
Abstract
Duchenne muscular dystrophy (DMD) is caused by mutations of the DMD gene that prevent the expression of functional dystrophin protein. BMN 351 is an antisense oligonucleotide (ASO) designed to induce skipping of exon 51 of dystrophin pre-mRNA and production of internally deleted but functional dystrophin. We determined whether extended-term BMN 351 dosing leads to exon skipping, dystrophin production, and improved motor function in hDMDdel52/mdx mice containing a human exon 52-deleted DMD transgene. Weekly intravenous doses of vehicle, 6 mg/kg BMN 351, or 18 mg/kg BMN 351 were administered for 25 weeks, and samples were analyzed 4 and 12 weeks post-dosing. BMN 351 produced dose-dependent exon skipping levels in the heart and quadriceps muscles, accompanied by dose-dependent increases in mean dystrophin levels of 17% to 55% 12 weeks post-dosing. Compared with vehicle-treated hDMDdel52/mdx mice, BMN 351 ameliorated DMD-related histopathologic changes in the gastrocnemius muscle and heart. Both BMN 351 doses preserved fine motor kinematics, which was worse in vehicle-treated hDMDdel52/mdx mice compared with wild-type 4 and 12 weeks post-dosing. Liver samples demonstrated findings consistent with ASO accumulation, to which mice are considered especially sensitive compared to humans and other non-clinical species. These results support further non-clinical and clinical development of BMN 351.
Introduction
Duchenne muscular dystrophy (DMD) is a rare, early-onset, degenerative, X-linked recessive neuromuscular disease characterized by progressive muscle weakness that predominantly affects boys (1 in 5000–6000 live male births).1–3 Individuals with DMD show impaired motor function by 1 to 3 years of age, including difficulty walking and climbing stairs. 1
DMD is caused by mutations in the DMD gene, which encodes the essential cytoskeleton-associated protein dystrophin.1,4 Because dystrophin is critical to coupling the sarcolemma with muscle fibrils and protecting cells from contraction-induced damage, lack of functional dystrophin leads to progressive skeletal muscle damage, cellular degeneration, and cardiomyopathy.5,6 A significant proportion of DMD-related mortality is associated with cardiovascular or respiratory failure. 1 Mutations that cause DMD result in minimal production of functional dystrophin; however, mutations of DMD commonly associated with Becker muscular dystrophy (BMD) lead to the production of internally deleted, partially functional dystrophin and a less severe disease course.4,7
DMD, the largest gene in the human genome, has 79 exons.2,8 Skipping a single exon may restore the DMD open reading frame in approximately 70% of the 68% of individuals with DMD due to exon deletions.9,10 One promising therapeutic approach is to use antisense oligonucleotides (ASOs) to skip one or more exons that contain disease-causing mutations to produce internally deleted yet functional dystrophin similar to that found in BMD.9,11 Exon 51 skip-amenable mutations account for approximately 14% of patients with DMD. 12 The only ASO for exon 51 skip-amenable DMD currently approved in the United States, eteplirsen, provides limited increases in dystrophin levels, likely due to poor tissue penetration and inefficient induction of exon skipping.13–15 To improve tissue penetration, eteplirsen was subsequently peptide-conjugated, resulting in SRP-5051 (vesleteplirsen).14,16 However, SRP-5051 retains the eteplirsen binding sequence; its efficacy is being investigated in a phase 2 trial (NCT04004065). The potential of ASOs to efficiently induce exon 51 skipping and near-full-length dystrophin production has yet to be realized.
BMN 351 is an ASO being developed for the treatment of individuals with DMD mutations amenable to exon 51 skipping.
17
BMN 351 has the same chemistries (including a phosphorothioate backbone, locked nucleic acids, and 2
Here, we extend this prior work by assessing the pharmacologic response and functional effects of BMN 351 treatment over a longer dosing period and with an additional dose level. hDMDdel52/mdx mice were dosed intravenously (IV) once weekly for 25 weeks with 6 mg/kg BMN 351, 18 mg/kg BMN 351, or vehicle and evaluated pharmacokinetics, tissue distribution, exon skipping, dystrophin expression, DMD biomarkers, and fine motor kinematics 4 and 12 weeks after dosing ended. We additionally evaluated the hematology, clinical chemistry, and histopathology of muscle and liver samples. The results support further non-clinical and clinical development of BMN 351.
Materials and Methods
Animals
Animal experiments were conducted at Charles River Discovery Research Services (Finland) according to the National Institutes of Health guidelines for the care and use of laboratory animals and were approved by the National Animal Experiment Board of Finland. Male and female homozygous hDMDdel52/mdx mice (n = 57) were bred at Charles River (UK) and genotyped by BioLytix (Switzerland). hDMDdel52/mdx mice contain murine DMD with a stop mutation in exon 23, harbor human DMD with exon 52 deleted, and have a dystrophic phenotype.18,19 Age-matched C57BL/6J mice (n = 18) were obtained from Charles River (Germany) to be used as wild-type (WT) controls. Mice were aged 5 weeks upon arrival at the testing facility.
Study design and dosing
BMN 351, a phosphorothioate 18-mer ASO with the same sequence and chemistries as the previously reported ASO AON-C12 with an additional 5′ triethylene glycol, 17 was manufactured by BioSpring GmbH (Frankfurt, Germany) and prepared in an aqueous vehicle (10 mM phosphate buffer, 0.7% NaCl, pH 6.9–7.1) by the testing facility. Beginning at 7 weeks of age, WT mice (n = 6 females/12 males) were dosed once weekly with the vehicle, and age-matched hDMDdel52/mdx mice were dosed with either vehicle (n = 6 females/12 males) or BMN 351 at 6 mg/kg (n = 6 females/12 males) or 18 mg/kg (n = 6 females/15 males) via tail vein injection (slow bolus manually for about 60 s; Fig. 1). Full-length product content, determined using ion pair reversed-phase high-performance liquid chromatography with ultraviolet mass spectrometry, was 78.9% of total drug substance mass; therefore, nominal doses of 6 and 18 mg/kg corresponded to 4.7 and 14.2 mg/kg (0.72 and 2.18 µmol/kg) of full-length product, respectively. Following each weekly injection, mice were carefully monitored for changes in behavior or condition, including position, movement, respiration, and seizures or convulsions.

