
Abstract
Amido-bridged nucleic acid (AmNA) and a 2′-O,4′-C-spirocyclopropylene bridged nucleic acid (scpBNA) are bridged nucleic acid analogs with high binding affinity toward complementary strands along with high nuclease resistance. AmNA and scpBNA have been developed to overcome phosphorothioate modified gapmer hepatotoxicity, while the mechanism of reducing hepatotoxicity still remains unknown. Here, we found that antisense oligonucleotides (ASOs) with combination of AmNA, scpBNA, and phosphodiester (PO) bonds could significantly reduce hepatotoxicity in mice. Histopathological findings of the periportal spaces of the liver were observed only in the locked nucleic acid and AmNA-scpBNA groups, but not in the AmNA-scpBNA-PO group. Furthermore, bioinformatics and histopathological analysis revealed that the reduced hepatotoxicity might be related to mitochondrial abnormalities, such as decreased expression levels of Atp5o and Sdhb genes. Taken together, the results of this study demonstrated that AmNA, scpBNA, and PO modification are able to reduce hepatotoxicity for improving the potential of ASOs.
Introduction
Antisense oligonucleotides (ASOs) are short, synthetic, and single-stranded deoxyribonucleotides regulating the target RNA expression through Watson-Crick base pairing. A gapmer-type ASO consists of a central region of deoxyribonucleotides (gap) flanked by 2′-modified sugar nucleotides region (wings), which can induce RNase H1-mediated target degradation. In a gapmer, the internucleotide linkages are modified with phosphorothioate (PS) bonds, where the nonbridging oxygen is replaced with a sulfur. The PS modification can improve nuclease stability and cellular/nuclear uptake and is therefore used as a favorable backbone modification in therapeutic ASOs. While PS gapmers are capable of improving metabolic stability and pharmacokinetics, they can also exhibit considerable cellular toxicity caused by increased tissue distribution and nonspecific protein interaction. Moreover, chemically modified nucleotides can strongly bind proteins, more than that by deoxynucleotides. 1 Cell surface proteins such as stabilin and integrin show binding specificity to the modified gapmers and play a key role in effective cellular uptake. Furthermore, the modified gapmers show nonspecific binding to numerous cytoplasmic and nuclear proteins because of the hydrophobicity and bridged sugar moieties.2–5 Locked nucleic acids (LNAs) contain a bridged structure in their ribose sugar, which improves thermodynamic stability and complementary strand binding affinity.6,7 Therefore, modification of ASOs with LNAs is a good strategy to enhance potency. 8 Although its potential is strong enough, there are as yet no FDA-approved LNAs and several rodent studies reported the safety concern of these modified oligonucleotides.9,10 One possible cause could be unintended off-target RNA knockdown by high affinity LNA gapmers, 11 Another possible cause could be robust nonspecific binding to proteins observed with LNA gapmers. Several modified gapmers form a paraspeckle protein ASO complex and subsequently delocalize to become nuclear paraspeckle protein in nucleoplasm, which causes nucleolar stress in cells.12,13 This nucleolar stress is known to activate caspases in vitro and cause hepatotoxicity in vivo. 13
Recently, several modified nucleic acids have been developed to reduce the toxicity of modified gapmer caused by protein binding. Incorporation of 2′-O-methyl (2′-OMe), mesyl-phosphoramidate bond and 5′-methyl DNA in the gap region lowers protein binding and cytotoxicity.13–15 All these modifications potentially overcome hepatotoxicity and improve therapeutic profile of the modified gapmer. Another strategy to mitigate the hepatotoxicity is an introduction of modifications toward wing regions of the modified gapmer. Amido-bridged nucleic acid (AmNA) is a modified nucleic acid with a hydrophilic amide bond bridge between the 2′ and 4′ carbons of the sugar16–18 , and 2′-O,4′-C-spirocyclopropylene bridged nucleic acid (scpBNA) is 2′-O,4′-C-methylene bridged nucleic acid (2′,4′-BNA/LNA) bearing a cyclopropane ring at the 6′-position. Both AmNA and scpBNA exhibit high affinity towards target RNA and extremely high nuclease resistance.19,20 Therefore, these are valuable replacement of LNA in the modified gapmer to overcome hepatotoxicity.
