
Abstract
Exon skipping with antisense oligonucleotides (ASOs) can correct disease-causing mutations of Duchenne muscular dystrophy (DMD) through RNA-targeted splice correction. This correction restores the reading frame and supports expression of near full-length dystrophin. First-generation exon 51-skipping ASOs targeted the same binding site, with limited clinical efficacy. We characterized a novel binding site within exon 51 that induced highly efficient exon skipping. A precursor ASO (AON-C12) and clinical ASO (BMN 351) were designed using 2′-O-methyl-modified phosphorothioate (2′OMePS) RNA and locked nucleic acids. hDMDdel52/mdx mice were given AON-C12 or BMN 351 for 13 weeks and evaluated for molecular and phenotypic correction of dystrophin deficiency. BMN 351 treatment induced durable, dose-dependent levels of exon skipping and dystrophin production in all muscles evaluated. In the heart, 8 weeks after the last BMN 351 dose at 18 mg/kg, exon-skipped transcripts remained at 44.3% of total, and dystrophin levels were 21.8% of wild type. BMN 351 reached higher tissue concentrations and percent exon skipping in the heart than a clinically relevant peptide-conjugated phosphorodiamidate morpholino oligomer comparator. BMN 351 also improved gait scores and clinical and anatomical muscle pathology parameters compared with vehicle-treated hDMDdel52/mdx mice. The pharmacologic activity and safety of BMN 351 warrant further nonclinical and clinical development.
Introduction
Duchenne muscular dystrophy (DMD) is an X-linked progressive neuromuscular disorder characterized by muscle weakness, 1 affecting 1 in 5000 to 6000 live male births.2,3 Symptoms manifest early, with delayed walking noticeable between 1 and 3 years and loss of ambulation by ages 8 to 14 years. 1 Most individuals will require assisted ventilation by 20 years of age, with eventual impairment of cardiac and diaphragmatic muscle representing the primary cause of morbidity and mortality in DMD.2,3
DMD is caused by mutations in the dystrophin gene (DMD) that create deficiency of dystrophin protein in muscle,1,2 which is required for myofiber function and structural integrity.2,3 In DMD, mutations can shift the open reading frame and introduce premature stop codons. Consequently, during protein translation, a shorter, nonfunctional dystrophin protein is produced. Mutations in the DMD gene can also maintain the open reading frame, thus producing an internally deleted protein that retains partial function. These mutations result in a milder disease called Becker muscular dystrophy (BMD). 3
The diversity of DMD-causing mutations makes it difficult to develop broadly applicable therapies. However, targeted therapies for groups of individuals with similar mutations are feasible. For example, the largest cohort of individuals with DMD have mutations amenable to exon 51 skipping. 4 Exon skipping with antisense oligonucleotides (ASOs) is a leading and promising strategy to restore the dystrophin reading frame using RNA-targeted splice modulation. 5 By restoring the open reading frame, ASO-based therapies produce an internally deleted dystrophin protein that retains functionality. 5 Comparisons of dystrophin protein levels in individuals with DMD and BMD suggest that restoring dystrophin levels to at least 10% of its content in healthy muscular tissue may restore a DMD phenotype to a BMD phenotype and therefore represents a real-world validated goal for dystrophin-restoring therapies. 6
Predominately, two types of ASOs have been tested in clinical trials for DMD: 2′-O-methyl phosphorothioate (2′OMePS)-modified RNA and phosphorodiamidate morpholino oligomer (PMO).7–11 Eteplirsen is a PMO that induces exon 51 skipping. Eteplirsen was well tolerated but had limited efficacy, especially in the heart, because of poor tissue penetration of unmodified PMOs.8,9,12 Drisapersen, another ASO targeting the same exon 51 binding site as eteplirsen, used 2′OMePS RNA. 11 However, the drisapersen clinical trials were terminated due to dose-limiting renal toxicity and insufficient efficacy. 5 To improve upon these ASOs, further modifications compatible with the 2′OMePS and PMO chemistries were implemented, including methylation of the nucleotides to improve ASO specificity, 13 addition of locked nucleic acids (LNAs) to improve nuclease resistance, 13 and bioconjugation of the ASOs to lipids, peptides, antibodies, or sugars to improve stability, cell targeting, and penetration. 14 For example, eteplirsen was conjugated to a cell-penetrating peptide that improves tissue penetration and exon skipping to produce SRP-5051 (vesleteplirsen), which is currently in a phase 2 clinical trial (NCT04004065).5,12
Here, we performed an in vitro tiling experiment that validated an alternative binding site within a splicing enhancer of exon 51 and an ASO (AON-C12) that induces exon skipping with high efficiency. 15 We then performed two 13-week in vivo studies to investigate the precursor ASO (AON-C12) and the intended clinical form (BMN 351), targeting the new site using the hDMDdel52/mdx mouse model. These novel ASOs were also evaluated alongside clinically relevant comparator ASOs designed with the sequence and chemistries corresponding to eteplirsen and SRP-5051. BMN 351 treatment in hDMDdel52/mdx mice induced exon skipping and dystrophin production that resulted in improved overall gait scores and pathology parameters. Performance improvement was generally equivalent to the comparator administered at 100 mg/kg once monthly, but BMN 351 administered at 18 mg/kg weekly demonstrated improved durability, especially in the heart and diaphragm.
Materials and Methods
ASO synthesis
AON-C12 is a PS 18-mer ASO that consists of LNAs and 2′OMe- and 5-methyl-2′OMe-C-RNA nucleosides. BMN 351 consists of the same chemistries as AON-C12 with an added 5′ triethylene glycol (TEG). AON-C12 and BMN 351 were synthesized by BioSpring GmbH (Frankfurt, Germany). Comparators 1 and 2 were synthesized by WuXi AppTec Co., Ltd (Tianjin, China) and produced using the sequence and chemistries corresponding to eteplirsen and SRP-5051, respectively. Comparator 1 and Comparator 2 were used as benchmark ASOs to evaluate the potential clinical relevance of AON-C12 and BMN 351. The mass of full-length product (FLP) in the BMN 351 lot was measured using an ion-pair reversed-phase high-performance liquid chromatography with ultraviolet mass spectrometry method.
In vitro screening
The ASO screen used an 18-base single-stranded oligonucleotide comprising 2′-O-methoxyethyl (2′MOE) PS RNA with the inclusion of 5-methylcytosines. ASOs were synthesized by Integrated DNA Technologies (Coralville, IA, USA). The ASOs were designed to cover the entirety of DMD exon 51, with 5 nucleotide offsets, and extended 50 bases into the upstream and downstream introns. In total, there were 64 ASOs in the screening study that spanned exon 51.
