDA is indeed planning a more flexible approach to genotoxicity assessment for ONTs. Where a candidate belongs to a class of ONT that is “well-characterized for genotoxicity,” the FDA is open to an assessment being informed by the characteristics of the class (provided that the size, structure, modifications, and impurity profile are consistent, with the only difference being the base sequence). 15 The EFPIA WG very much welcomes this more nuanced position communicated by the FDA, who acknowledge the very low genotoxicity risk for the ONT modality (based on published literature and the experience within the agency). 15
This article reports the results of an industry survey conducted by the WG to obtain additional details on the approaches to genotoxicity evaluation of ONTs and collective testing results. Additionally, the survey afforded the opportunity to gather information on carcinogenicity strategy for ONTs, given the relatedness of this topic to genotoxicity assessment. These data should provide greater clarity on the relevance of genotoxicity testing for ONTs and the point at which risk can be considered discharged for a given chemical modification or class. Based on the results of the survey, we suggest an ICH Q&A to address whether revision of ICH S2(R1) to cover specific considerations for ONT drugs is warranted. In addition, these findings may also provide valuable input to the ICH new topic proposed jointly by the EMA and PMDA on “Nonclinical Safety Studies for Oligonucleotide-Based Therapeutics”, for which an expert working group has now been formed.16,17 A provisional timeline for the ICH S13 guidance being available by November 2027 was communicated in the recently ratified final concept article. 18
Methods
Eleven EFPIA members assigned one or more representatives (see Author List and Acknowledgments), with experience in the nonclinical safety assessment of ONTs and/or genotoxicity testing. Additionally, representatives from the nonclinical safety discipline of three biotechnology companies that work on therapeutic ONTs joined as non-EFPIA members to ensure a diverse representation of opinions and expertise across the industry (see Author List and Acknowledgments).
During the design of the survey by the WG, feedback on the content and structure of the questions was received from PDEG and recognized industry Key Opinion Leaders outside of the WG (see Acknowledgments). The survey was created using a commercial online application (Survey Monkey [SM]) and administered by the EFPIA secretariat (see Acknowledgments) to secure blinding of the respondents’ identity. WG member companies were also invited to participate. A pilot version of the SM questionnaire was tested among the WG members to check for question relevance, logical operators, overall acceptability of information volume/time to complete, and typographical errors.
For the purposes of the survey, only ONT classes that target RNAs or proteins to exert their primary pharmacology were considered “in scope.” This included ONTs formulated in complex delivery systems (e.g., lipid nanoparticles [LNPs]) and as targeted conjugates. ONTs that target genomic DNA were “out of scope” as the WG considered the theoretical risk for genotoxicity associated with such approaches could be different. Similarly, RNA and DNA vaccines and gene therapies as defined in Footnote #1 in the FDA Guidance for Industry on “Human Gene Therapy Products Incorporating Human Genome Editing” were considered out of scope, as these entities are regulated as biologics. 19
The primary focus of the survey was on gathering data and industry experience related to the assessment of genotoxicity of ONTs in definitive tests (i.e., majority GLP tests) as per ICH S2(R1). An additional section containing questions on the related topic of carcinogenicity testing was also included. The survey did not ask for commercially sensitive information, such as therapeutic target, sequence, specific indication, or compound name/number. Participants were encouraged to contribute as much information and specific project examples as possible, to enable the fullest picture to be developed. However, participants were free to exclude from their survey responses any sensitive information related to specific assets that could be identified easily.
The final version of the survey was structured as follows:
Background, level of experience, and ONT classes covered by respondents:
Company type and broad disease areas they are working in. Size and type of ONT development phase project portfolio. ONT classes covered, including delivery systems used. Strategy for genotoxicity evaluation with only GLP studies considered:
Genotoxicity tests used with oligonucleotide class and chemistries specified. Approaches for confirming test item exposure in vitro and in vivo. Examples of bespoke or non-GLP investigations. Investigations of genotoxic (i.e., positive) results. Managing triplex formation risk. Other experience and approaches:
Regulatory interactions. Impurity/degradant evaluation. Carcinogenicity assessment strategy. Optional case studies: Free-text options allowed respondents to share specific experience on:
Positive genotoxic findings. Testing waiver requests.
Thirty-nine companies (including those of the WG members) operating in the United States, Europe, or Japan were invited to participate in the survey between 20 September and 11 November 2022. Single-point contacts working at each company collated information/data and completed the survey response on behalf of their employer. In the interests of preventing duplicate responses, where candidate ONTs were/are being developed as part of an alliance between two or more companies, it was made clear that the “current sponsor” should include those examples in their responses. The results were analyzed, discussed, and summarized by the WG members. The anonymized raw data were shared directly with survey participants through the EFPIA secretariate in December 2022, which was a commitment made by the WG. An Excel version of the final raw data file is included in the Supplementary Data S1 of this article (rawdata.xlsx), along with the survey and case study forms for reference (surveyandforms.pdf).
Results
Twenty-nine anonymized completed questionnaires were received, equating to 74% of the companies invited to participate. No case study forms detailing “positive genotoxic findings” or “testing waiver examples” were received. Although two responding companies indicated that they were yet to conduct GLP genotoxicity studies for ONTs in their portfolio, the WG took the decision to include their responses to maximize the perspectives collected on questions related more to overall strategy.
Respondent statistics
The majority of respondents (55%) were “Large Pharma,” with 38% “Biotech” and 7% “Mid Pharma” (Table 1). Note: The authors did not define these categories within the survey, leaving it the respondents to “self-classify.” Approximately half of the respondents had small ONT portfolios, and only four companies reported large ONT portfolios (Table 1). Three respondents did not have ONT projects in GLP development; however, they were included in the total when deriving percentages. Just over half of respondents had projects for “rare diseases,” 14% for oncology and the majority (86%) for “other” indications. The most common ONT classes were siRNAs and RNase H-dependent ASOs (Fig. 1), with five Large Pharma companies reporting experience with both of these ONT classes. Four or fewer companies reported genotoxicity testing experience for each of the steric blockers, antagomir ASOs, RNA-editing, or CpG/immunostimulatory ONT classes. No experience was reported for miRNA mimics, DNAzymes, or aptamers (Fig. 1).