Dosing regimen and study design. Mice were hDMDdel52/mdx unless otherwise specified. The red blood drop denotes when blood draws were performed in live mice (saphenous vein) or collected at terminal time points via cardiac puncture. The nominally administered doses of 6 and 18 mg/kg BMN 351 corresponded to 4.7 and 14.2 mg/kg (0.72 and 2.18 µmol/kg) full-length products, respectively. QW, once weekly; Veh, vehicle; WT, wild-type C57BL/6J.
Baseline fine motor kinematics were tested 1 week prior to dosing and again 4 weeks after the end of dosing (study week 29); a subset of 12 mice/group was additionally tested at 12 weeks after the end of dosing (study week 37). At each time point, mice were euthanized by anesthetization with 180 mg/kg sodium pentobarbital (Mebunat, Orion Pharma, Finland) and subjected to cardiac puncture to collect whole blood for pharmacokinetics and clinical pathology analyses before transcardial perfusion with phosphate-buffered saline and tissue sample collection (Supplementary Table S1). Samples reserved for pharmacokinetics/pharmacodynamics analyses were flash-frozen in isopentane chilled with liquid nitrogen, sectioned on a cryostat, and stored at −80°C; samples used for histopathology were stored in formalin at room temperature.
Plasma pharmacokinetics and tissue biodistribution analyses
Saphenous vein blood samples were collected for analysis of plasma pharmacokinetics prior to and after dosing on weeks 1 (day 1) and 25 (day 169) at 2 to 3 min, 20 min, 1 h, 3 h, and 8 h following each injection. Plasma was separated by centrifugation and shipped to Ardena Bioanalysis (Assen, Netherlands), where BMN 351 concentrations were quantified using a hybridization enzyme-linked immunosorbent assay (hELISA; Supplementary Data). For quantification of BMN 351 concentration in target tissues, samples were homogenized at room temperature and underwent quantitative hELISA.
Exon skipping
Exon skipping efficiency was determined by reverse transcriptase droplet digital polymerase chain reaction performed by Precision for Medicine (Houston, TX; Supplementary Data). Percent exon 51 skippings were calculated as follows:
Dystrophin and exploratory biomarkers
Dystrophin and exploratory biomarkers were assessed by Biognosys AG (Schlieren, Switzerland) using liquid chromatography parallel reaction monitoring (LC-PRM; Supplementary Data). A panel of exploratory biomarkers relevant to DMD, selected based on differential protein expression in the quadriceps muscle between WT and DMD mouse models, was used to calculate a biomarker score for each individual animal as follows:
Mean composite biomarker scores for vehicle-treated WT and hDMDdel52/mdx mice were designated as 100 and 0, respectively. 20
Clinical chemistry and histopathology
Non-hemolyzed serum was analyzed for clinical chemistry parameters including alanine aminotransferase (ALT), aspartate aminotransferase (AST), lactate dehydrogenase (LDH), and creatine kinase (CK) activities by Movet Oy (Kuopio, Finland) using standard procedures.
Necropsies were performed on mice allocated to the 4- and 12-week post-dosing time points, during which heart, skeletal muscle (gastrocnemius), and liver samples were collected along with a panel of other tissues as part of a general survey (kidney, tail/injection site, spleen, gallbladder, and mesenteric lymph node). Tissues were formalin-fixed and routinely processed as ∼4 µm hematoxylin and eosin-stained glass slides. Slides were evaluated microscopically, selecting descriptive terminologies and subjective severity scores for any observations made.
Fine motor kinematics
Fine motor kinematics were evaluated using the MotoRater system (TSE Systems, Berlin, Germany) using walking behaviors (step, stride, stance, swing analyses, and limb coordination) as previously described.21,22 SimiMotion (Simi Reality Motion Systems, Unterschleissheim, Germany) was used to track points on the body (eg, limb joints) in relation to the ground. Gait patterns and movements were analyzed using a custom-made automated analysis system. Principal component analysis was used to reduce data dimensionality by identifying sets of highly correlated gait parameters. The resulting eigenvectors underwent orthogonality-preserving varimax rotation. A discriminant direction vector based on the differential principal component scores between WT and vehicle-treated hDMDdel52/mdx mice was identified. Overall gait scores were then derived by projecting normalized parameter data from each mouse on the discriminant direction vector; thus, the overall gait scores describe the overall kinematic effects of a pharmacologic agent.
Statistics
Hypothesis testing (α = 0.05) was performed using GraphPad Prism (San Diego, CA). Data are represented as mean ± standard error of the mean (SEM) unless otherwise noted. Differences in exon skipping and dystrophin protein were assessed using a 2-way analysis of variance (ANOVA) with a Šidák correction for multiple comparisons; data were log-transformed in cases of unequal variance. Overall gait score was analyzed using a Greenhouse-Geisser-corrected mixed model fit using restricted maximum likelihood (REML) as implemented in GraphPad Prism with animals matched at baseline, 4 weeks post-dosing, and 12 weeks post-dosing; Fisher’s least significant differences post hoc test were used to compare vehicle-treated hDMDdel52/mdx mice with vehicle-treated WT mice at each time point, and Dunnet’s multiple comparisons post hoc test were used to compare vehicle-treated hDMDdel52/mdx mice with 6 and 18 mg/kg BMN 351-treated hDMDdel52/mdx mice at each time point. Mean body weight differences were analyzed using the same REML-based mixed-effects model with animals matched at each week from week 7 to 43; vehicle-treated hDMDdel52/mdx mice were compared with vehicle-treated WT mice using Šidák’s post hoc test for multiple comparisons, and vehicle-treated hDMDdel52/mdx mice were compared with 6 and 18 mg/kg BMN 351-treated hDMDdel52/mdx mice using Dunnet’s multiple comparisons test. Biomarker scores were analyzed using a 1-way ANOVA with Tukey’s post hoc test for multiple comparisons (each treatment group compared with every other treatment group) separately 4 and 12 weeks post-dosing. Clinical chemistry parameters were analyzed using Welch’s 2-sample t-test (vehicle-treated hDMDdel52/mdx vs. vehicle-treated WT) and 1-way ANOVA with Dunnett’s multiple comparisons test (vehicle-treated hDMDdel52/mdx vs. 6 and 18 mg/kg BMN 351-treated hDMDdel52/mdx mice) separately for each time point.