Here, we report the reduction of hepatotoxicity by incorporation of AmNA, scpBNA, and partial phosphodiester (PO) bond into wing regions of a toxic modified gapmer by using scavenger receptor class B member 1 (SR-BI, Scarb1) as our target. Scarb1 is known for its role in reverse cholesterol transport as it takes up cholesterol esters from high-density lipoprotein (HDL). 21 Shen and colleagues demonstrated that the constrained ethyl bridged nucleic acid (cEt) gapmer ISIS 449093 targeting Scarb1 is highly toxic and causes hepatotoxicity in mice. 13 On the other hand, 2′-O-methoxyethyl gapmer has been reported to have low hepatotoxicity and we considered it to be a suitable compound for evaluating the effects of modification on toxicity. 22 Thus, we used this Scarb1 gapmer as a tool compound. We found that AmNA and scpBNA, alone or in combination, in the gapmer can reduce hepatotoxicity effectively, and incorporation of PO bonds would further reduce hepatotoxicity. Notably, the combination of AmNA and scpBNA with partial PO bonds retained activity which was comparable to that without PO bonds. Our results provide new insights into the toxicity mechanisms of modified gapmers. Furthermore, combination of AmNA, scpBNA, and partial PO bond modifications can be a new scaffold to enhance therapeutic profiles and bring findings to understand biochemistry of modified gapmer toxicity.
Material and Methods
Oligonucleotide synthesis and purification
ASOs targeting Scarb1 sequence were designed using LNA, AmNA, and scpBNA as shown in Fig. 1A and B. Scarb1 gapmers were synthesized on a 10 μmol scale using an NS-8II Synthesis System (Genedesign, Inc.) according to the standard phosphoramidite protocol. 5-[3,5-bis(trifluoromethyl)phenyl]−1H-tetrazole (Activator 42®) was used as the activator, and Cap mix A (10% acetic acid in tetrahydrofuran) and Cap mix B (10% 1-methylimidazole in tetrahydrofuran/pyridine) were used as the capping agents. 0.05M DDTT Sulfurizing Reagent [3-((dimethylaminomethyliden)amino)−3H-1,2,4-dithiazol-3-thione] (ChemGenes) in pyridine/acetonitrile was used for thiolation and 0.02M iodine in tetrahydrofuran/water/pyridine was used for oxidation. The standard synthesis cycle (trityl off mode) was used for assembly of the reagents and synthesis of the oligonucleotides, except that the coupling time was extended to 30 min for the AmNA, scpBNA monomers. The phosphoramidites were dissolved in anhydrous acetonitrile at 0.1M concentration. Unylinker CPG-solid supports from Chemgenes were used. After synthesis, the synthesized oligonucleotides were treated with 20% diethylamine in acetonitrile for 1 h and cleaved from the solid support and deprotected by treatment with triethylamine:methanol:water (1:1:2, v/v/v) at 65° C for 15 h. The oligonucleotides were purified by ion-exchange chromatography on a Akta pure 25 using aqueous buffer with 20 mM Tris-HCl (pH 8.0) and 2M NaClO4. Pure fractions were desalted on sephadex G-25 column (cytiva Hiprep 26/10 desalting) and lyophilized. Purity and mass of oligonucleotides were determined using ion-pair LCMS.