An immortalized KM571 human myoblast cell line containing an exon 52 deletion was used for in vitro screening. On day 1, cells were nucleofected with 1.4 µM of individual ASOs for screening, and the solution was diluted 10-fold with skeletal muscle growth media (12.5% heat-inactivated bovine serum and 100 units/mL penicillin/streptomycin) and transferred to collagen-coated tissue culture plates. On day 4, cells were harvested and RNA extracted for cDNA synthesis. Samples were analyzed by quantitative polymerase chain reaction (PCR) with the following primer sets: skip forward, 5′-AAAGCAGCCTGACCTAGCT-3′; skip probe, 5′-ACCACTATTGGAGCCTTTGAAAGA-3′; and skip reverse, 5′-CTTGTACTTCATCCCACTGATTCTGA-3′. A double-stranded DNA fragment of the skipped amplicon (exon 50 spliced to exon 53) was used as a control and to quantify copy numbers in unknown samples (reported as Skip Copies e50-53).
Animal husbandry
All animal experiments were conducted at Charles River Discovery Research Services, Finland, according to the National Institute of Health guidelines 16 and were approved by the National Animal Experiment Board, Finland. Homozygous hDMDdel52/mdx mice 17 were bred at Charles River, United Kingdom, and age-matched C57BL/6J mice were bred at Charles River, Germany, for use as wild-type (WT) controls.
Study design
Vehicle, AON-C12, and BMN 351 were administered by intravenous (IV) tail vein injections (approximately 1-min slow bolus) for 13 weeks. In the AON-C12 study, a total of 124 mice were used, and AON-C12 was administered weekly (QW) at 4.5, 9, and 18 mg/kg (0.7, 1.4, and 2.9 μmol/kg) of body weight. An additional dosing regimen included IV injections once every 2 weeks (Q2W) at 18 mg/kg. Comparator 1 was administered QW at 18 mg/kg (1.8 μmol/kg) of body weight. Mice received their first dose at 7 weeks of age, and dosing continued until study week 13. At study weeks 15, 17, and 21 (2, 4, and 8 weeks after the last dose), blood was collected for clinical pathology investigations. Then, after euthanasia, tissue collection and anatomical pathology evaluations were performed. Cohorts of mice were also assessed in fine motor kinematic testing at 6 weeks of age (baseline) and 1 and 4 weeks after the last dose.
In the BMN 351 study, 72 mice were used, and BMN 351 was administered QW at 18 mg/kg (2.8 μmol/kg) of body weight. The nominal 18 mg/kg dose of BMN 351, adjusted by mass for FLP using a molecular weight of 6,517 g/mol, was 14.2 mg/kg (2.2 µmol/kg). Comparator 2 was administered once every 4 weeks (Q4W) at 100 mg/kg (8.8 μmol/kg) of body weight. Similar dosing and assessment regimens were used as in the previous study.
Plasma pharmacokinetic and in vivo efficacy analysis
On the first and last (13 weeks) injection days, 120 µL of saphenous vein blood (lithium heparin anticoagulant) was collected for pharmacokinetic analysis from three different mice per time point at 0 (within 2–3 min of dosing), 20 min, 1 h, 3 h, and 8 h postdose; blood samples were also collected via cardiac puncture at euthanization. After transcardial perfusion with phosphate-buffered saline, tissue samples were collected and snap frozen using liquid nitrogen. Plasma and a portion of tissue samples were used for ASO measurements using a two-step hybridization enzyme-linked immunosorbent assay (Ardena Bioanalysis, Assen, Netherlands). Briefly, biotin-labeled oligonucleotide capture probes were bound to streptavidin-coated plates. Samples were pipetted into the 96-well plates, and the respective target ASOs were hybridized to the biotin-labeled capture probe. After washing to remove unbound material, a digoxigenin-labeled detection probe was added to the plates. The plates were washed once again, and an anti-digoxigenin antibody conjugated with horseradish peroxidase was added to catalyze color development from the 3,3′, 5,5′-tetramethylbenzidine (TMB) dihydrochloride substrate. Samples for quality control were added at 3.13 nM, 25.0 nM, and 150 nM. Calibration curves for detection of BMN 351 and Comparator 2 can be found in Supplementary Tables S1 and S2.
Exon skipping
Exon skipping in target tissues was quantified by reverse-transcriptase droplet digital PCR (RT-ddPCR) using a method previously described (PRECISION for medicine, Houston, Texas).
18
Briefly, RNA was extracted with Quick RNA Miniprep kits (Zymo Research, Irvine, CA) and quantified with a Qubit RNA HS Assay kit (ThermoFisher, Waltham, MA). RT-ddPCR was performed with a QX200 Droplet Digital PCR system (Bio-Rad, Hercules, CA) using the One-Step RT-ddPCR Advanced kit for probes (Bio-Rad). Separately, for quantifying the percent of DMD exon 51 skipping, the following equation was used:
To investigate whether ASO binding to the target sites could potentially reduce reverse transcription or PCR amplification efficiency and thereby lead to overestimation of exon skip efficiency, the measured number of nonskipped DMD transcripts was plotted for each sample and compared across treatment groups.
Proteomic analysis
Dystrophin levels and exploratory biomarker levels were evaluated by a targeted proteomics approach using liquid chromatography parallel reaction monitoring (LC-PRM; BIOGNOSYS AG, Schlieren, Switzerland). Briefly, tissues samples were homogenized with ceramic beads and lysis buffer. Then, 200 µg of protein per sample was transferred to a 96-well plate, digested overnight with sequencing-grade trypsin at a protein-to-protease ratio of 50:1, and trypsin was removed using a C18 MIDI spin plate according to the manufacturer’s instructions. Samples were spiked with known concentrations of stable isotope-labeled reference peptides. For the LC-PRM measurements, 1 µg of the sample was analyzed using a C18 column on a Thermo Scientific Q Exactive mass spectrometer equipped with a nano-electrospray source. Solvent A was 1% acetonitrile in water with 0.1% formic acid, and solvent B was 15% water in acetonitrile with 0.1% formic acid. LC used a gradient of 5% to 40% solvent B over the course of 60 min, followed by 90% B for 10 min and 1% B for 5 min. The data acquisition window was 8 min for each peptide.