Results
Pharmacokinetics and tissue biodistribution analyses
Systemic exposure to BMN 351 was roughly proportional to the dosages examined at week 25 (Table 1, Supplementary Fig. S1). The accumulation ratios in an area under the plasma analyte concentration versus time curve from the time of dosing to the time of the last sample with quantifiable drug (AUClast [h•nM]), quantified as week 25 AUClast/week 1 AUClast, were 1.19 and 1.32 for 6 and 18 mg/kg BMN 351-treated mice, respectively, after 25 weeks of once-weekly dosing.
Plasma Pharmacokinetics Parameters in hDMDdel52/mdx Mice
n = 14–17 mice/group. The accumulation ratio was determined by dividing AUClast on week 25 by AUClast on week 1. Mice were hDMDdel52/mdx unless otherwise specified. The nominal doses of 6 and 18 mg/kg corresponded to 4.7 and 14.2 mg/kg (0.72 and 2.18 µmol/kg) of full-length product, respectively.
AUCinf, an estimate of the area under the plasma concentration versus the time curve from the time of dosing to infinity; AUClast, area under the plasma analyte concentration versus the time curve from the time of dosing to the time of last sample with quantifiable drug; CL, clearance; Cmax, maximum concentration; NA, not applicable; QW, once weekly; RAUC, accumulation ratio; t1/2, half-life; Tmax, time of maximum concentration; Vz, the volume of distribution; W, week.
We next assessed BMN 351 tissue concentrations, which were higher in all examined tissues from mice treated with 18 mg/kg BMN 351 compared with those treated with 6 mg/kg BMN 351 4 and 12 weeks post-dosing (Fig. 2). Four weeks after dosing, mean BMN 351 concentrations in the quadriceps muscle and heart were 0.489 nmol/g and 2.19 nmol/g, respectively, in the 6 mg/kg BMN 351 group and 2.13 nmol/g and 12.4 nmol/g, respectively, in the 18 mg/kg BMN 351 group. Twelve weeks after dosing, the mean BMN 351 concentration in the quadriceps decreased to 0.234 nmol/g in 6 mg/kg-treated mice but increased to 2.94 nmol/g in 18 mg/kg-treated mice. In the heart, mean concentrations 12 weeks post-dosing decreased in both 6 and 18 mg/kg BMN 351-treated mice to 1.20 nmol/g and 4.85 nmol/g, respectively. BMN 351 concentrations were highest in the liver, followed by the kidney, diaphragm, heart, gastrocnemius, and quadriceps.
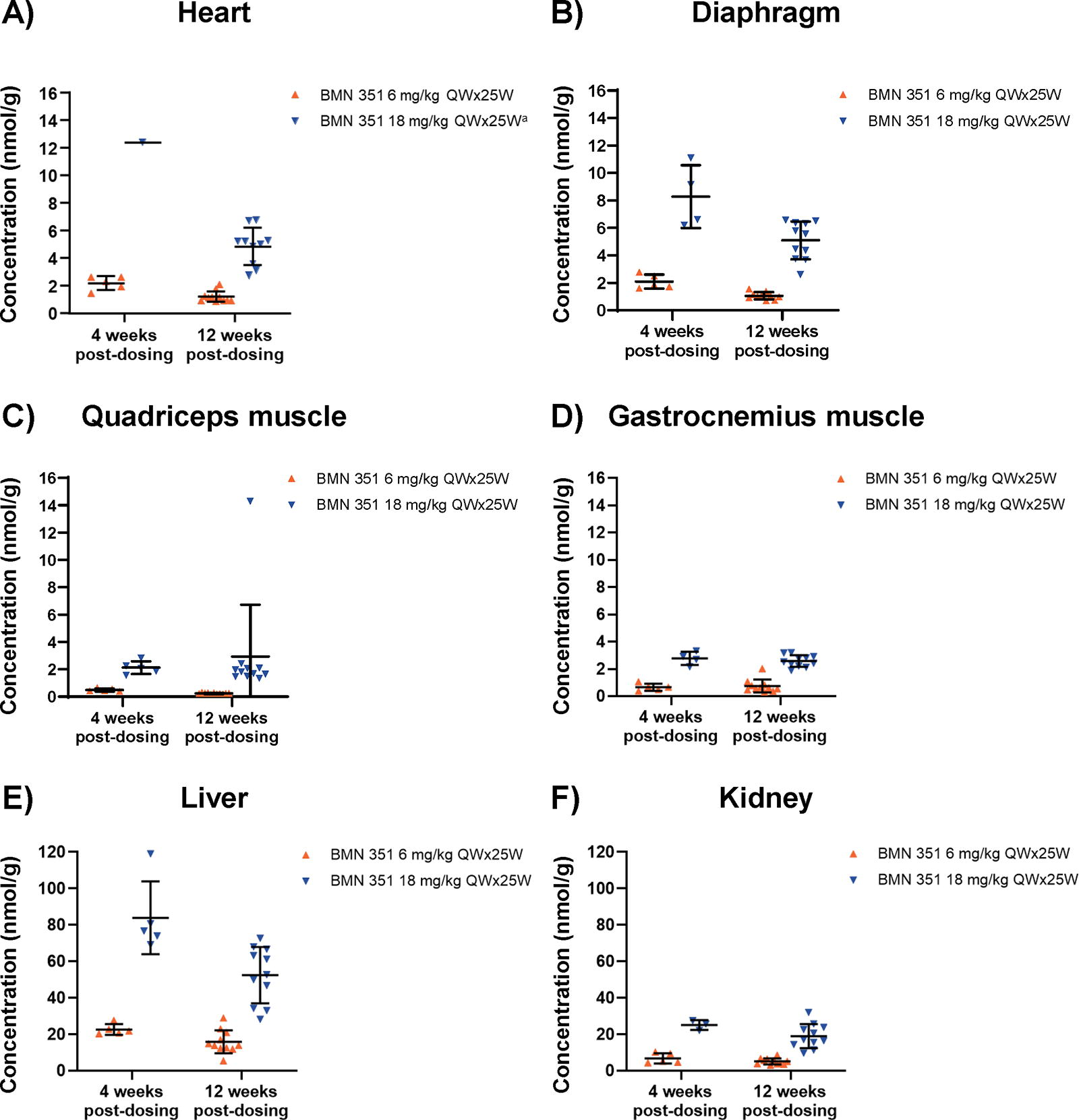
BMN 351 concentration in the
Exon skipping in target tissues
We assessed BMN 351-induced exon 51 skipping of the hDMDdel52 transgene mRNA in target tissues. Mean levels of exon 51 skipping (reported as the percentage of hDMDdel52 mRNA transcripts missing exon 51) were dose-dependent both 4 weeks and 12 weeks post-dosing in the heart, diaphragm, quadriceps muscle, and gastrocnemius muscle (P < 0.0001; Fig. 3A–D). Exon skipping levels significantly increased between weeks 4 and 12 post-dosing in the heart and gastrocnemius for the 6 and 18 mg/kg groups (P < 0.001). In vehicle-treated mice, mean exon 51 skipping levels were minimal (0.1%–0.3%) across all time points and tissues.