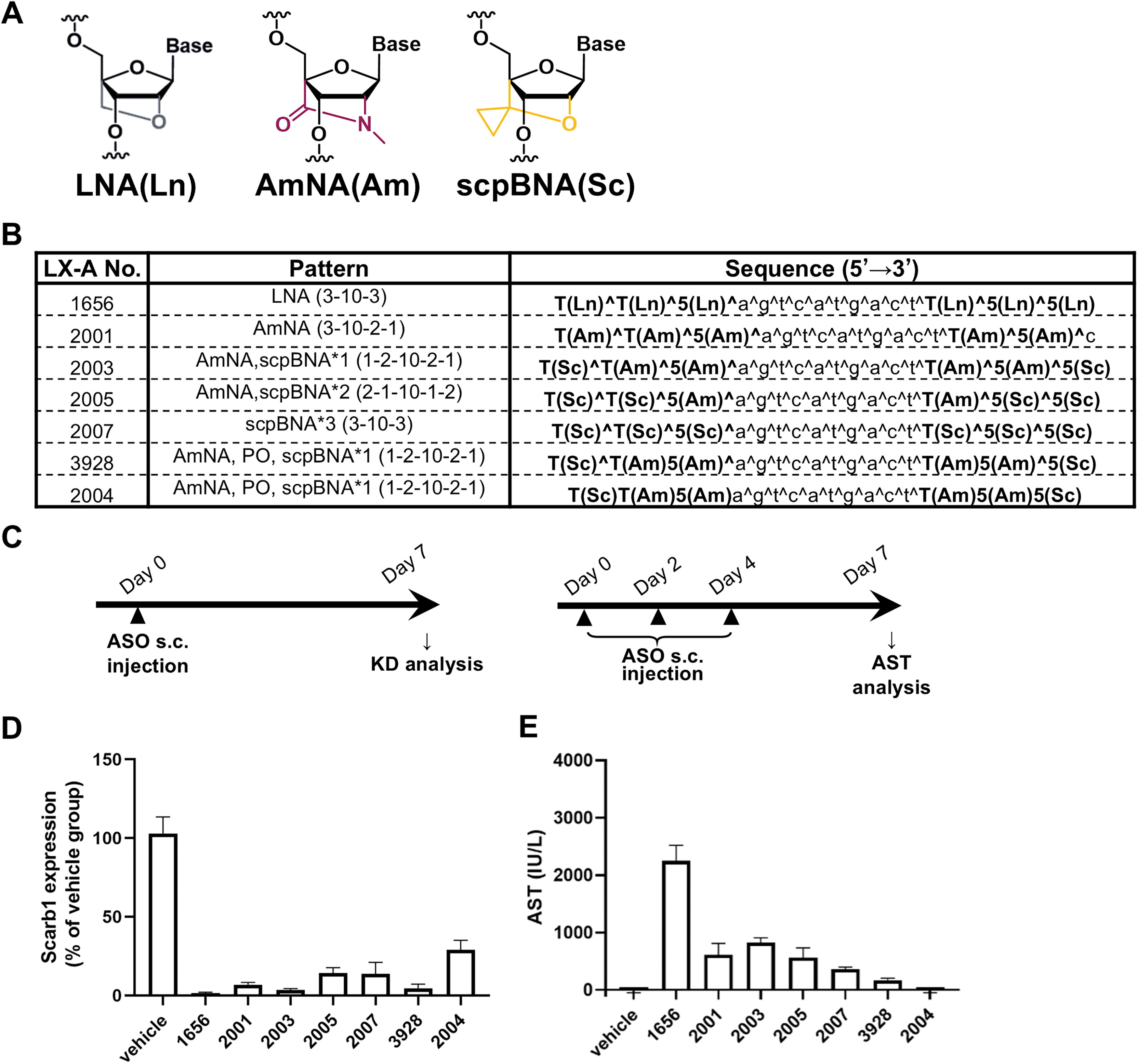
AmNA and scpBNA suppressed elevation of AST level.