Composite exploratory biomarker panel
The exploratory biomarker panel included 3 housekeeping proteins (glyceraldehyde-3-phosphate dehydrogenase, obscurin, and titin) and 49 target proteins associated with the dystrophin glycoprotein complex, muscle contraction, RNA processing, carbohydrate/energy metabolism, and immune response that were selected based on their relevance to DMD (Supplementary Table S3). A composite biomarker score was then calculated for each sample based on the average normalized protein level from 39 proteins that were statistically different between vehicle-treated hDMDdel52/mdx and C57BL/6J WT mice in the quadriceps as follows, where X represents the relative intensity for each of the 39 proteins included in the composite score:
Fine motor kinematic testing
Baseline fine motor kinematic assessments (MotoRater system, TSE Systems, Berlin, Germany) were conducted as previously described19,20 before dosing, as well as multiple times after the last dose. The assessment examined behavioral tasks associated with walking, such as step, stride, stance, swing analyses, and limb coordination. The limb joints and tail were marked to facilitate data analysis. Movement data were captured using cameras from three different positions. Videos of each mouse were analyzed using SimiMotion software (Simi Reality Motion Systems, Unterschleissheim, Germany) to track the movement of the marked points in relation to the ground. Observed gait patterns were analyzed using a custom-made automated system that evaluates the general gait pattern, body posture, balance, and fine motor skills of the mice. An overall gait analysis score was created using a principal component analysis. Differences between principal components measured for the hDMDdel52/mdx groups and C57BL/6J vehicle group were used to create a “fingerprint” for the disease model; kinematic effects of ASOs were then evaluated by comparing against the fingerprint.
Clinical pathology and anatomical histopathology
Blood was sampled on the day of euthanasia for analysis of clinical chemistry parameters, including alanine aminotransferase (ALT), aspartate aminotransferase (AST), lactate dehydrogenase (LDH), and creatine kinase (CK) activities. Designated animals were submitted to necropsy, and the weights of the heart, liver, spleen, and kidneys were recorded. Tissue samples of the heart, kidney, liver, mesenteric lymph node, gastrocnemius muscle, spleen, and tail (injection site) were collected, formalin-fixed, paraffin-embedded, and cut into 3- to 4-µm-thick sections that were stained with hematoxylin and eosin and submitted for microscopic evaluation.
Statistical methods
All statistical analyses were performed using GraphPad Prism (version 9.45.1; San Diego, CA). Body weight differences were assessed using a Greenhouse–Geisser-corrected mixed-model fit using restricted maximum likelihood (REML). Vehicle-treated groups (hDMDdel52/mdx and WT) were compared using a Šidák’s test for multiple comparisons, and then vehicle-treated and ASO-treated hDMDdel52/mdx mice were analyzed with a Šidák’s test (AON-C12 study) or a Dunnett’s test (BMN 351 study) for multiple comparisons. Exon skipping and dystrophin protein differences were assessed using a two-way analysis of variance (ANOVA) with Dunnett’s post hoc test for multiple comparisons (AON-C12 study) and a two-way ANOVA with Šidák correction for multiple comparisons (BMN 351 study). The significance of biomarker scores was tested separately at each time point using a one-way ANOVA with a Tukey’s post hoc test for multiple comparisons. An unpaired t test (AON-C12 study) and a Welch’s two-sample t test (BMN 351 study) were used for comparison of clinical chemistry parameters between vehicle-treated hDMDdel52/mdx and WT mice, and then vehicle-treated and ASO-treated hDMDdel52/mdx mice were analyzed using a one-way ANOVA with a Dunnett’s multiple comparison test. The overall gait scores were also analyzed using a Greenhouse–Geisser-corrected mixed-model fit using REML; vehicle-treated hDMDdel52/mdx mice were compared with vehicle-treated WT control mice at each time point using a Šidák’s test (AON-C12 study) or a Fisher’s least significant difference post hoc test (BMN 351 study), and vehicle-treated and ASO-treated hDMDdel52/mdx mice were analyzed at each time point using a Šidák’s test (AON-C12 study) or a Dunnett’s multiple comparisons test (BMN 351 study). Pharmacokinetic parameter summary statistics were performed using Phoenix WinNonlin by Certara (v.8.2 or later).
Results
Validation of ASOs targeting an alternative exon 51 binding site
Human KM571 myoblast cells with a DMD exon 52 deletion were used for nucleofection of 64 ASOs spanning the entirety of exon 51. A broad region was identified by ASOs 10 to 27 capable of inducing exon skipping that overlapped with the binding site of drisapersen and eteplirsen. However, ASO 36, corresponding to an ASO first reported by van Deutekom et al., 15 bound to a unique splicing enhancer site, distinct from prior ASOs targeting DMD exon 51, capable of inducing exon 51 skipping with high efficiency (Fig. 1).
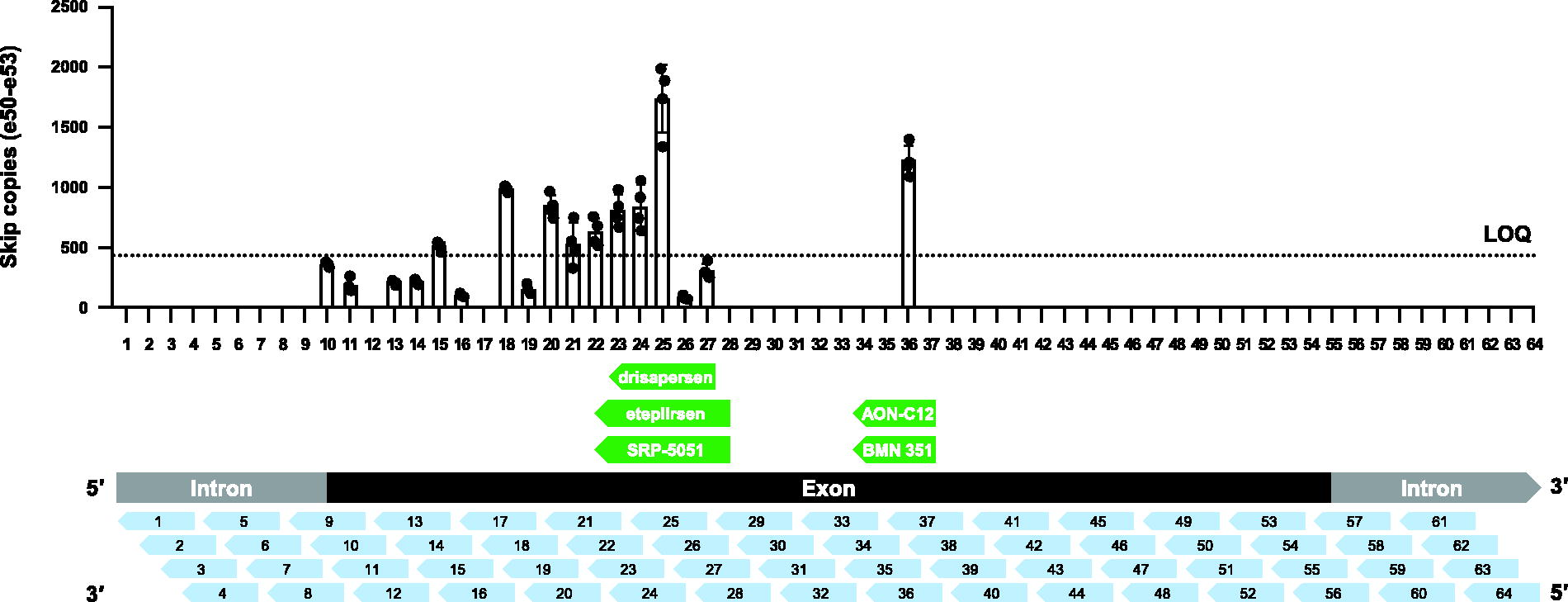
In vitro ASO tiling of DMD exon 51. For the presented results in this article, Comparator 1 corresponds to an ASO matching the chemistries and binding site of eteplirsen, and Comparator 2 corresponds to an ASO matching the chemistries and binding site of SRP-5051. AON-C12 and BMN 351 use the same chemistries and bind to the same unique site. They only differ in the addition of a 5’ triethylene glycol to BMN 351. ASO, antisense oligonucleotide; DMD, dystrophin gene; LOQ, limit of quantitation.