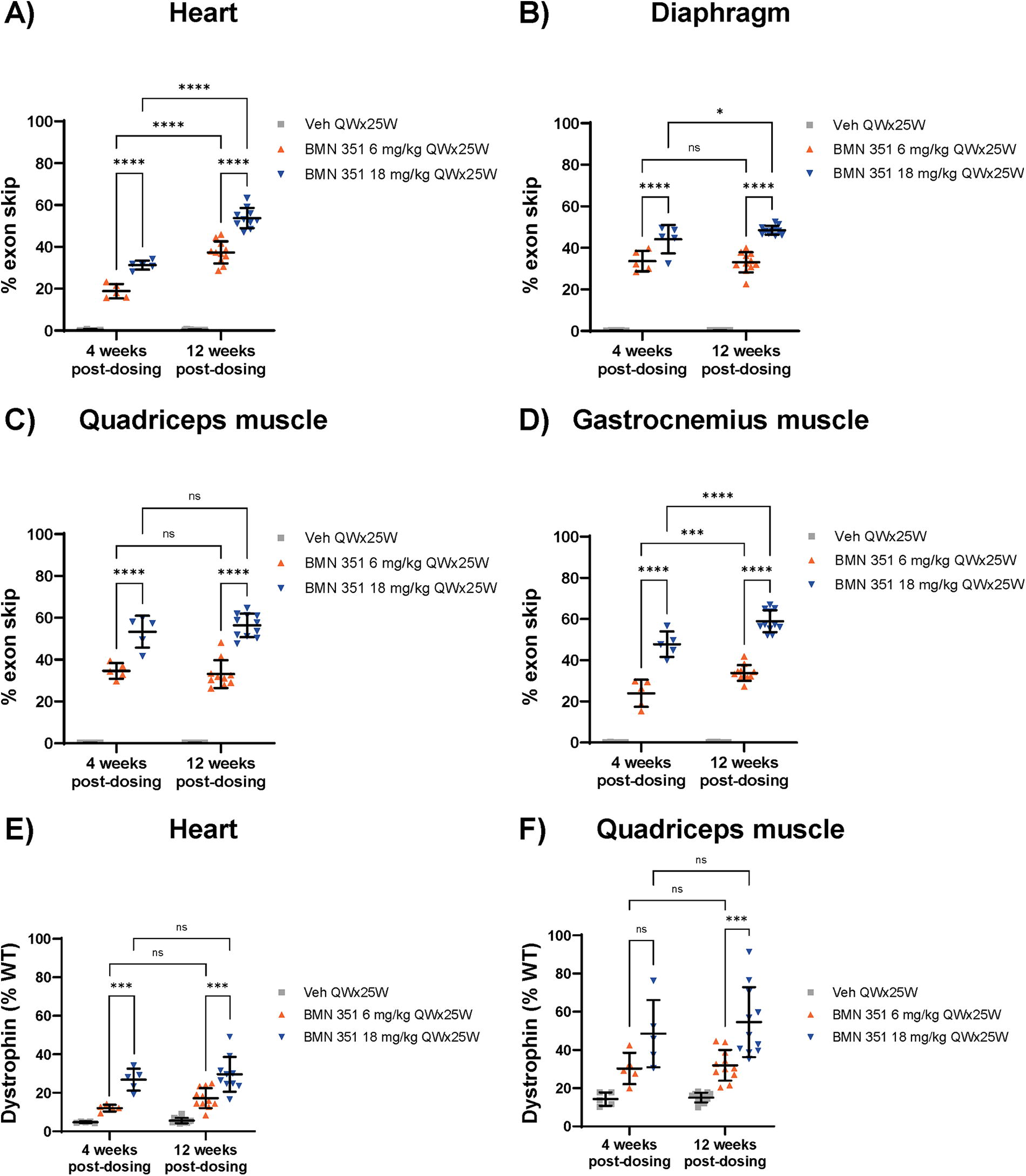
Exon 51 skipping of DMD mRNA and dystrophin protein levels in critical tissues 4 and 12 weeks after dosing. Exon 51 skips as a percentage of total DMD transcripts in the
Dystrophin levels in target tissue
Next, we evaluated dystrophin protein expression in the heart and quadriceps muscle of BMN 351-treated hDMDdel52/mdx mice. Dystrophin levels were dose-dependent. In the heart and quadriceps 4 weeks after dosing, mean dystrophin levels were 12.1% and 30.3% of WT, respectively, in mice treated with 6 mg/kg BMN 351, and 26.8% and 48.5% of WT, respectively, in 18 mg/kg BMN 351-treated mice (Fig. 3E and F). Mean dystrophin production for both 6 mg/kg BMN 351-treated mice (heart, 17.2% of WT; quadriceps, 32.0% of WT) and 18 mg/kg BMN 351-treated mice (heart, 29.6% of WT; quadriceps, 54.6% of WT) was sustained 12 weeks after dosing. Vehicle-treated hDMDdel52/mdx mice had 5.6% and 15.1% WT dystrophin levels in the heart and quadriceps, respectively, 12 weeks post-dosing. Taken together, these results suggest that BMN 351 treatment leads to sustained dystrophin production in target tissues up to 12 weeks post-dosing.
Histopathology
In the myocardium of hDMDdel52/mdx mice examined 4 and 12 weeks post-dosing, there were foci of minimal to mild myofiber necrosis, atrophy, and fibrosis accompanied by mononuclear cell infiltrates. These observations, which were not noted in the myocardium of WT mice, were reflective of the untreated hDMDdel52/mdx mouse phenotype. The incidence and/or severity of these findings were decreased in hDMDdel52/mdx mice treated with 18 mg/kg BMN 351 compared with those treated with 6 mg/kg BMN 351 or vehicle 12 weeks after dosing (Fig. 4A; Supplementary Table S2).