In vivo studies in mice
Animal experiments were conducted in accordance with a protocol approved by the Animal Care and Use Committee at Luxna Biotech. Male 6-week-old mice (BALB/cCrSlc) were purchased from CLEA Japan and allowed to acclimatize for 1 week before the administration. In the 1st experiment, modified gapmers were administered via subcutaneous single injection at a dose of 30 mg/kg (Day 0) for RNA expression analysis and three times injection at a dose of 30 mg/kg (Days 0, 2, and 4) for toxicity testing. Tissues were collected 7 days after administration. Laparotomy was performed for all animals under isoflurane anesthesia (1.5%–3%). After macroscopic examination for abdominal organs and tissues, blood samples were collected from the inferior vena cava using a heparin-added syringe. After the blood sampling, animals were euthanized by exsanguination and then the livers were dissected. Liver samples were subsequently saved in RNAlater Solution for RNA expression analysis. In the 2nd experiment, modified gapmers were administered via subcutaneous single time (Day 0), two times (Days 0 and 2), or three times (Days 0, 2, and 4) at a dose of 30 mg/kg. Tissues were collected 1 day after single administration (Day 1), 1 day after two times administration (Day 3), or 3 days after last administration (Day 7). Blood samples were collected and livers were dissected as described above. The left lateral lobe was separated from each liver. For the histopathological examination, a piece of transversal tissue was obtained from the left lateral lobe of the liver by a razor with about 2 mm thickness and it was immersed in 10 vol% neutral buffered formalin and fixed at room temperature. For the electron microscopic examination, 3–5 pieces of cubic tissue were obtained from the left lateral lobe of the liver by a razor with about 1 mm3 size and they were immersed in 2.5 w/v% glutaraldehyde/0.1 mol/L phosphate buffer in cold storage. For the transcript analysis, a piece of tissue was obtained from the right lateral lobe of the liver and they were immersed in RNAlater Stabilization Solution (Thermo Fisher Scientific).
RNA extraction and quantitative polymerase chain reaction analysis
RNA extraction from tissues was according to manufacturer instructions following beads homogenization using Tissue Lyser II (Qiagen) in TriPure isolation reagent (Roche). TaqMan qRT–PCR was performed using QuantiNova PCR Kits (Qiagen) and TaqMan Gene Expression Assays (Thermo Fisher Scientific). In brief, reverse transcription was performed at 50°C for 10 min; then the reactions were denatured at 95°C for 5 min and 40 cycles of PCR reactions were performed at 95°C for 15 s and 60°C for 30 s, using CFX Connect Real-Time PCR detection system (Bio-rad). RNA expression levels were analyzed by CFX Maestro Software (Bio-Rad).
Clinical pathology
Blood samples were centrifuged at 7,500 × g for 10 min at room temperature to obtain plasma. The values of total cholesterol (T-Cho), triglyceride (TG), urea nitrogen (UN), creatinine (Cre), aspartate aminotransferase (AST), alanine aminotransferase (ALT), glutamate dehydrogenase (GLDH), and total bile acid (TBA) were determined with an automated blood chemistry analyzer (LABOSPECT008, Hitachi High-Technologies Corporation, Tokyo, Japan) and standard reagents. The analysis methods were shown in Supplementary Fig. S1A.
Histopathology
The liver tissues for the histopathological examination were embedded in paraffin, sectioned at a thickness of ∼4 μm, stained with hematoxylin and eosin (HE), and examined microscopically.
Electron microscopy
The liver tissues for the electron microscope examination were rinsed in 0.1 mol/L phosphate buffer with 6 w/v% sucrose (pH 7.2–7.4) and then postfixed with 1 w/v% osmium tetroxide and embedded in epoxy resin. After fields were selected from the toluidine blue-stained sections, ultra-thin sections were made and stained with uranyl acetate and lead citrate, and were examined by electron microscopy (H-7600, HITACHI). The electron microscope examination was conducted for the 3-times dosing groups.
Immunohistochemistry for modified gapmer
The paraffin embedded liver tissues for histopathology were sectioned at a thickness of ∼4 μm and mounted on a coated glass slide (Matsunami, Cat No. CRE-1). The method for immunohistochemistry was shown in Supplementary Fig. S1B.
Library preparation and RNA-sequencing
Sequence library was prepared using the Ion AmpliSeq Transcriptome Mouse Gene Expression Kit (Thermo Fisher Scientific) according to the manufactures’ instruments. Then sequence libraries were sequenced on the Ion S5 Sequencer (Thermo Fisher Scientific). Base calling, quality filtering, read mapping, and gene expression quantification were performed by the Ion Torrent Browser plugin.