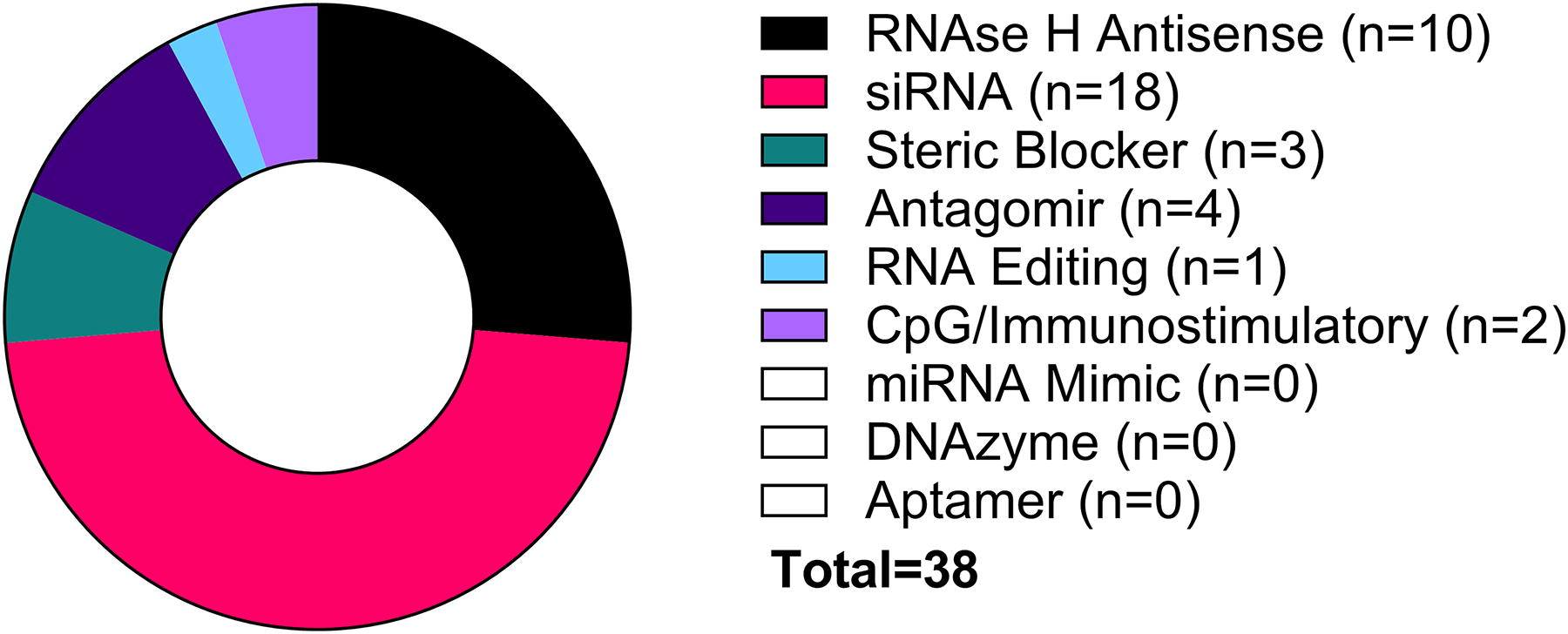
Which oligonucleotide mechanisms do you assess for genotoxicity? Respondents were able to select multiple options (RNase H = 10; siRNA = 18; Steric Blocker = 3; Antagomir = 4; RNA editing = 1; CpG/Immunostimulatory = 2), with none having development experience with miRNA mimic, DNAzyme, or aptamer classes.
Background Information of Responding Companies
The authors did not define these categories within the survey, leaving it to the respondents to “self-classify.”
Three responding companies had not progressed ONT projects through GLP genotoxicity testing. The percent figures expressed relate to the proportion of all 29 responding companies in this table.
Companies were able to give multiple answers to the question regarding therapeutic areas they were active in, hence the percent total exceeding 100%.
GLP, Good Laboratory Practice; ONT, oligonucleotide therapeutics.
Nearly all companies have been using aqueous solutions for formulating their candidate ONTs, with a much smaller proportion also having experience using complex delivery systems (Table 2). The majority of companies use conjugated ONTs for targeted delivery in at least one of their projects, although almost a quarter of respondents use nontargeted delivery (Table 2). To maintain respondent anonymity, further details on targeting methods were not collected as part of the survey.
Formulation and Delivery Approaches
28 (of 29) companies responded to this question.
All these companies also used simple aqueous formulation approaches.
Company did not use other approaches for formulation.
Genotoxicity evaluation approaches
All companies with ONT projects in development confirmed that they conduct GLP genotoxicity testing on candidates for nononcology indications (see raw data file in Supplementary Data S1). Of the in vitro tests outlined in ICH S2(R1) standard battery, the Ames or bacterial reverse mutation test, mammalian chromosomal aberration, and mammalian micronucleus tests (MNTs) were the most commonly reported (Fig. 2). The mouse lymphoma Tk test has been employed less frequently. For in vivo testing, the rodent MNT is the overwhelming favorite, with just single companies having evaluated at least one candidate ONT in an in vivo chromosomal aberration test (a steric blocker ONT) or comet test (an antagomir ONT) (Fig. 2).
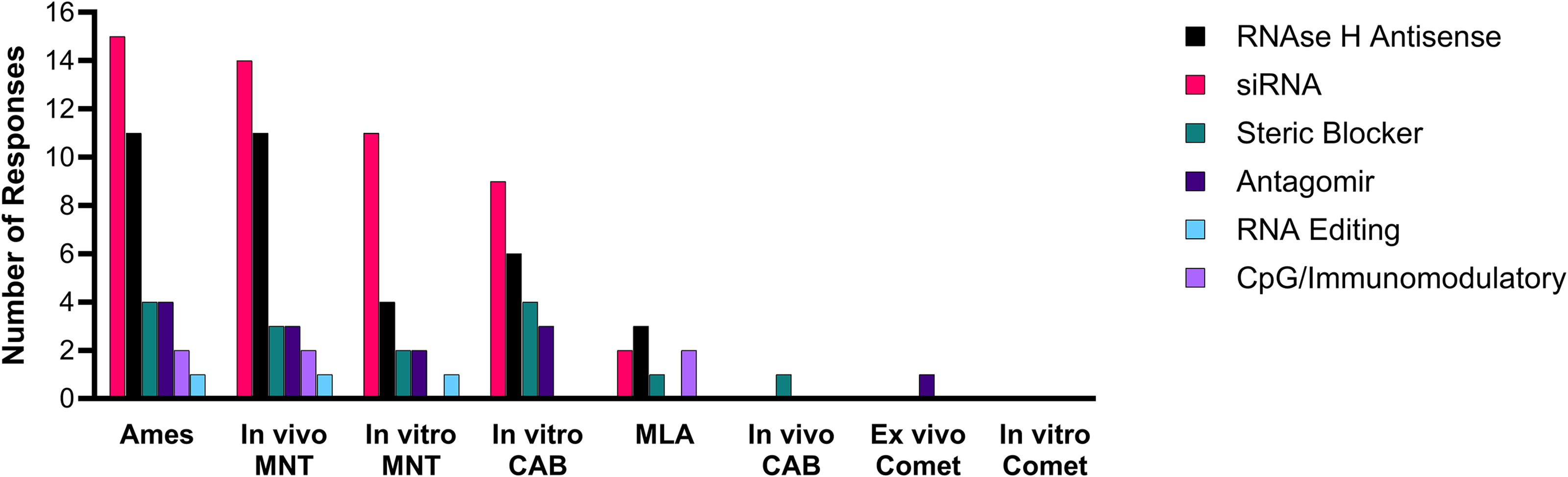
Which GLP tests have you used to characterize the genotoxic potential of each oligonucleotide mechanism? CAB, chromosomal aberration assay; GLP, Good Laboratory Practice; MNT, micronucleus test; MLA, mouse lymphoma Tk6 assay.
The survey also sought to understand whether companies had diverged from ICH S2 (R1) guidance, 6 for example, by undertaking nonstandard tests to characterize the genotoxicity potential of an ONT candidate. Three companies (11%) provided examples, two of which indicated they were addressing potential safety concerns for the therapeutic target, one citing an RNA-editing mechanism. The third company conducted a nonstandard genotoxicity test (specifically, the gH2AX test 20 ) to assess a confirmed hybridization-mediated off-target effect that carried a theoretical genotoxicity risk, which was discharged.