Building on this indication of efficient exon skipping, we conducted preliminary dose range, finding and toxicity studies with a panel of ASOs of varying chemistries targeting the splicing enhancer site identified by ASO 36 (data not shown). These experiments showed stronger induction of exon skipping by ASOs with more optimized chemistries. From these studies, AON-C12 was selected for further development using hDMDdel52/mdx mice to assess pharmacologic activity in a DMD disease-relevant model.
Plasma pharmacokinetics and tissue distribution
IV tail vein injections were used to dose hDMDdel52/mdx mice for 13 weeks with AON-C12 at 4.5, 9, or 18 mg/kg body weight QW or 18 mg/kg Q2W (Fig. 2A). For these studies, Comparator 1 was administered at 18 mg/kg QW and used as a clinically relevant benchmark. Mice were euthanized at week 17 or 21 (4 or 8 weeks, respectively, after the last ASO injection at week 13) for tissue collection. Systemic exposure to AON-C12 increased dose proportionally and, with weekly administration, was slightly higher at week 13 compared with week 1 for all doses evaluated (Table 1). For AON-C12, concentrations were still quantifiable in plasma up to 8 h postdose (Supplementary Fig. S1). Regardless of dose, dosing regimen, or sampling time, the plasma half-life ranged from 0.8 to 1.9 h postadministration. At weeks 4 and 8 postdosing, AON-C12 was detected in the heart and quadriceps muscle (Fig. 2B, C) as well as in the diaphragm, gastrocnemius muscle, liver, and kidney; Comparator 1, an unconjugated PMO, was only detected at low levels in the kidney (Supplementary Fig. S2).
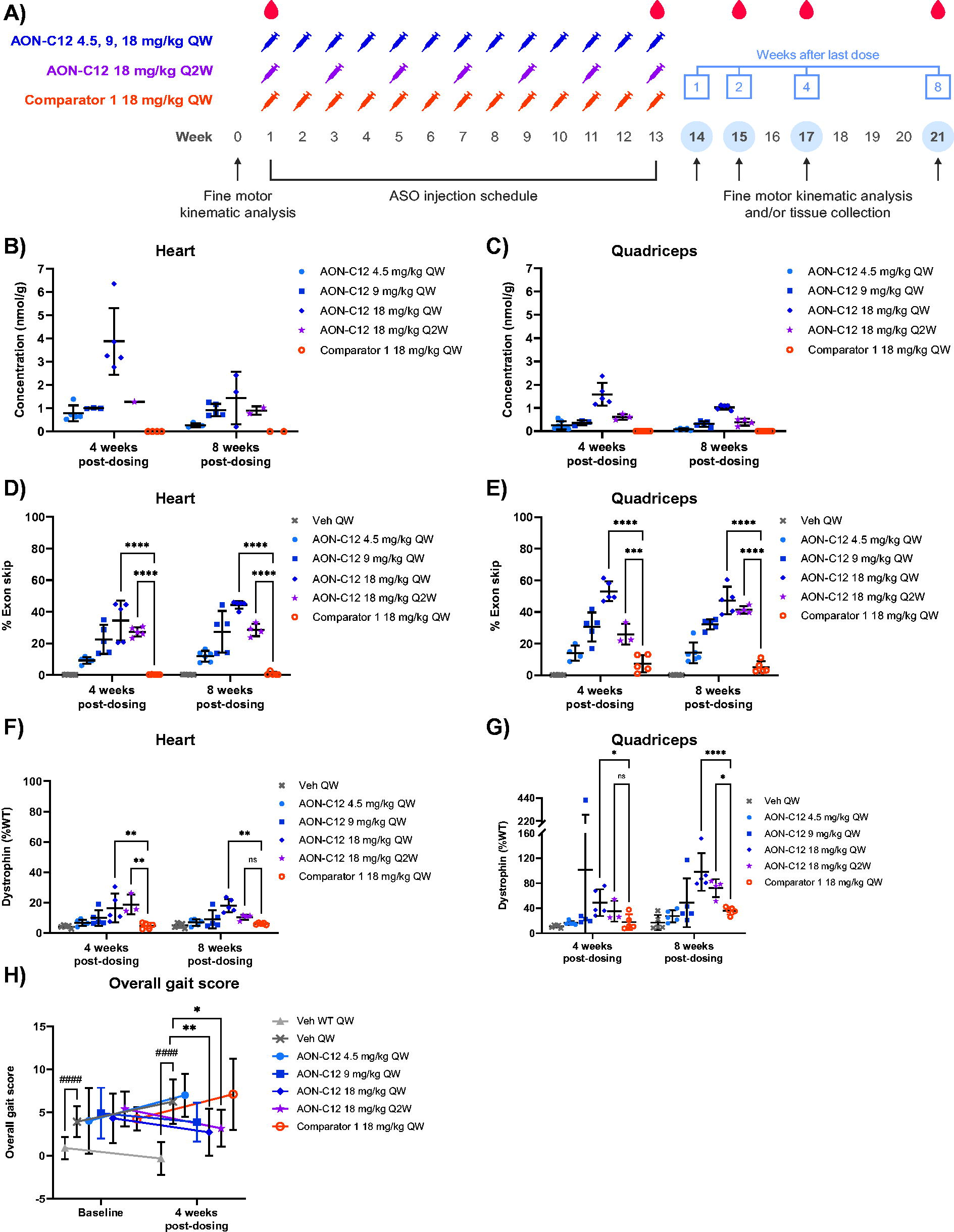
In vivo efficacy evaluation of AON-C12 and Comparator 1.