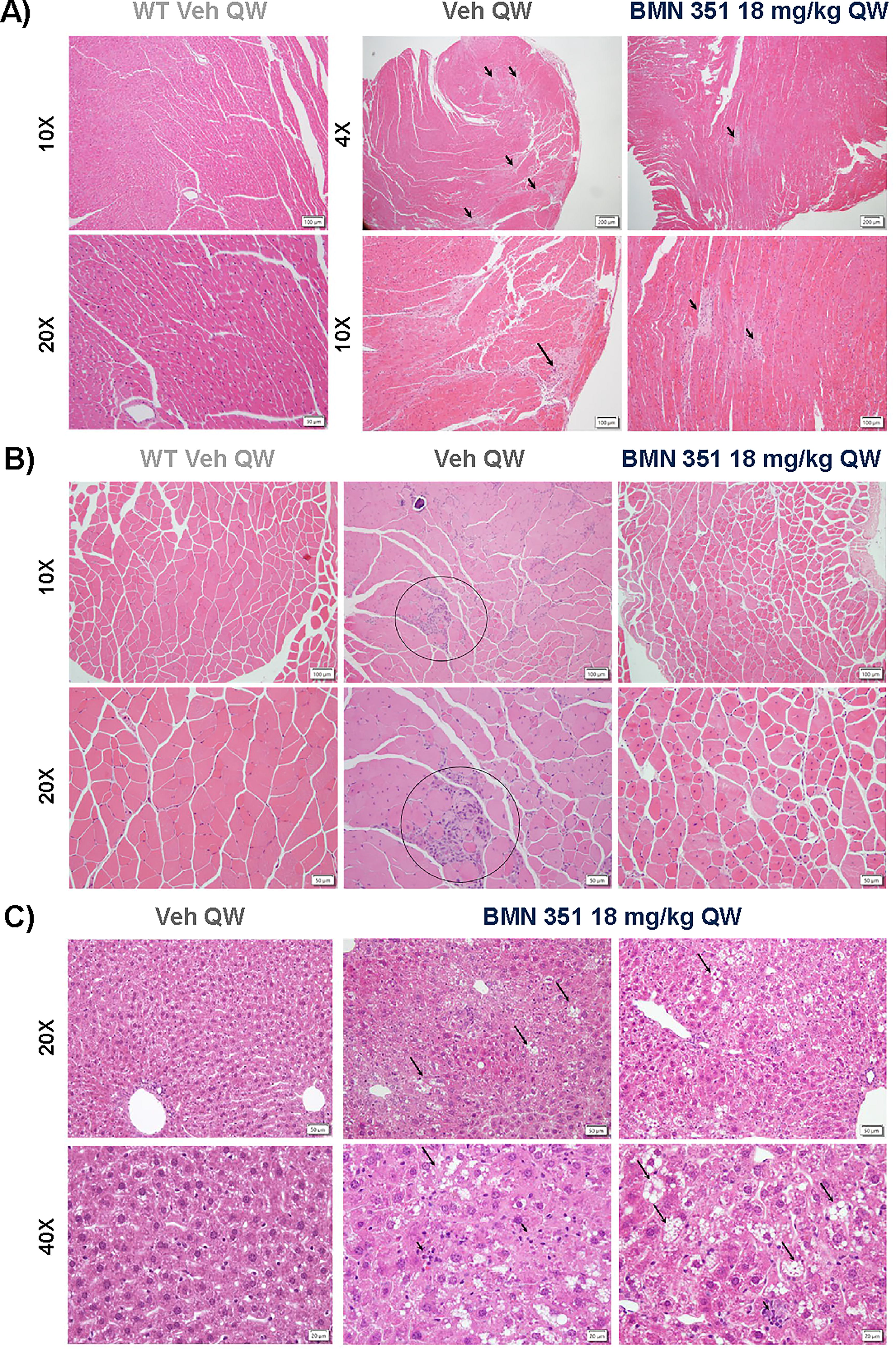
Histological images in WT and hDMDdel52/mdx mice of the
Similarly, in the gastrocnemius muscle, hDMDdel52/mdx mice had low-grade myofiber atrophy, necrosis, regeneration, fibrosis, and mononuclear cell infiltration 4 and 12 weeks post-dosing (Fig. 4B; Supplementary Table S3). At both time points, these findings showed a dose-dependent trend toward lower incidence and severity in BMN 351-treated mice. In 18 mg/kg BMN 351-treated mice, only minimal myofiber atrophy was apparent by 12 weeks post-dosing.
Four weeks after dosing, there was minimal to mild hepatocellular vacuolation, mononuclear inflammatory cell infiltration, and minimal cell necrosis in the liver of 18 mg/kg BMN 351-treated, but not 6 mg/kg BMN 351- or vehicle-treated, hDMDdel52/mdx mice (Supplementary Table S4). These results are generally consistent with findings recognized as background changes in this species. 23 In the context of slightly elevated ALT activity without concurrently elevated CK, they may also suggest mild hepatocellular effects, although the origin of liver and muscle biomarkers was not specifically interrogated. Twelve weeks after dosing, BMN 351-related liver findings were still observed at a similar severity and included minimal or mild hepatocellular vacuolation and mononuclear cell infiltration with 6 and 18 mg/kg BMN 351 treatment, minimal hepatocellular cytoplasmic rarefaction with 18 mg/kg BMN 351 treatment, and minimal single cell necrosis with 6 mg/kg BMN 351 treatment (Fig. 4C). Minimal mononuclear cell infiltration was observed in 2 vehicle-treated hDMDdel52/mdx mice.
Mesenteric lymph nodes were also examined as part of a general survey. Scattered macrophages with faintly basophilic granular/vacuolated cytoplasm were observed in a few mice treated with 6 or 18 mg/kg BMN at one or both time points (data not shown). There were no BMN 351-related microscopic changes in the kidney, tail, spleen, or gallbladder at either time point.
Fine motor kinematics
We evaluated the functional benefit of BMN 351 treatment by evaluating fine motor kinematics as measured by an overall gait score, which characterizes the overall kinematic effects of a pharmacologic agent. Based on BMN 351-induced exon skipping and increases in dystrophin protein levels, we expected BMN 351 treatment to decrease the overall gait score, indicating reduced impairment. Vehicle-treated hDMDdel52/mdx mice had significantly worse overall gait scores at baseline, 4 weeks post-dosing, and 12 weeks post-dosing compared with WT, consistent with recapitulation of DMD symptomatology (P < 0.05; Fig. 5, Supplementary Table S5). BMN 351 treatment at both 6 and 18 mg/kg significantly improved overall gait scores in hDMDdel52/mdx mice compared with vehicle treatment at both time points (P < 0.05; Supplementary Table S6). Overall gait scores for hDMDdel52/mdx mice treated with BMN 351 were comparable to WT mice 4 weeks post-dosing with the 18 mg/kg dose and 12 weeks post-dosing with the 6 mg/kg dose.