Chemical modifications tested—no evidence of genotoxicity in GLP and non-GLP tests
A key finding of the survey was that, when asked whether they had ever had a positive signal for an ONT candidate in any genotoxicity test, whether GLP or non-GLP, all 27 companies that answered this question responded in the negative, that is, with a “no” (note: the two companies that did not respond to this question had also indicated they had no ONT candidates that had been assessed in GLP genotoxicity tests at the time the survey was conducted). The rationale for non-GLP testing being included in this question was to maximize the chances of uncovering the experience of companies observing genotoxicity with ONTs. If an ONT candidate was shown to be genotoxic in an early test/screen, it would very likely be deselected and not progress to GLP testing. Due to the way data were captured, it is not possible for the WG to derive the total number of ONTs that tested negative for genotoxicity across the various in vitro and in vivo tests. We are able to report only on the number of different ONT candidates in which a given modification had been tested.
Respondents were asked to indicate which ONT backbone, ribose sugar, and base modifications they had evaluated in GLP genotoxicity tests within the context of individual candidates using predefined options (refer to Fig. 3 for definitions of abbreviations). A “free-text” response option was also included to allow companies to submit data for modifications not listed in the survey (with one company doing so). To simplify data acquisition, the tests were categorized as (1) Ames = GLP bacterial mutation test, (2) in vitro = any mammalian cell in vitro GLP genotoxicity test, and (3) in vivo = any in vivo rodent GLP genotoxicity test.
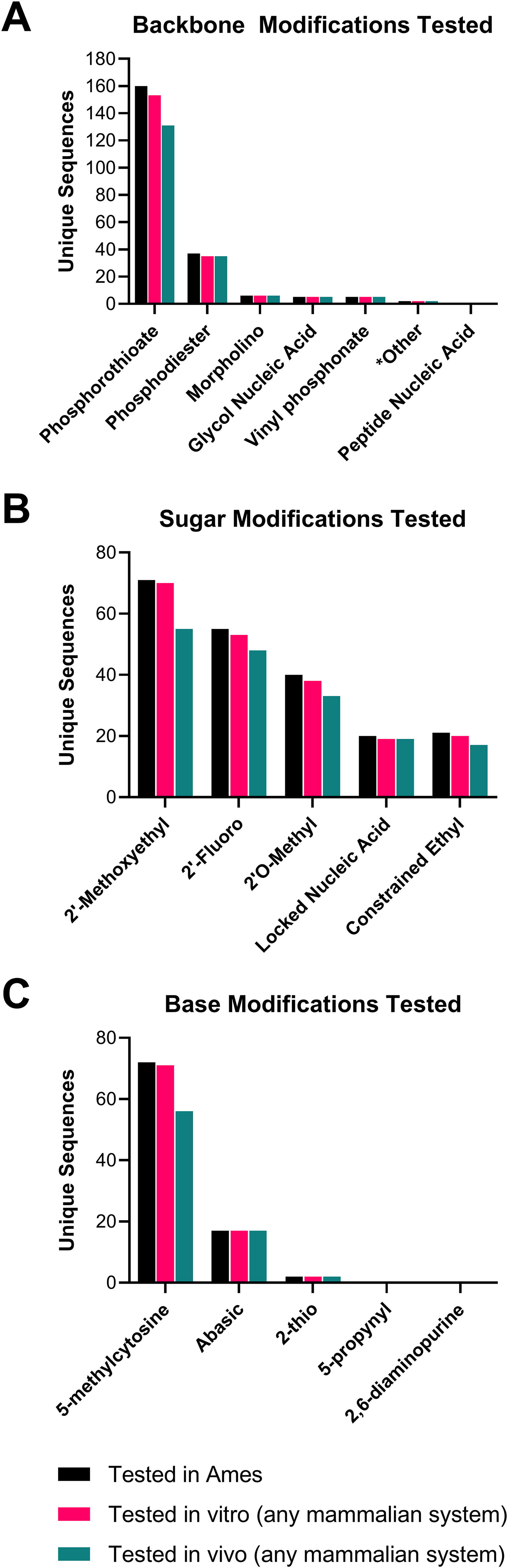
Backbone
For backbone modifications (Fig. 3A), the PS linkage was predictably the most frequently tested (across >130 different candidates in the Ames, in vitro, and in vivo). Morpholino, glycol nucleic acid (GNA), and vinyl phosphonate (VP) backbones have all been assessed in ≥5 different candidates, and two candidates with the phosphoryl guanidine (PN) modification have been evaluated. Across responding companies, there were no examples of the peptide nucleic acid (PNA) backbone modification being assessed in GLP genotoxicity tests. Of the sugar modifications (Fig. 3B), 2′-methoxyethyl (2MOE), 2′-fluoro (2F), and 2′-O-methyl (2OMe) have been assessed on >30 independent occasions each, and LNA and constrained ethyl (cEt) on >15 occasions each. For base modifications (Fig. 3C), 5-methylcytidine (5mC) and abasic residues have been evaluated most frequently, with >50 and >15 different candidates, respectively. The 2-thio base modification was tested in two candidates, and no responding company had evaluated 5-propynyl or 2,6-diaminopurine bases in GLP tests at the time of the survey.
Critically, responding companies confirmed that 100% of ONT candidates (and by association, the backbone, sugar, and base modifications contained within them) had all tested negative in the GLP in vitro and in vivo standard battery of tests for genotoxicity (see raw data file in Supplementary Data S1). Given the findings outlined above, the following question was considered redundant and elicited no responses: “Where a sequence(s) was identified as genotoxic, do you think a specific nucleotide/nucleoside was responsible?”
Companies were asked if they had tested individual monomer modifications for genotoxicity. Only two indicated they had, with phosphodiester (PO) and PS backbone and 2MOE ribose modifications confirmed as negative in the standard test battery (Table 3) when tested.
Chemical Modifications Confirmed as Negative in GLP Genotoxicity Tests as Monomers
Tested by one company.
Tested by two companies.
PO, phosphodiester; PS, phosphorothioate.
Approaches for targeting platforms and complex delivery agents
Of the 20 companies employing targeted delivery such as GalNAc conjugation (that answered relevant follow-on questions), all confirmed testing the intact drug candidate. A single company had also tested each component (i.e., ONT, linker, and targeting moiety) separately; none had evaluated a targeting platform (i.e., linker attached to targeting moiety) alone. When asked whether in vivo assessment was tailored to the targeted tissue, 18 companies indicated defaulting to standard tissue assessments (e.g., bone marrow in the rodent MNT). Reasons for this approach included “alignment to regulatory guidance” and “exposure of the bone marrow to GalNAc-targeted ONTs is shown in the literature.” A single company indicated conducting the liver comet test for GalNAc-targeted ONTs (refer to raw data file in Supplementary Data S1).
Only four companies answered a question on approaches used for assessing the genotoxicity of ONTs formulated in complex delivery agents. Three companies confirmed testing the delivery agent formulated with the candidate ONT (i.e., the drug product). Two of these companies tested only drug product and not the ONT alone. Two companies had tested each individual component within the delivery agent separately, with one also confirming evaluation of all components together on a single occasion. No respondents indicated adopting a “platform-based approach” by testing the delivery agent in the context of an exemplar ONT (refer to raw data file in Supplementary Data S1).
Confirming ONT exposure in genotoxicity tests
The survey explored approaches taken to confirm exposure of the genotoxicity test systems to the test item/ONT (Table 4). Testing up to the ICH S2 (R1)-defined limit concentrations/doses 6 was by far the most common approach, although five companies also rely on published data to infer exposure. One company measured exposure for a single exemplar oligonucleotide (their only approach), and one company measured exposure for each ONT candidate.