Plasma Pharmacokinetic Parameters for AON-C12 and Comparator 1 on Day 1 and Week 13 (Day 85)
AON-C12 and Comparator 1 were administered for a total of 13 weeks, and the table presents plasma pharmacokinetic parameters following administration on the first and last day.
ASO, antisense oligonucleotide; AUC, area under the plasma concentration–time curve; AUC0-inf, AUC from time zero extrapolated to infinity; AUC0-t, AUC from start of dose administration to time after dosing at which the last quantifiable concentration was observed; Cmax, maximum plasma concentration; CL, total body clearance of parent test item; NA, not applicable; QW, once every week; Q2W, once every other week; RAUC, the AUC0-t after repeat dosing divided by the AUC0-t during the initial dosing interval; t1/2, terminal elimination phase half-life; VZ, volume of distribution of parent test item based on the terminal elimination phase.
Exon skipping
Exon skipping was quantified by RT-ddPCR and is expressed as the percentage of DMD transcripts with exon 51 skipped relative to total DMD transcripts at weeks 4 and 8 postdosing. AON-C12 treatment at doses of 4.5 to 18 mg/kg induced dose-dependent increases in exon skipping in the heart and quadriceps muscle, with the mean maximum percentage reaching 44.0% and 53.1%, respectively (Fig. 2D, E). Percent exon skipping with AON-C12 treatment in the gastrocnemius and diaphragm muscles also increased dose proportionally (Supplementary Fig. S3). Comparator 1 induced <1% exon skipping in the heart and 7.3% in the quadriceps muscle. The number of nonskipped DMD transcripts in each sample was plotted for each treatment group to assess whether ASO binding interfered with PCR amplification, and no dose-dependent trends were observed for reduced nonskipped copies compared with vehicle-treated controls (Supplementary Fig. S4).
Dystrophin levels
AON-C12 treatment produced dose-dependent increases in dystrophin protein in the heart and quadriceps muscle, with a mean maximum dystrophin expression of 18.0% and 102.0% of WT mouse dystrophin levels, respectively (Fig. 2F, G); dose-dependent increases were also observed in the gastrocnemius and diaphragm muscles (Supplementary Fig. S5). Dystrophin levels for Comparator 1 in the heart were approximately equal to vehicle-treated controls and, in the quadriceps muscle, reached 36.1% of WT mouse levels.
In-life observations and pathology
On average, mice in all treatment groups either gained or maintained weight throughout the AON-C12 study (Supplementary Fig. S6), suggesting that the dose regimens were well tolerated. A total of 12 mice died or were euthanized due to welfare issues in treatment cohorts receiving AON-C12 at doses ≥9 mg/kg of body weight; however, a direct relationship to treatment could not be found despite a detailed pathology examination of 9 of these animals.
Compared with vehicle-treated hDMDdel52/mdx mice, AON-C12-treated mice at the 4- and 8-week postdose intervals had a reduction in skeletal muscle (gastrocnemius) myofiber atrophy, necrosis, and inflammation, corresponding to decreases in muscle-associated serum biomarkers (ALT, AST, CK, and LDH) that surpassed those seen with Comparator 1 in a dose-related manner. Minimal periportal hepatocellular vacuolation and cytoplasmic basophilic granules within the lymph node macrophages were also noted microscopically. Despite this, the liver-associated biomarkers were improved in AON-C12-treated mice across all doses compared with animals treated with vehicle or Comparator 1; however, there is the potential for overlap between liver and muscle origin for ALT, AST, and LDH. In Comparator 1-treated mice, the serum enzyme activities for ALT, AST, CK, and LDH generally remained similar to vehicle-treated hDMDdel52/mdx mice (Supplementary Fig. S7).
Fine motor kinematics
The functional benefit of ASOs was evaluated using fine motor kinematics. For the assessment, an overall gait analysis score was created based on a general gait pattern, body posture, balance, and fine motor skills of the mice. A baseline fine motor kinematic assessment was conducted before dosing as well as at 4 weeks after the last dose. As expected, vehicle-treated hDMDdel52/mdx mice exhibited significantly impaired overall gait scores during fine motor kinematic testing relative to WT controls at all time points (Fig. 2G). At 4 weeks postdosing, treatment with AON-C12 at ≥9 mg/kg significantly improved overall gait scores compared with vehicle (P < 0.05). In Comparator 1-treated mice, overall gait scores remained similar to vehicle-treated mice.
In vivo validation of BMN 351, an optimized ASO to induce exon 51 skipping
The encouraging in vivo pharmacologic activity of AON-C12 triggered further development of ASO candidates targeting the AON-C12 binding site within a splicing enhancer of exon 51. Based on preliminary exposure and toxicity studies (data not shown), a 5′ phosphate-bonded TEG form of AON-C12, BMN 351, was chosen for in vivo validation. BMN 351 and Comparator 2, a peptide-conjugated PMO, were evaluated in hDMDdel52/mdx mice and WT control mice (Fig. 3A). Furthermore, the mass of FLP was measured in the BMN 351 lot to support translation of preclinical results to future clinical applications where dose concentrations will be based on FLP content. For the nominally administered 18 mg/kg BMN 351 dose, 78.9% of the total administered drug mass was the full-length BMN 351 product, with the remaining balance comprising sodium, water, and truncated or altered BMN 351. Accordingly, the dose of BMN 351 used in this study, adjusted by mass for FLP using a molecular weight of 6,517 g/mol, was 14.2 mg/kg (2.18 µmol/kg).