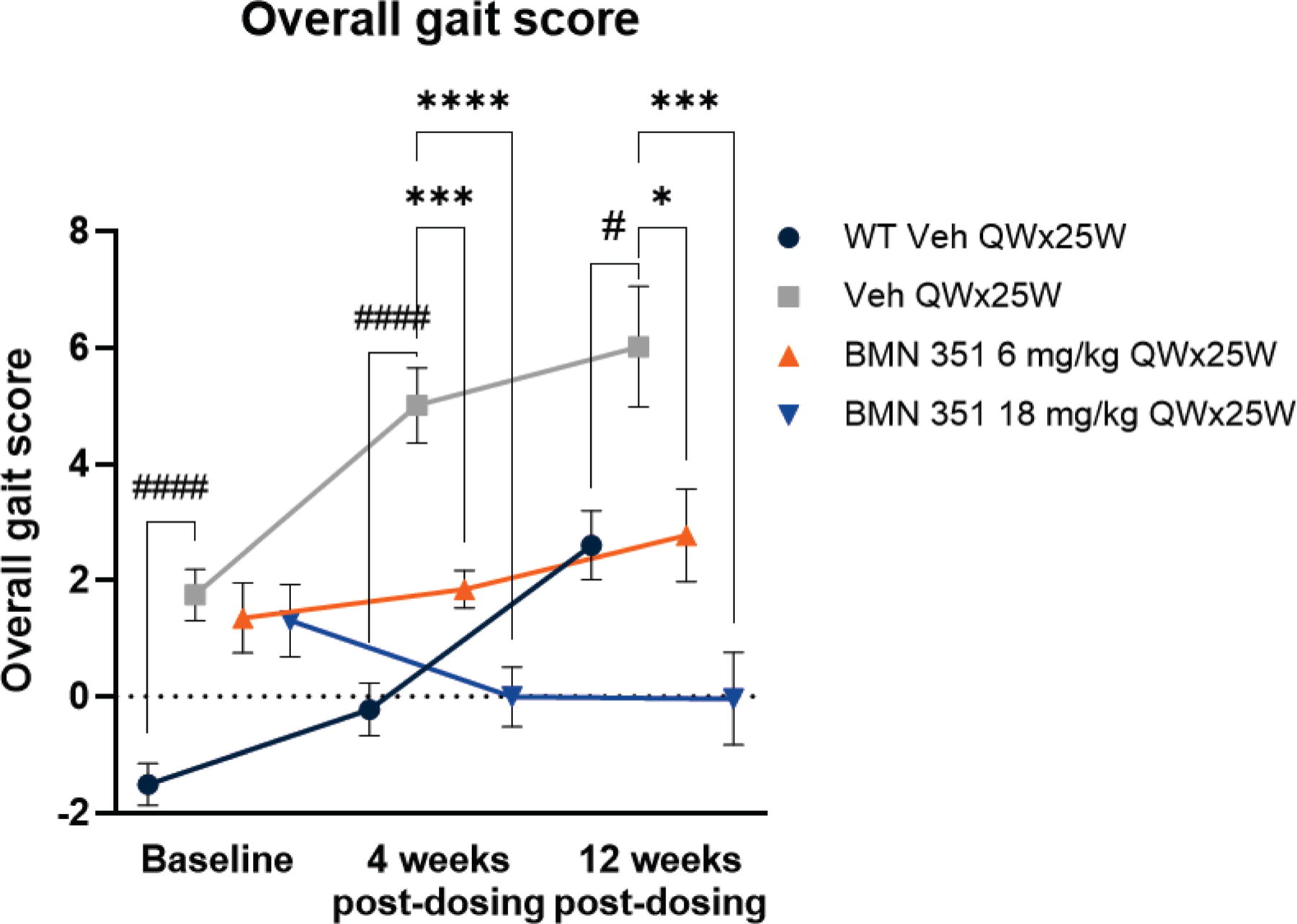
Overall gait scores compared among vehicle-treated WT, vehicle-treated hDMDdel52/mdx mice, and BMN 351-treated hDMDdel52/mdx mice. A lower overall gait score indicates better fine motor kinematic function. Mean ± SEM; n = 10–18 mice/group. #P < 0.05, ####P < 0.0001 for vehicle-treated C57BL/6J mice (WT Veh) versus vehicle-treated hDMDdel52/mdx mice (Veh) assessed with a mixed model fit using REML and uncorrected Fisher’s least significant differences post hoc test; *P < 0.05, ***P < 0.0005, and ****P < 0.0001 for Veh versus BMN 351 6 and 18 mg/kg assessed with a mixed model fit using REML and Dunnett’s post hoc test for multiple comparisons. Mice were hDMDdel52/mdx unless otherwise specified. The nominally administered doses of 6 and 18 mg/kg BMN 351 corresponded to 4.7 and 14.2 mg/kg (0.72 and 2.18 µmol/kg) full-length products, respectively. QW×25W, once weekly for 25 weeks; REML, restricted maximum likelihood; SEM, standard error of the mean; Veh, vehicle; WT, wild-type C57BL/6J.
Exploratory biomarkers and dystroglycan complex proteins
A panel of 39 disease biomarkers that were previously established as significantly different in the quadriceps muscle between WT and hDMDdel52/mdx mice 20 was measured and used to calculate a composite biomarker score (Supplementary Table S7). These proteins are involved in broad functional activities in muscle including the dystrophin glycoprotein complex, muscle contraction, RNA processing, energy metabolism, and immune response. Median biomarker scores in hDMDdel52/mdx mice were > 50% higher with BMN 351 treatment at both doses compared with vehicle both 4 and 12 weeks after dosing (P < 0.05 for all; Fig. 6), suggesting broad BMN 351-induced reduction of muscle disease. Accordingly, higher biomarker scores correlated with better overall gait scores (Supplementary Fig. S2).