General Approaches Taken to Confirm Exposure to ONTs in Genotoxicity Tests
Companies were asked to select all scenarios that applied, and some adopt multiple approaches.
Five of 29 companies responding to the survey did not answer this question.
Single company, but also uses published data and testing up to ICH S2 (R1) limits 6 to justify approach.
Single company, which does not rely on any other approach.
Of the nine companies that generate experimental data to confirm exposure for in vitro tests (Table 5), most did so by demonstrating cytotoxicity in the in vitro test system. Only two indicated measuring intracellular concentrations of the ONT candidate although, as mentioned above, one of these companies takes a platform-based approach by measuring the intracellular concentration of one representative exemplar oligonucleotide. No company evaluates target engagement in vitro as a means of confirming exposure.
Technical Approaches Used to Confirm Exposure in In Vitro Genotoxicity Tests
Companies were asked to select all scenarios that applied; none selected multiple approaches.
Nine companies responded to this question.
Fourteen companies responded to the question related to how exposure is confirmed in vivo (Table 6), with three adopting multiple approaches. Measuring systemic exposure was the most common, with fewer companies having assessed exposure in the same organ that the genotoxicity endpoint was assessed in, or by demonstrating dose-limiting toxicity. None measured target engagement, consistent with in vitro test responses.
Technical Approaches Used to Confirm Exposure in In Vivo Genotoxicity Tests
Companies were asked to select all scenarios that applied; two companies assess both ONT systemic exposure and tissue levels in the organ the genotoxicity endpoint is tested in. A single company additionally demonstrates dose-limiting toxicity.
Fourteen companies responded to this question.
Assessing the potential risk of triplex formation
Companies were asked how they discharge the theoretical risk of triplex DNA formation, as previously related to ONTs.7,8 Of the 26 companies that responded to this question, approximately two-thirds indicated not assessing this risk at all, with the remainder conducting a paper-based assessment that may include an assessment of sequences associated with triplex formation (Table 7). None of the responding companies took an experimental approach to discharging this risk.
Determining the Potential Risk of Triplex Formation for ONT Candidates
Twenty-six companies responded to this question; no companies selected multiple options.
Regulatory interactions regarding genotoxicity testing
Twenty-seven companies answered the question “have you ever requested a waiver for genotoxicity assessment of an ONT?” The majority (25) answered “no” and only two companies indicated that they had requested a waiver (refer to raw data file in Supplementary Data S1). Unfortunately, these two companies did not submit case study forms to provide more information on their examples. Separately, two companies indicated having submitted nonclinical safety packages to support first-in-human (FIH) studies that did not contain genotoxicity data (excludes oncology projects). One of these two companies had also submitted a genotoxicity package that was non-compliant with ICH S2(R1) 6 to support FIH studies. Neither company was asked by regulatory agencies to “backfill” missing genotoxicity testing, although one of them elected to conduct a full genotoxicity battery prior to Phase 2 clinical trials (as per ICH S2[R1] 6 ).
Impurity and degradant evaluation
When asked whether genotoxicity testing was performed to support impurity qualification for ONT development, the majority of respondents answered “never,” with a much smaller proportion (4 of 22) conducting it on a case-by-case basis and one company conducting it for every project (Table 8).
Genotoxicity Testing of ONT-Related Degradants or Impurities
27 companies answered this question.
Companies were able to elaborate on the circumstances under which they conducted this testing. Two of which did, indicating:
“Synthetic intermediates that were potential degradants—linked to route of production. Identified during the routine genotoxicity risk assessment and concerns from an occupational toxicology perspective. Tested eight in total and all were negative in Ames.” The respondent also confirmed this response pertains to an RNase H ASO candidate. “We have not conducted any to date, but we would consider an Ames test if we received a positive in silico flag and did not have an adequate control strategy in place.”
Carcinogenicity strategy
Of the 28 companies responding to the question “Have you conducted rodent carcinogenicity studies on ONTs?” 11 (39%) had to date (Fig. 4). This subset of companies was further asked to specify which of four approaches to carcinogenicity testing they had undertaken, being asked to select all that applied in terms of their experience. Ten (of 11) had conducted studies in two rodent species, with 7 conducting studies as a postmarketing commitment and 5 achieving a partial waiver to conduct the assessment in a single species only (Fig. 4). One company indicated that they had received a full waiver from one health authority, based on a “weight of evidence” approach for a locally administered ONT in a severe and life-threatening indication.
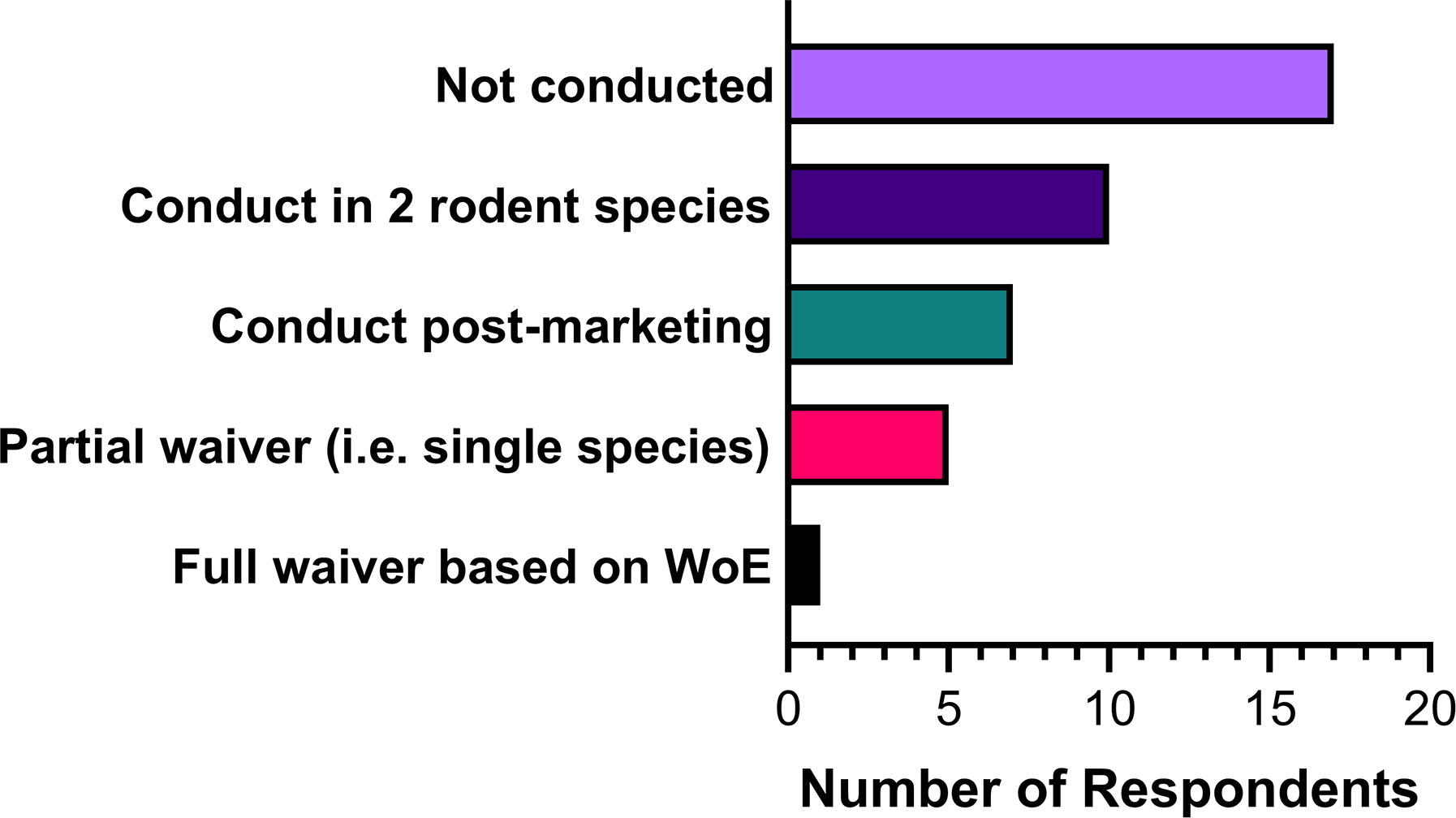
Carcinogenicity testing experience for oligonucleotides and strategy taken by companies who had experience (n = 28).