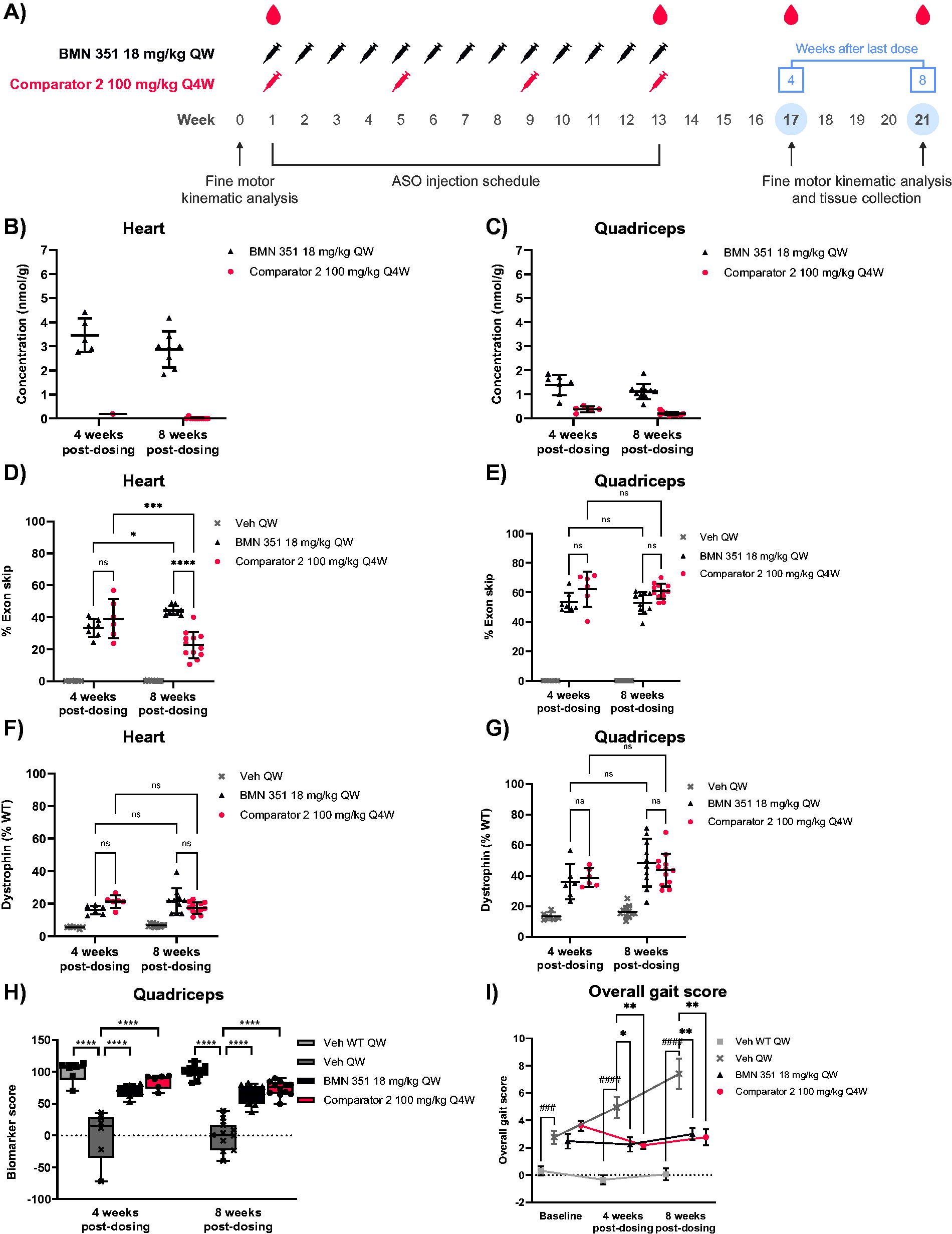
In vivo efficacy evaluation of BMN 351 and Comparator 2.
Plasma pharmacokinetics and tissue distribution
Following ASO administration, the half-life in plasma ranged from 1.2 to 1.6 h for BMN 351 (18 mg/kg QW) and 1.2 to 1.3 h for Comparator 2 (100 mg/kg Q4W; Table 2). Low concentrations of BMN 351 and Comparator 2 remained quantifiable in plasma up to 8 weeks after the last dose (Supplementary Fig. S8). The tissue distribution of BMN 351 at 18 mg/kg QW and Comparator 2 at 100 mg/kg Q4W was evaluated at weeks 17 and 21 (4 and 8 weeks after the last dose). Mean concentrations of ASO in the heart at weeks 4 and 8 postdosing were 3.46 and 2.87 nmol/g for BMN 351 and 0.19 and 0.01 nmol/g for Comparator 2 (Fig. 3B). Mean concentrations of ASO in the quadriceps muscle at weeks 4 and 8 postdosing were 1.39 and 1.12 nmol/g for BMN 351 and 0.38 and 0.20 nmol/g for Comparator 2 (Fig. 3C). Tissue concentrations of BMN 351 and Comparator 2 suggest that BMN 351 has a longer half-life in multiple target muscle tissues than Comparator 2, whereas Comparator 2 shows a similar accumulation to BMN 351 in the kidney (Supplementary Fig. S9).
Plasma Pharmacokinetic Parameters for BMN 351 and Comparator 2 on Day 1 and Week 13 (Day 85)
BMN 351 and Comparator 2 were administered for a total of 13 weeks, and the table presents plasma pharmacokinetic parameters following administration on the first and last day. The nominally administered dose of 18 mg/kg for BMN 351 corresponded to 14.2 mg/kg (2.18 µmol/kg) of full-length product.
ASO, antisense oligonucleotide; AUC, area under the plasma concentration–time curve; AUC0-inf, AUC from time zero extrapolated to infinity; AUC0-t, AUC from start of dose administration to time after dosing at which the last quantifiable concentration was observed; Cmax, maximum plasma concentration; CL, total body clearance of parent test item; RAUC, the accumulation ratio in AUC0-t calculated by AUC0-t after repeat dosing at week 13 divided by the AUC0-t after the initial dosing on day 1; t1/2, terminal elimination phase half-life; VZ, volume of distribution of parent test item based on the terminal elimination phase.
Exon skipping
Mean percent exon skipping in the heart at weeks 4 and 8 postdosing was 33.4% and 44.3% for BMN 351 at 18 mg/kg QW and 39.1% and 22.7% for Comparator 2 at 100 mg/kg Q4W, with BMN 351 inducing significantly higher levels of exon skipping relative to Comparator 2 at 8 weeks postdosing (P < 0.0001; Fig. 3D). In the quadriceps muscle at weeks 4 and 8 postdosing, mean percent exon skipping was 53.4% and 52.9% for BMN 351 and 62.2% and 60.9% for Comparator 2 (Fig. 3E). BMN 351 and Comparator 2 induced similar levels of exon skipping at 4 weeks postdosing in the gastrocnemius muscle and diaphragm, but by 8 weeks postdosing, similar to in the heart, the percentage of exon skipping in the diaphragm declined for Comparator 2 (P < 0.0001; Supplementary Fig. S10). Therefore, exon skipping significantly decreased by 8 weeks postdosing in both the heart and diaphragm for Comparator 2, while it remained constant for BMN 351. These results suggest that BMN 351 had a more durable primary pharmacologic effect than Comparator 2, and the percentage of exon skipping generally correlated with the tissue concentrations of the respective ASO.