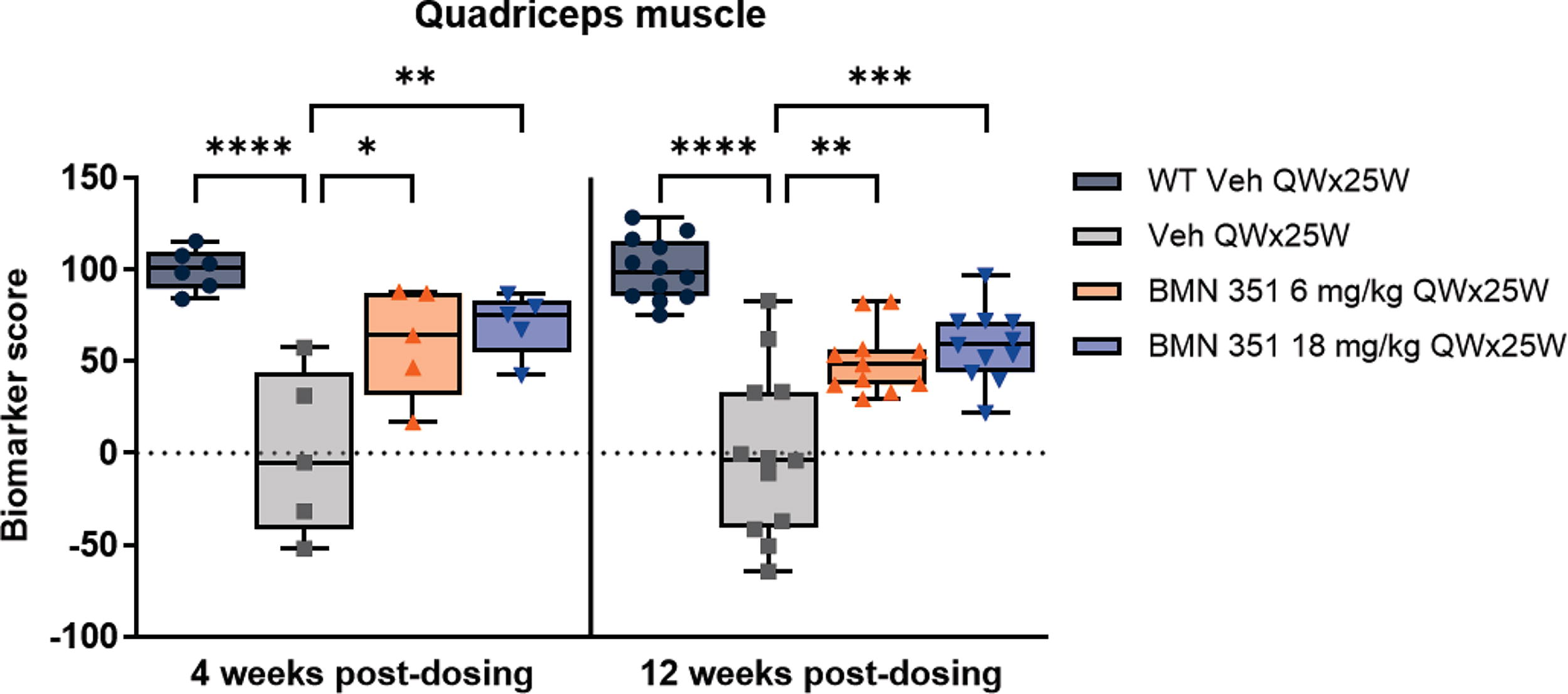
Biomarker scores in the quadriceps muscle 4 and 12 weeks after BMN 351 treatment. Interquartile range (boxes), median (horizontal line), individual range (whiskers), and individual animal scores (points). n = 5–12 mice/group. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 for the comparison indicated by the bracket using a 1-way ANOVA with Tukey’s post hoc test for multiple comparisons. Only the significance for the comparisons of each group with Veh QWx25W is shown. Mean composite biomarker scores for vehicle-treated WT and hDMDdel52/mdx mice were designated as 100 and 0, respectively. Mice were hDMDdel52/mdx unless otherwise specified. The nominally administered doses of 6 and 18 mg/kg BMN 351 corresponded to 4.7 and 14.2 mg/kg (0.72 and 2.18 µmol/kg) full-length products, respectively. ANOVA, analysis of variance; QW×25W, once weekly for 25 weeks; Veh, vehicle; WT, wild-type C57BL/6J.
The dystroglycan complex, which links the extracellular matrix to cytoskeletal elements in skeletal muscles, is unstable without dystrophin. 24 We, therefore, examined the effect of BMN 351 treatment on relative intensities of dystroglycan complex proteins in the quadriceps of hDMDdel52/mdx mice. Expression of these proteins approached WT levels, particularly in mice dosed with 18 mg/kg BMN 351, at both 4 and 12 weeks post-dosing (Supplementary Fig. S3).
In-life observations
There were no differences in mean body weight between vehicle-treated WT and vehicle-treated hDMDdel52/mdx mice (Supplementary Fig. S4). While there was no main group effect between treatment with BMN 351 and vehicle, post hoc comparisons found that 18 mg/kg BMN 351-treated hDMDdel52/mdx mice had lower body weights than vehicle-treated hDMDdel52/mdx mice at weeks 14 through 36 (P < 0.05). No other in-life findings were attributable to BMN 351 treatment.
Clinical chemistry
ALT, AST, LDH, and CK were elevated in vehicle-treated hDMDdel52/mdx mice relative to vehicle-treated WT mice 4 and 12 weeks post-dosing, consistent with muscle injury and expected phenotypes in mice lacking dystrophin (P < 0.05; Supplementary Fig. S5).25–27 In 6 mg/kg BMN 351-treated hDMDdel52/mdx mice, AST, LDH, and CK activities were significantly lower compared with vehicle-treated mice 4 weeks after dosing, while only CK activity was significantly lower 12 weeks after dosing (P < 0.05). The 18 mg/kg BMN 351-treated hDMDdel52/mdx mice had significantly lower AST, LDH, and CK activities relative to vehicle-treated mice 4 weeks post-dosing (P < 0.05), and ALT, AST, LDH, and CK activities were all significantly lower compared with their vehicle-treated counterparts 12 weeks post-dosing (P < 0.05). The generally dose-dependent trend of these declines indicates that BMN 351 treatment ameliorates hDMDdel52/mdx-related muscle injury. However, elevated ALT activity 4 weeks after dosing without concurrently elevated CK activity in mice treated with 18 mg/kg BMN 351 suggests minor hepatocellular effects.