In terms of the specific approaches to testing, almost all companies with experience conducting carcinogenicity studies on ONT candidates had conducted testing in both rat and mouse, with the 6-month transgenic mouse study conducted more frequently than the 2-year mouse study (Table 9). A smaller proportion had run a mouse 6-month transgenic study only, and no companies had adopted a “rat only” approach. However, companies were reporting only on studies conducted to date; thus, the survey results do not reflect future studies required to meet regulatory expectations.
Types of Rodent Carcinogenicity Packages Conducted for ONT Candidates
Eleven companies answered this question, with some selecting multiple responses.
Use of a rodent surrogate (also known as an animal-active analogue) may be warranted in order to assess the carcinogenic potential related to on-target pharmacological activity, particularly if the ONT candidate is not pharmacologically active in the rat or mouse. 21 Companies were asked whether they employ the use of rodent surrogate ONTs in their carcinogenicity studies. The responses were relatively evenly split among the nine companies that provided answers, with four answering “No” and five answering “Yes” (refer to raw data file in Supplementary Data S1). It is important to note the survey did not contain any follow-up questions on this topic; therefore, it is not known whether testing of a surrogate was warranted.
A series of questions that allowed “free-text” responses were included in the survey to better understand which types of approaches to carcinogenicity risk assessment had (and had not) been successful, in terms of regulatory engagement (Table 10). A “weight of evidence” approach to justify not conducting rodent carcinogenicity studies had been explored by only a single company, with the EMA accepting their position and the FDA rejecting it. A single-species approach was a more successful alternative, with the majority of agencies accepting justifying positions, although not for every submission. Based on responses, a single-species approach was more likely to be accepted when there were technical limitations with chronic administration, exposure issues upon chronic administration, and/or the target was well validated in terms of carcinogenicity risk. However, one company indicated that a single-species approach had been rejected where the candidate was pharmacologically active in both the mouse and rat. There were also multiple programs for which companies had successfully made the case for conducting the carcinogenicity studies as a postmarketing commitment, particularly where the candidate was indicated for a serious and life-threatening disease. However, the rejection of such an approach was more likely for an ONT that contained a novel (unprecedented) chemical modification.
Summary of Carcinogenicity Risk Assessment Strategies That Differed from the Standard Approach (i.e., Two Species Reported at the Time of Filing)
This response pertains to a waiver submitted to multiple agencies for the same ONT candidate.
EMA, Medicines Agency; FDA, US Food and Drug Administration; MEB, Medicines Evaluation Board, Netherlands; MHRA, Medicines and Healthcare Products Regulatory Agency, UK; PMDA, Pharmaceuticals and Medical Devices Agency, Japan.
Discussion
The WG, sponsored by the PDEG committee of EFPIA, conducted a survey of pharmaceutical and biotechnology companies actively developing ONTs, focused on understanding approaches to genotoxicity testing and carcinogenicity strategies for this modality. Companies based in Europe, Japan, and the United States were invited to participate, with the 29 respondents (74% of those invited) representing a broad cross-section of industry. Over half of the responding companies self-classified as “Large Pharma” (Table 1), an increase in relative proportion compared with this WG’s previous industry survey, for which only a third of companies self-classified into this category. 4 This shift may be indicative of the maturation of ONTs as a modality over the last ∼5 years and/or the addition of ONTs to Large Pharma pipelines. Most respondents have 1–3 ONT projects in their pipelines (Table 1) regardless of the size of company, which suggests that ONTs still comprise a small overall proportion of the pipelines for Large Pharma. Among companies indicating they had medium (4–9 projects) or large (≥10 projects) ONT portfolios, Biotech was more heavily represented (as a relative proportion of respondents).
Requirements (or timing) for genotoxicity testing for oncology indications 22 (and for individualized ONTs 14 ) differ compared with those for other diseases; however, most companies had ONT projects outside of these areas (Table 1). Thus, the collective experience in evaluating the genotoxicity of ONTs is extensive. The overwhelming majority of responding companies were working with ONTs in simple aqueous formulations for delivery, with only a small proportion having experience in genotoxicity testing of ONTs formulated in complex delivery systems, such as LNPs. This reflects the approved ONTs, as only one of the 20 marketed medicines is formulated in a complex delivery system (i.e., patisiran/Onpattro). 23
The majority of companies had experience developing siRNAs or RNase H ASOs, although small numbers were also working on other single-stranded ASO classes (e.g., steric blockers, antagomirs, and RNA editors), as well as CpG/immunostimulatory ONTs (Fig. 1). The survey population had no experience evaluating the genotoxicity of miRNA mimic, DNAzyme, or aptamer ONTs. However, two aptamer ONTs have been approved (pegaptanib, indicated for neovascular [wet] age-related macular degeneration [AMD], and avacincaptad pegol, for geographic atrophy secondary to AMD) and the parent molecules, which contain 2′-O-methyl and/or 2′-fluoro modifications, as well as the 2′-fluoro modified monomers contained in pegaptanib are all nongenotoxic in vitro and in vivo.24–26 That these ONTs were not captured within the survey data set illustrates that the findings of the survey do not represent the totality of data for the modality, despite the best efforts of the WG in extending invitations to participate.
Approaches to evaluating genotoxicity of ONTs
All companies developing ONTs for indications outside of oncology confirmed they conduct GLP genotoxicity testing to enable clinical development. This is in line with the outcome of this WG’s previous survey 4 and demonstrates that ICH M3(R2)-aligned approaches 12 are taken for this modality.
Of the standard battery tests detailed in ICH S2(R1), 6 the bacterial reverse mutation test (Ames), in vitro mammalian chromosomal aberration or in vitro MNT, and in vivo rodent MNT were by far the most commonly used (Fig. 2). The in vitro mouse lymphoma Tk and in vivo comet tests are much less frequently utilized (Fig. 2), which in general aligns with the small molecule experience. 27 Only one company reported having performed the in vivo comet test in the liver for a GalNAc-targeted candidate. Disposition studies with unconjugated and GalNAc-conjugated ONTs indicate that these drugs do distribute to the bone marrow (which is the most frequently used tissue for evaluating genotoxicity endpoints in in vivo), albeit at much lower levels relative to the main tissues of distribution, which includes the liver.3,28 Overall, results of the survey indicate that the in vivo genotoxicity testing approach adopted for ONTs is no different from that for small molecule candidates, that is, in vivo genotoxicity assessment in the bone marrow (or peripheral blood) MNT with systemic toxicokinetic data to demonstrate exposure as described in ICH S2(R1). 6
There was limited evidence of companies diverging from standard battery genotoxicity testing as defined in ICH S2(R1). 6 Only three companies indicated applying nonstandard tests to understand either specific target concerns (e.g., theoretical risk related to the RNA-editing class of ONT) or a genotoxicity risk associated with a confirmed hybridization-mediated off target (discharged successfully). These examples provide evidence of companies modifying their approaches appropriately to investigate specific genotoxicity concerns for candidate ONTs that may not have been characterized adequately by the standard battery tests.