Dystrophin levels in target tissues
Dystrophin levels were greater in BMN 351- and Comparator 2-treated hDMDdel52/mdx mice compared with vehicle-treated controls. At 4 and 8 weeks postdosing, mean dystrophin levels in the heart were 16.1% and 21.8% with BMN 351 at 18 mg/kg QW and 21.3% and 17.4% with Comparator 2 at 100 mg/kg Q4W, respectively (Fig. 3F). Mean dystrophin levels in the quadriceps muscle at 4 and 8 weeks postdosing were 36.0% and 48.5% with BMN 351 and 38.7% and 43.7% with Comparator 2 (Fig. 3G). For BMN 351-treated animals, dystrophin levels remained generally constant until the last evaluation at 8 weeks postdosing, suggesting a sustained impact on dystrophin synthesis over time. A similar effect was seen for Comparator 2-treated animals in the quadriceps muscle and heart. However, greater differences between the ASOs on their ability to produce dystrophin protein were likely masked by the long half-life of dystrophin protein and the comparatively short observation period of the study.
Exploratory biomarker analysis
In addition to measuring dystrophin, a panel of exploratory biomarkers was evaluated in the quadriceps muscle to determine if other protein levels generally improved with treatment. The biomarker panel was selected based on unpublished data demonstrating the levels of the proteins were significantly different between WT and mdx4cv mice 21 (an alternative DMD mouse model) and their relevance to muscle function and disease. In the panel, 39 of 49 biomarkers were used to calculate a composite biomarker score (Supplementary Table S3). Mean composite biomarker scores for vehicle-treated WT and hDMDdel52/mdx mice were designated as 100 and 0, respectively. Mean composite biomarker scores at 4 and 8 weeks postdosing improved to 68.0 and 64.5 for BMN 351 and 78.9 and 73.6 for Comparator 2 (Fig. 3H). Importantly, dystrophin glycoprotein complex proteins, which aid dystrophin in mechanical stabilization and signaling by mediating interactions between the cytoskeleton, membrane, and extracellular matrix, were partially restored in hDMDdel52/mdx mice with BMN 351 and Comparator 2 treatment (Supplementary Fig. S11). Overall, improvements in biomarker scores and dystrophin glycoprotein complex proteins were similar between BMN 351 and Comparator 2.
In-life observations and pathology
On average, mice in all treatment groups either gained or maintained body weight throughout the study (Supplementary Fig. S12). Two animals treated with BMN 351 18 mg/kg died in events unrelated to treatment. Compared with vehicle-treated hDMDdel52/mdx controls, observations in BMN 351- and Comparator 2-treated mice reflected less pronounced skeletal and heart muscle injury. Microscopically within the gastrocnemius, there was a decline in muscle fiber atrophy, necrosis, and inflammation; lower severity or absence of myofiber regeneration; and an absence of fibrosis (Supplementary Table S4, Supplementary Fig. S13), while in heart muscle by 8 weeks postdosing, myofiber atrophy and fibrosis were at a lower severity in BMN 351- and Comparator 2-treated mice (Supplementary Table S5, Supplementary Fig. S14).
In the liver of BMN 351- and Comparator 2-treated mice, there were mild mononuclear cell infiltrates at 4 and 8 weeks postdosing (Supplementary Table S6). In the kidney (week 4 postdosing only), cytoplasmic basophilic granules, considered to reflect intracytoplasmic accumulation of the ASO, 22 were occasionally observed in macrophages at the mesenteric lymph node of BMN 351-treated mice and in renal tubular epithelial cells of Comparator 2-treated females. Serum biomarkers of muscle and liver injury (ALT, AST, CK, and LDH) were also decreased by week 4 postdosing in BMN 351- and Comparator 2-treated mice compared with vehicle-treated hDMDdel52/mdx animals (Supplementary Fig. S15). No BMN 351- or Comparator 2-related gross abnormalities or organ weight changes were observed at either time point.
Fine motor kinematics
For the fine motor kinematic testing at 4 and 8 weeks postdosing, BMN 351 and Comparator 2 treatment significantly improved overall gait scores compared with vehicle-treated hDMDdel52/mdx controls (P < 0.05; Fig. 3I). The quadriceps muscle biomarker scores were also positively correlated with overall gait scores (Supplementary Fig. S16). Collectively, these results provide a functional validation of the dystrophin protein restoration observed with BMN 351 treatment.
Discussion
We present a combination of in vitro and in vivo drug candidate optimization and comparator experiments that expands upon a recently identified site within DMD exon 51 to induce exon skipping. 15 Multiple ASOs were evaluated to optimize chemistries, and BMN 351 was selected for further nonclinical and clinical development. BMN 351 is a fully modified 18-mer with 2′OMe- and 5-methyl-2′OMe-C-modified PS RNA conjugated to 5′ TEG and stabilized against enzymatic degradation with LNA modifications. It binds to exon 51 of the dystrophin pre-mRNA, inducing exon 51 skipping, and restores the open reading frame, leading to the production of an internally deleted but functional dystrophin protein.
We used therapeutically relevant benchmark comparator ASOs to assess the clinical significance of BMN 351 and its precursor. We chose a dose of Comparator 1 that is below the clinical dose to match, on a per-molecule basis, the middle dose of our experiments with the BMN 351 precursor. This study design included a dose of AON-C12 equivalent to Comparator 1 as well as a dose above and below for our lead optimization experiments. In the second 13-week study, we compared the optimal dosing regimen of BMN 351 (18 mg/kg QW) versus a relevant dose of Comparator 2 in mice (100 mg/kg Q4W).
The hDMDdel52/mdx mouse model was used to explore the pharmacokinetics, pharmacodynamics, and disease-related phenotypic impact of BMN 351 and its precursor AON-C12 at doses ≤18 mg/kg administered for 13 weeks. Importantly, for the assessments of exon skipping induced with AON-C12, which contains LNAs, there was a report suggesting that ASOs with LNA modifications have the potential to overestimate exon skipping levels due to the binding affinity of LNAs to RNA. 23 However, our results suggest that at least for the specific ASOs investigated in this study, there was no interference with reverse transcription or ddPCR. The promising results from AON-C12, including improvements in clinical chemistry parameters over Comparator 1, warranted further evaluation and led to the development of BMN 351. Detection of BMN 351 was confirmed in all target tissues evaluated, including the heart, diaphragm, quadriceps, and gastrocnemius muscles. Dose-proportional DMD mRNA exon 51 skipping and increased dystrophin protein production were observed at all evaluated doses. Importantly, pharmacologic effects continued throughout the last observation period at 8 weeks postdosing. Compared with vehicle, BMN 351-treated mice had improved clinical chemistry and exploratory disease-related biomarkers, the gastrocnemius muscle and heart had less microscopic evidence of injury, and overall gait scores improved. Collectively, these changes indicate that BMN 351 achieved excellent tissue concentrations in the heart and diaphragm, muscle groups directly related to the morbidity and mortality of DMD,3,24 and restored muscle-related function in a disease-relevant mouse model.