Discussion
DMD is a progressive, fatal disease with devastating effects on daily life caused by muscle loss. 27 Even with holistic care, people with DMD have reduced life expectancy due to cardiac or respiratory failure. 2 ASOs can be used to skip DMD-causing mutations of dystrophin pre-mRNA and may enable the production of functional dystrophin at levels high enough to alleviate impairments and improve prognosis. 11 Here, we evaluated BMN 351, an ASO for exon 51 skip-amenable DMD mutations that contain chemical modifications to improve stability and tissue penetration and target a novel splicing enhancer site in DMD exon 51. BMN 351 aims to ameliorate the disease course of DMD by allowing the production of internally deleted yet functional dystrophin. After dosing hDMDdel52/mdx mice once weekly for 25 weeks, we found that BMN 351 persisted in critical tissues such as the heart and quadriceps muscle. This effect was associated with exon skipping, increased dystrophin levels, and preserved fine motor kinematics through 12 weeks post-dosing.18,19
Myocardial penetration of ASOs is critical for improving cardiac function. Some current ASO chemistries may be limited by low exposure to critical tissues.11,14,28 BMN 351 was designed with locked nucleic acids, 2
While a benchmark for dystrophin-restoring therapies has not been established, dystrophin levels at 10% of healthy muscle may be considered a target. 29 In our study, 18 mg/kg BMN 351-treated hDMDdel52/mdx mice had 29.6% of WT dystrophin in the heart and 54.6% of WT dystrophin in the quadriceps up to 12 weeks post-dosing, though care must be taken in interpreting these values in the context of background dystrophin levels observed in vehicle-treated hDMDdel52/mdx mice. Importantly, in addition to restoring dystrophin levels, ASOs for DMD treatment are designed to produce functional dystrophin structurally similar to the forms found in people with BMD with a mild phenotype. The severity of hDMDdel52/mdx histological phenotypes (eg, myocardial fibrosis and myofiber necrosis) was reduced 12 weeks after dosing in the heart and gastrocnemius muscle of 18 mg/kg BMN 351-treated animals, suggesting that the form of dystrophin produced with BMN 351 treatment is functional. Furthermore, the test material in this study contained 4.7 and 14.2 mg/kg (0.72 and 2.18 µmol/kg) full-length product, indicating that BMN 351 may be more potent than the respective nominal doses of 6 and 18 mg/kg BMN 351.
DMD causes difficulty with propulsion and stability during movement, and people with DMD show differential kinematics while walking at an early age. 30 Consistent with DMD-like fine motor kinematics deficits, hDMDdel52/mdx mice performed significantly worse on a battery of walking tasks compared with WT mice. BMN 351 treatment for 25 weeks at both dose levels prevented altered gait parameters, including maintaining intralimb coordination and hindlimb trajectory in line with WT mice, through 12 weeks post-dosing. This provides strong evidence that BMN 351-mediated exon 51 skipping produces functional dystrophin levels sufficient to maintain motor function in mice lacking endogenous dystrophin.
BMN 351 was generally well tolerated in this study, with some exacerbation of liver findings also noted in WT controls. These observations represent common changes among different ASO categories to which mice may be especially sensitive. 31 Correlating biomarkers were not specific to the tissue of origin, with common sources in the muscle as a function of disease phenotype and in the liver as a function of treatment-related toxicity. Elevations in untreated hDMDdel52/mdx exceeded those in treated animals irrespective of any contribution from hepatic sources, which likely occurred to some degree at the highest dose. In this study, functional endpoints were prioritized—rigorous preclinical toxicity and safety evaluations were performed prior to human dosing.
This study was limited by the lack of an active comparator, which may have provided context to the results of long-term BMN 351 dosing in this mouse model. However, a previous study that directly compared BMN 351 with a clinically relevant comparator with the same sequence and chemistry as SRP-5051 after a 13-week dosing regimen found that BMN 351 reached higher tissue concentration. 20 In addition, because dystrophin levels continued to increase roughly proportionally to exon skipping through 12 weeks post-dosing, it is possible that steady-state dystrophin levels were not reached—a longer duration may have allowed us to determine the durability of the pharmacodynamic effect of BMN 351. Finally, the LC-PRM method used to quantify dystrophin resulted in a background signal in vehicle-treated hDMDdel52/mdx mice that likely represented truncated, non-functional dystrophin; however, because BMN 351 corrects out-of-frame transcripts, we expect that background signal represents a small proportion of the dystrophin levels observed in BMN 351-treated hDMDdel52/mdx mice. Although both LC-PRM and capillary western blotting measured similar mean dystrophin levels in quadriceps samples from ASO-treated hDMDdel52/mdx mice, we chose LC-PRM because it offered greater precision (unpublished data). The clinical relevance of this study benefited from the use of hDMDdel52/mdx mice, which obviate the need for surrogate ASOs due to the inclusion of an exon 52-deleted human DMD transgene. 18
Conclusions
BMN 351 has desirable, dose-dependent pharmacologic effects in mice when administered once weekly for 25 weeks, with the lowest effective dose for maintenance of fine motor kinematic function being 6 mg/kg BMN 351, corresponding to 4.7 mg/kg (0.72 µmol/kg) of full-length product. The sustained dystrophin production and magnitude of motor deficit amelioration associated with BMN 351 treatment in mice support further evaluation of BMN 351. A phase 1/2 study (NCT06280209) will assess the safety and tolerability of BMN 351 in patients with DMD with single ascending and multiple ascending doses administered IV once weekly for up to 56 weeks.
Footnotes
Acknowledgments
The authors thank Britta Handyside of
Data Sharing Statement
The data that underlie the results reported in this article (including text, Tables, Figs., and appendices) will be made available for noncommercial, academic purposes. Additional supporting documents may be available upon request. Investigators will be able to request access to these data and supporting documents via a data-sharing portal beginning 6 months and ending 2 years after publication. Data associated with any ongoing development program will be made available within 6 months after approval of the relevant product. Requests must include a research proposal clarifying how the data will be used, including the proposed analysis methodology. Research proposals will be evaluated relative to publicly available criteria available at ![]() to determine if access will be given, contingent upon the execution of a data access agreement with
to determine if access will be given, contingent upon the execution of a data access agreement with
Author Disclosure Statement
At the time these studies were conducted, all authors were employees and/or shareholders of
Funding Information
This work was funded by BioMarin Pharmaceutical Inc.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.