For conjugated ONTs, which would comprise the ONT, linker, and a targeting moiety, genotoxicity testing was conducted consistently on the intact molecule. This approach is very much aligned with that recommended by the OSWG Genotoxicity subcommittee 8 and represents the most clinically relevant test item. Only a single company had also tested the individual components alone, although that was in addition to testing the intact ONT conjugate. Most companies also focus on evaluating genotoxicity endpoints in vivo in standard surrogate tissues (e.g., bone marrow or peripheral blood), rather than tailoring assessment to the tissue or cell type being targeted by the ONT (see earlier discussion on assessment of GalNAc-targeted ONTs for the validity of this approach). It is possible that this approach is partly driven by a lack of validated methods and/or limited historical control database for assessing genotoxicity in other tissue types for some tests. 29 Survey responses indicated limited experience in assessing the genotoxicity of ONTs formulated in complex delivery systems, like LNPs. For those that had conducted testing, the formulated ONT was used as the test item, an approach analogous to that taken for conjugated candidates.
No evidence of genotoxicity across ONT classes and chemistries
A critical finding of this survey is that, across the responding companies, there was not a single example of a positive genotoxic result (in vitro or in vivo) for an ONT candidate, whether tested in GLP standard battery or non-GLP/nonstandard tests. The latter observation infers consistently negative results for ONT candidates, including some that would not necessarily have progressed to clinical development. These represent a population of tested ONTs for which regulatory agencies may not have had data when considering requirements for continued genotoxicity testing for this modality. These results are consistent with, and build upon, previously published data8–11 and reflect the recently communicated FDA experience. 15 Moreover, together with the observation that all approved ONTs have been demonstrated to be nongenotoxic, the collective knowledgebase provides a weight of evidence to reconsider the need for the genotoxicity assessment of ONTs containing precedented chemistries. In this regard, such an approach appears to be broadly aligned to the principles outlined in the FDA’s draft guidance for industry on “Platform Technology Designation Program for Drug Development” 30 and the agency’s assertion that a candidate belonging to a class of ONT that is “well-characterized for genotoxicity” may be informed by the class experience. 15
Overall, the chemical modifications that had been assessed most frequently in GLP genotoxicity tests (Fig. 3) were in line with the WG expectations and the work of others.8–10 The PS backbone represents the cornerstone modification of single-stranded ONTs for the last 30 years 31 and has tested negative reproducibly (>130 ONT candidates) in the Ames test, and in in vitro and in vivo mammalian cell tests. This figure includes not only ASOs but also siRNAs in which the PS modification is used to enhance in vivo stability. 32 Other backbone modifications have also tested negative reproducibly, although on fewer occasions, that is, ≥5 ONT candidates, each covering the morpholino, GNA, and VP modifications. The more recently developed PN 33 backbone modification has been tested only twice (at the time the survey was conducted). None of the responding companies had tested candidates with a PNA backbone. Although clinical experience with this modification is limited relative to the others mentioned above, 34 clinical trials (outside of oncology) have been initiated on at least one PNA-containing candidate, 35 which suggests that this modification is nongenotoxic. Ribose sugar modifications have also been tested extensively (i.e., >15 ONT candidates each) and shown to be nongenotoxic; these include 2MOE, 2OMe, 2F, LNA, and cEt ribose modifications. For base modifications, 5mC (employed to mitigate CpG motif-driven immunostimulatory effects 36 ) and abasic sites have also tested negative with >15 ONT candidates reported.
Based on the data (Fig. 3), slightly fewer ONT candidates have been tested in vivo versus in bacterial or in vitro mammalian tests. However, the WG does not consider this finding to be indicative of divergence from ICH S2(R2) guidance (i.e., Option 1 or 2 approaches). 6 Rather, it is more likely to reflect a greater number of ONT candidates in pre-FIH development (when in vitro tests are required) versus pre-Phase 2 development (at which time the in vivo test is required).
Just two respondents indicated that they had evaluated the genotoxicity of ONT modifications as individual monomers, with PS and 2MOE the only synthetic modifications being tested; both were confirmed as negative (Table 3). In addition, as mentioned above, the 2′-fluoro modified monomers contained in pegaptanib are nongenotoxic. 24 Modified monomers could theoretically cause genotoxicity through nucleotide pool imbalance or incorporation into nuclear DNA.37–39 These hypothetical concerns also apply to naturally occurring nucleotides, although it is important to note that the PMDA guidance on the nonclinical safety evaluation of ONTs indicates that genotoxicity testing of candidates containing only naturally occurring nucleotides is not expected. 13 Nonetheless, modified nucleotide monomers from siRNAs administered at suprapharmacological doses in rats did not exceed levels of endogenous ribonucleotide and deoxynucleotide pools. 40 Additionally, modified mononucleotide components of ONTs are neither designed nor intended to interact with DNA or RNA processing and they are generally poor substrates for endogenous mammalian kinases or polymerases.40,41 Should any modified nucleotides be incorporated into DNA, the potential to cause strand breakage or mutation would have been assessed adequately using the standard genotoxicity test battery 6 on the intact ONTs.8,10,40,41
Confirming ONT exposure in the test system
A clear recommendation of the OSWG Genotoxicity subcommittee was the demonstration of ONT exposure in cells or tissues used for genotoxicity assessment. 8 Evidence of exposure may be generated using relevant tool ONTs (i.e., molecules with the same chemistry/design, including conjugated targeting platform) or in in vitro or in vivo models comparable to those being employed for genotoxicity testing, conferring validity on the experiment.
Based on the results of this survey, it is clear that the vast majority of companies assume that exposure occurs at the ICH S2(R1)-defined limit doses 6 for in vitro and in vivo genotoxicity testing (Table 4). Given the number of ONTs in clinical development, it appears that this approach is generally accepted by regulatory agencies, although companies may have been asked to justify their position. However, it is quite possible that regulatory agencies accept the published literature demonstrating exposure of genotoxicity test systems to ASOs and siRNAs.8–10,41 A smaller proportion of companies also rely on published relevant evidence for each test. A subset of these indicated that they also generate some experimental evidence of exposure in vitro (Tables 5) and in vivo (Table 6). For in vitro tests, demonstrating cytotoxicity was the most favored approach, which is in alignment with established practices for small molecule drugs. 8 Assessing intracellular levels of the ONT candidate was undertaken less frequently.