One difference between BMN 351 and the AON-C12 precursor is the addition of a 5′ TEG. This additional modification was selected to help reduce toxicity. For example, in an in vitro drug candidate optimization experiment, the addition of a 5′ TEG may have contributed to reduced complement activation (unpublished data). The potential improvement of BMN 351 over other ASOs is derived from novel chemical modifications to resist endonucleases and improve durability. This impact is best exemplified by the ASO tissue concentrations observed in target muscles at 4 and 8 weeks postdosing. The last doses of BMN 351 (18 mg/kg QW) and Comparator 2 (100 mg/kg Q4W) were administered at week 13, and despite the higher dose of Comparator 2, tissue concentrations were greater for BMN 351 in all target muscle groups at 4 and 8 weeks postdosing. Furthermore, the total dose of BMN 351 used over the course of the 13-week study (234 mg/kg; 185 mg/kg corrected for FLP) was less than Comparator 2 (400 mg), suggesting a possible superiority at a milligram level. By extension, to maintain therapeutic levels of dystrophin protein, BMN 351 may offer the potential benefit of reduced dosing frequency due to its improved durability. The difference in tissue concentrations between BMN 351 and Comparator 2 was most notable in the heart, where BMN 351 levels were relatively stable throughout the study, while Comparator 2 was barely detectable at 8 weeks postdosing. Improved tissue retention of BMN 351 in the heart likely contributed to the percentage of exon skipping remaining constant, or even increasing slightly at 8 weeks postdosing, whereas Comparator 2 levels were beginning to decline. Although BMN 351 induced more exon skipping in the heart, there were comparable levels of dystrophin protein observed for both ASOs at 8 weeks postdosing.
The similar levels of dystrophin protein observed with BMN 351 and Comparator 2 at 8 weeks postdosing could reflect differences in exposure–response relationships previously described between 2′OMe-modified PS-based and PMO-based ASOs. 25 Accordingly, despite higher tissue concentrations of BMN 351, the lower levels of Comparator 2 may be able to induce similar levels of exon skipping and dystrophin expression. An alternative interpretation is that these results identified a weakness of the presented analysis and suggest that the beneficial effect of BMN 351 extends beyond the duration of follow-up in this study. The analysis of ASO concentrations in various tissues at 4 and 8 weeks postdosing effectively captured the improved durability of BMN 351 over Comparator 2. However, the declines in Comparator 2-induced exon-skipped mRNA are just beginning to emerge in the heart and diaphragm. Therefore, resulting declines in dystrophin protein, which has a longer half-life, are not expected until time points beyond those included in the present study. To better characterize the effects of an ASO with improved durability, a long-term study with BMN 351 administered for 25 weeks and a 12-week postdose analysis were also performed. 26 For both studies, dystrophin protein was detected using an LC-PRM method in place of the traditional capillary western blot. The LC-PRM method was chosen because in a prior study to compare the detection of dystrophin in quadriceps samples from ASO-treated hDMDdel52/mdx mice, the LC-PRM and capillary western blot methods yielded similar mean dystrophin levels, but the LC-PRM method offered greater precision (unpublished data). However, a limitation of these analyses was the use of an N-terminal peptide (relative to the ASO binding site) for dystrophin quantitation that led to detection of background levels of dystrophin protein in vehicle-treated hDMDdel52/mdx mice, which likely represents truncated, nonfunctional dystrophin protein produced without exon skipping. Alternatively, with ASO treatment, the truncated form of dystrophin as a percent of total dystrophin is likely reduced, and therefore, background levels of dystrophin observed in vehicle-treated controls cannot simply be subtracted from ASO-treated mice, as it would underrepresent the treatment effect.
A further limitation is that FLP was only quantified for BMN 351, and corresponding data are not available for AON-C12 or the comparator ASOs. The FLP analysis of BMN 351 was conducted to support alignment with future good laboratory practice toxicology and clinical studies where dose concentrations are based on FLP content. The results also indicate that the potential efficacy of BMN 351 may be higher than what is presented here, considering that the dose corrected for FLP was approximately 21% lower than the intended target.
The effects of ASOs on restoration of dystrophin are transient; thus, to maintain efficacy, lifelong administration of the ASOs is required. Renal clearance of ASOs and repeated ASO injections has raised concerns about off-target accumulation and renal toxicity. The improved pharmacologic durability of BMN 351 supported greater tissue retention in target muscles, such as the heart and diaphragm, without a concomitant increase in kidney accumulation compared with Comparator 2. This could be advantageous for the long-term implications of repeated BMN 351 administrations but requires further investigation. Microscopic findings in macrophages from the liver and lymph node were not pronounced and represent common changes among different ASO classes, to which mice may be especially sensitive with respect to liver effects. 22
Conclusion
BMN 351 is a chemically modified 18-base 2′OMePS RNA oligonucleotide containing a 5′ TEG modification that induces durable exon 51 skipping by targeting a novel binding site. In an hDMDdel52/mdx mouse model, it induced dystrophin production with high efficiency in skeletal muscles as well as in the heart and diaphragm. BMN 351 could potentially provide clinically meaningful improvement over current DMD therapies and is currently being evaluated in a phase 1/2 trial to assess the safety and tolerability in individuals with DMD (NCT06280209).
Footnotes
Acknowledgments
Medical writing support was provided by Tony Sallese, PhD, of AlphaBioCom, a Red Nucleus company, and funded by BioMarin Pharmaceutical Inc. The authors thank Britta Handyside of BioMarin Pharmaceutical Inc. for her contributions to the development of the biomarker panel.
Data Sharing Statement
The data that underlie the results reported in this article (including text, tables, figures, and appendices) will be made available for noncommercial, academic purposes. Additional supporting documents may be available upon request. Investigators will be able to request access to these data and supporting documents via a data-sharing portal beginning 6 months and ending 2 years after publication. Data associated with any ongoing development program will be made available within 6 months after approval of the relevant product. Requests must include a research proposal clarifying how the data will be used, including a proposed analysis methodology. Research proposals will be evaluated relative to publicly available criteria available at ![]() to determine if access will be given, contingent upon execution of a data access agreement with
to determine if access will be given, contingent upon execution of a data access agreement with
Author Disclosure Statement
At the time these studies were conducted, all authors were employees and/or shareholders either of BioMarin Pharmaceutical Inc. or Charles River and its affiliates.
Funding Information
This work was funded by BioMarin Pharmaceutical Inc.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.