Evaluation of target engagement was not undertaken to confirm exposure either in vitro or in vivo (Tables 5 and 6). Although assessing mRNA knockdown by an ASO or siRNA in a mammalian cell line is technically straightforward, it is dependent on the expression of the intended target in the test system, which may not be the case for cell types validated for use in genotoxicity tests. Not using target engagement to confirm in vivo exposure may be for the same reasons, as the rodent species may not be pharmacologically relevant for ONTs designed to target human RNA sequences. Additionally, ICH S2(R1) does not specify target engagement as a method of confirming exposure, 6 so it is not surprising that companies have not gone beyond approaches within that guidance. For in vivo genotoxicity testing, most respondents rely on measuring systemic exposure. Smaller proportions assess ONT concentrations in the tissue being tested for genotoxicity or demonstrating dose-limiting toxicity. Given the predictable nature of ONT biodistribution in vivo,42–44 it seems unnecessary to assess every candidate though, once data are available for a given platform. Based on the published data for ASOs and siRNAs8–10,41 the WG proposes that further assessment of genotoxicity test system exposure to these classes of ONT is no longer warranted. The FDA does address the issue of exposure of the test system in their draft guidance for industry on the nonclinical safety assessment of oligonucleotide-based therapeutics. 15 This document currently states that “the test batteries recommended in ICH S2(R1) are generally considered appropriate, provided data are available to support cellular uptake of the ONT into the cells analyzed.”
The potential for genotoxicity induced by triplex formation is not a prominent concern
The potential for ONTs to interact directly with double-stranded genomic DNA through sequence-dependent means, forming a triple helix or “triplex,” was highlighted previously by the EMA. 7 Such interactions can produce site-specific mutations. 45 The EMA flagged the concern that the standard battery genotoxicity tests would not detect such interactions, and that the risk should be considered for ONTs. Subsequently, the OSWG Genotoxicity subcommittee included a thorough review of this theoretical risk in their white paper. 8 They concluded that the stringent sequence and biochemical conditions required for triplex formation are not necessarily compatible with ONT design, and that the risk was low. However, they did recommend that the potential for triplex formation be assessed for ONTs based on sequence analysis, without the need to address the risk experimentally.
Based on the survey responses, the risk of triplex formation is not considered by about two-thirds of companies (Table 7), with the remainder conducting a paper-based assessment using the literature and ONT sequence assessment. The fact that such a large proportion do not even consider this risk implies that regulatory agencies generally view triplex formation not to be a significant concern for ONTs. Indeed, the potential for triplex formation is not mentioned in the genotoxicity sections of the PMDA and (draft) FDA guidance documents.13,15 Should companies face questions on this topic during clinical trial submissions or marketing applications, then the principles outlined by the OSWG Genotoxicity subcommittee 8 should address these concerns adequately.
Regulatory interactions
Based on survey responses, it was clear that divergence from ICH S2(R1) 6 when evaluating the genotoxicity of ONTs is uncommon, with only two companies indicating that they had requested testing waivers. However, neither of these two companies submitted an optional “testing waiver request” case study form; thus, it was not possible to understand the rationale underlying their requests, or whether these had been accepted/rejected by regulatory agencies. It was notable that two (different) companies had submitted FIH-enabling nonclinical safety packages without genotoxicity data, outside of the context of oncology or individualized ONT projects, and that neither had been asked to backfill data at that point by regulatory agencies. Overall, it is not possible to draw any conclusions on whether regulatory agencies are open to waiving genotoxicity testing of ONTs. However, based on the agency-specific “reflection” or guidance documents that are available, there is evidence of divergent opinions among agencies.7,13–15 The upcoming ICH guidance on “Nonclinical Safety Studies for Oligonucleotide-Based Therapeutics” (ICH S13)16,18 should provide an opportunity for more harmonized approaches to be defined.
Impurity and degradant evaluation
The genotoxicity of impurities or anticipated degradants of ONTs were not tested routinely by the majority of respondents, although about a fifth of companies do some form of testing (predominantly on a case-by-case basis) (Table 8). ONT-related impurities will typically be structurally similar to the parent (or its metabolites), or contain moieties found in native genetic material and, therefore, present low risk for genotoxicity. 8 Where novel structures have been identified in ONT-related impurities, these have not been predicted to be genotoxic based on the absence of structural alerts. 8 No doubt companies are applying the principles of the ICH M7(R2) guidance 46 when assessing the genotoxic potential of process- and drug-related impurities to their ONT programs. However, the findings of this survey suggest that limited downstream testing of ONT-related impurities has been required. Where testing has been undertaken, the Ames test has been favored, and there is no evidence that ONT-related impurities have been associated with genotoxicity. A recent draft EMA guideline on the “development and manufacture of oligonucleotides” 47 emphasizes that ONTs are not within scope of ICH M7(R2) and outlines principles for qualification of various classes of ONT-related impurities. Notably, this draft guideline does cross reference the EMA reflection article on the assessment of the genotoxic potential of ASOs, 7 stating this document should be considered (if appropriate).
Carcinogenicity strategy
The results of the survey show that most companies with ONT programs beyond early development have followed the recommended approach of using two rodent species for carcinogenicity assessment, with the majority utilizing the 6-month transgenic mouse study and a 2-year rat study (Table 9). The different approaches may be explained in part by the timing of the programs, which likely preceded the wider adoption of the transgenic mouse model in some cases (i.e., where 2-year studies in the mouse and rat were conducted). The survey did not include a question as to whether the ONT was active in the species being used, so it is unclear whether these studies assessed on-target pharmacodynamic-based carcinogenicity or only chemistry-related risk. This is important because one of the key aspects of the carcinogenicity risk assessment is the understanding of on-target-related carcinogenicity risk. The use of a rodent surrogate ONT to assess on-target-related carcinogenicity risk varied among the companies, which may reflect the variation in pharmacological activity of the candidates in rodents. For example, if the ONT candidate targeted an mRNA sequence that is not expressed in wild-type rodents, the use of a surrogate is not warranted. Unfortunately, the survey did not address this granularity with follow-up questions. The decision to use a rodent surrogate ONT should be based on a case-by-case evaluation of the scientific rationale and the available data.
When respondents reported which types of approaches to carcinogenicity assessments other than the 2-species approach have been successful (Table 10), the results indicate that both a weight-of-evidence approach for a full waiver or a single-species approach have been employed. The EMA appears to take the least conservative stance on these alternative strategies, approving full or partial waivers in all five cases. All regulatory agencies were more likely to accept justification for a single-species approach when robust scientific justification was provided as to why a species would not provide value to overall carcinogenicity assessment. A key example is the exacerbation of chronic progressive nephropathy in rats during chronic administration of ASOs.48,49 In this case, the respondents were able to use only the mouse in a 2-year study due to the rat-specific nephrotoxicity and inability to complete a 2-year rat study. However, this justification was not accepted by all agencies and a respondent was required to attempt a 2-year rat study despite this limiting toxicity on multiple programs. The postmarketing commitment approach was generally accepted, particularly for serious and life-threatening indications (consistent with approaches taken for approved ONTs). However, for novel chemistries and mechanisms of action, agencies were more likely to request that the full carcinogenicity assessment be completed for final submission.
It should be noted that this survey was conducted around the time of finalization and implementation of the ICH S1B(R1), 5 which provides a framework for waiving the 2-year rat study based on a comprehensive assessment of the carcinogenicity risk. 5 This revised carcinogenicity guidance may provide an opportunity for companies developing ONTs to streamline their carcinogenicity testing paradigms and reduce the use of animals, as well as the time and cost of development. However, ICH S1B(R1) 5 does not provide a formal mechanism for waiving the mouse carcinogenicity study, which may explain in part why there were no examples of companies conducting the assessment in a 2-year rat study only. The carcinogenicity section of the draft FDA guidance does indicate that it can be appropriate to assess this risk in a 2-year study of a single species. 15 However, it is unclear in the current draft whether this would be acceptable in either the rat or mouse, or mouse only (as per ICH S1B[R1] 5 ). It is also worth noting that a carcinogenicity waiver was granted for pegaptanib and avacincaptad pegol (the aptamers discussed previously, that are clearly absent from the survey data set) because these ONTs are locally administered to the eye and result in limited systemic exposure.24,26 Lastly, the current survey did not ask if any tumors that were considered clinically relevant were detected in completed rodent carcinogenicity studies. However, the authors understand that the OSWG Carcinogenicity subcommittee will address this in their upcoming white paper (Personal Communication, Cindy Berman, September 2024).
Concluding Remarks
The survey provides valuable insights into industry practices and regulatory expectations for the genotoxicity and carcinogenicity assessment of ONTs. The results highlight a consistent absence of genotoxic findings across a diverse range of ONT classes and chemistries and support the conclusion that well-established chemistries have been robustly evaluated and are not of concern from a genotoxicity perspective. The findings are supportive of the position that continued genotoxicity testing of established classes, which contain only well-precedented modifications, is unlikely to yield positive results or generate data that would further inform human/patient risk assessment in a meaningful way. However, due to regulatory expectations, companies continue to adhere to the standard genotoxicity testing guidelines, with limited divergence from the ICH S2(R1) standard battery tests. 6 The WG concludes that the survey data indicate a very low risk associated with this modality and, therefore, advocates for greater flexibility in the genotoxicity testing approach for ONTs. With the more nuanced approach to genotoxicity testing detailed in the recently published FDA “draft guidance for industry on the nonclinical safety assessment of ONTs,” 15 it would be interesting to reassess industry experience with achieving genotoxicity testing waivers for new candidate ONTs that belong to classes considered well characterized in one to two years time.
Based on the findings of this survey and relevant previously published data,8–11 the WG considers that well-established backbone (i.e., PS, GNA, VP, morpholino), sugar (i.e., 2OMe, 2MOE, 2F, LNA and cEt), and base (i.e., 5mC and abasic) modifications can be considered “precedented,” in terms of negligible hazard based on in vitro and in vivo genotoxicity testing. Given the body of evidence, the WG proposes that additional genotoxicity testing of new ONT candidates that contain only these “precedented” modifications is unwarranted as long as other potential mechanisms of genotoxicity have clearly been excluded. These other mechanisms would include any concerns over specific mode of action/class of ONT, primary pharmacology, confirmed hybridization-mediated off target effects that carry a genotoxicity risk, or the potential for triplex formation. The WG considers this approach to be broadly aligned with the principles proposed previously by the EMA for PS ONTs 7 and the recent FDA’s draft guidance for industry on “Platform Technology Designation Program for Drug Development” 30 although there are subtle differences relative to the “well-characterized class” approach outlined by the FDA recently. 15
In addition to specifying the “precedented” ONT modifications, the WG proposes a pathway through which this status could be achieved for novel/proprietary chemistries. Based on the current body of evidence for precedented ONT modifications (i.e., consistently testing nongenotoxic in vitro and in vivo) and likely ‘structural read-across’ for novel modifications to those that are precedented, the WG proposes that a novel modification and/or class be demonstrated as nongenotoxic within the context of five different ONTs in order to be considered “precedented” from a genotoxicity risk perspective. This testing should be conducted in compliance with GLP and include appropriate in vitro and in vivo standard battery tests, as outlined in ICH S2(R1). 6 Additionally, experimental evidence for an exemplar ONT, or an appropriate paper-based assessment, of test system exposure should be provided. This robust data package could be presented to regulatory agencies as supportive evidence to justify a “genotoxicity testing waiver,” prior to initiating clinical studies on a new ONT candidate. The WG considers this proposed pathway to be a balanced approach, ensuring patient safety with regard to genotoxicity for novel ONT chemistries/classes (by demonstrating they are nongenotoxic on multiple independent occasions), while accounting for the substantial preexisting evidence indicating a very low risk across the wider modality. Such an approach also has the potential to reduce the usage of animals and finite resources on tests that add little further value to human risk assessment, enabling these to be better focused on accelerating delivery of medicines to patients.
Companies are encouraged to share such data through peer-reviewed manuscripts in order to continue to grow the publicly available knowledgebase. Alternatively, the authors are aware that the OSWG have had some preliminary discussions on the idea of a cross-industry database for sharing ONT genotoxicity data, which could potentially be accessed by regulatory agencies too. Both approaches would enable other companies to utilize this prior evidence when developing genotoxicity waiver positions for their ONTs.
In terms of carcinogenicity assessment, the survey indicates that most companies follow the recommended approach of using two rodent species, although alternative strategies, such as single-species testing, can be accepted by regulatory agencies under certain circumstances (additionally evidenced by the carcinogenicity position outlined by the FDA recently 15 ). However, because carcinogenicity studies for serious and life-threatening diseases are often conducted as postmarketing commitments, only a limited number of ONT candidates have completed carcinogenicity testing compared to the much larger dataset for genotoxicity testing. Therefore, it will be important for industry to share experiences across ONT chemistries/classes to further build understanding and facilitate design of optimal approaches. In this regard, the authors look forward to the future OSWG Carcinogenicity subcommittee white paper on this very topic.
Overall, the findings of this survey underscore the need for a more tailored approach to the nonclinical safety assessment of ONTs, taking into account the unique properties and low genotoxicity risk of these compounds. The results also highlight the potential for the ICH S1B(R1) 5 to streamline carcinogenicity testing paradigms for ONTs. The WG suggests that it is time to consider a future revision of ICH S2(R1), 6 via development of supplementary questions and answers, to cover considerations specific to ONTs. Additionally, we hope that the findings of this survey provide valuable input to the ICH S13 topic on ‘Nonclinical Safety Studies for Oligonucleotide-Based Therapeutics’, which commenced in the second half of 2024 16 and is provisionally scheduled for finalization in November 2027. 18
Footnotes
Acknowledgments
The authors thank the EFPIA secretariat (specifically, Silvia Garcia, Fatumata Seck, and Katarina Nedog) for executing survey administration and the PDEG committee of EFPIA for their sponsorship, advice, and guidance. Key opinion leaders who supported the project, providing invaluable feedback on the survey design and/or thoughtful review of the draft article: Cindy Berman (industry consultant and former chair of OSWG Genotoxicity and member of the Carcinogenicity subcommittees), Jeff Foy (PepGen and former OSWG chair), Scott Henry (Ionis Pharmaceuticals and member of the OSWG Carcinogenicity subcommittee), and Maja Janas and Joe Dybowski (Alnylam Pharmaceuticals). Yutaka Tonomura (Nippon Shinyaku) for facilitating participation of Japan Pharmaceutical Manufacturers Association member companies and his review and valuable comments on the draft article. Former EFPIA oligonucleotide WG members: Cathaline den Besten (ProQR Therapeutics), Jonathan Moggs (Novartis), Kai Schaefer (AbbVie), and Lene Diness Jensen (Novonordisk). The 29 respondents who took the time and effort to complete the survey.
Author Disclosure Statement
The authors declare no conflict of interest given the precompetitive nature of this work, and their respective employers during the conduct and analysis of the survey, as well as at the time of publication are detailed in the affiliations. The views and opinions expressed in this article are those of the authors and do not necessarily represent the views, opinions, and/or policies of their employers or EFPIA.
Funding Information
No funding was received for this work, other than the publication fees, which were covered by GSK R&D (employer of the WG Chair).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.