Bioinformatics analysis
Gene expression count data were normalized to counts per million and logarithmic transformed using R v3.6.2 software. After filtering out the low expressed genes with the mean of value under −1, gene expression was normalized by using the TMM (Trimmed mean of M values) method on edgeR v3.28.1 package. Principal component analysis was performed using the procomp function in R software. Differential gene expression analysis was carried out using voom function in limma v3.42.2 package. Differentially expressed genes (DEGs) compared with PBS were defined as genes which yielded P value <0.05 and fold change >2 or fold change <0.5. Functional enrichment analysis of upregulated or downregulated DEGs using Gene Ontology or Reactome database was conducted by cluster Profiler v3.14.3 and ReactomePA v1.30.0 packages, respectively. P value of upregulated DEGs and that of downregulated DEGs were combined by Fisher′s method and we extracted significantly enriched GO terms or pathways that yielded combined P value <0.05. We constructed the gene co-expression network using WGCNA v1.70–3 package and clustered genes into co-expression modules. Functional enrichment analysis of genes in each co-expression module were performed. Hybridization-dependent off-target candidate genes were searched using GGGenome.
Statistical analysis
Statistical analysis was performed on the clinical pathology in the second experiment. Each value from the treatment groups were compared with the corresponding control group for each sampling point (Days 1, 3, or 7) by Dunnett test. The test was conducted at the significance level of 0.05 and 0.01. Analyses were performed using SAS version 9.3 (SAS Institute Inc.).
Results
Potency of AmNA and scpBNA gapmer in vivo
The effects on modified gapmer hepatotoxicity by the incorporation of AmNA have been already reported. 18 Here, we tested the toxicity reduction effect on modified gapmer by the combination of AmNA and scpBNA. We used ISIS 449093 modified sequence as a toxic modified gapmer, replacing cEt with LNA. The modified gapmers were designed substituting LNA with AmNA and, capping both ends with scpBNA. The PS bonds of the wing regions were partially made PO to avoid protein interaction (Fig. 1A, B). All modified gapmers were tested in BALB/c mice after single-dose and three-dose subcutaneous administrations to evaluate modified gapmer potency and hepatotoxicity (Fig. 1C). The three doses were intended for pathological analysis of time- and dose-dependent changes. In the single-dose study, target scarb1 gene expression was strongly suppressed with LX-A1656 and AmNA-scpBNA gapmers except LX-A2004 (Fig. 1D). In three-dose study, significant elevation in AST was observed in LX-A1656 treated animals on Day 7 (Fig. 1E). On the other hand, AmNA-scpBNA gapmer treatment suppressed elevation of AST level. The hepatotoxicity-reducing effect of AmNA-scpBNA was demonstrated at the maximum dose. Modified gapmer distribution in liver was measured and found that content of full-length PO gapmers (LX-A3928, LX-A2004) was lower compared with that of PS gapmers (Supplementary Fig. S2). Focused on LX-A1656 and LX-A3928, IC50 of on-target reduction of Scarb1 mRNA and EC50 of AST elevation were acquired. The on-target knockdown activity of LX-A3928 was ∼1.5 times weaker than that of LX-A1656. On the other hand, AST elevation was improved by ∼2.5 times (Supplementary Fig. S3A–C). These effects were also observed in sequences other than the Scarb1 gapmer (Supplementary Fig. S3D–G). Taken together, these results suggest that combination of AmNA, scpBNA, and PO bonds can reduce modified gapmer hepatotoxicity accompanied by similar gene knockdown activities.
AmNA and scpBNA mitigated modified gapmer hepatotoxicity
We found that our modified gapmers showed significant reduction in hepatotoxicity with the strong suppression of the target gene (Fig. 1D, E). Therefore, we further investigated hepatotoxicity reduction profiles of LX-A2003 and LX-A3928 in detail in-vivo. We evaluated the hepatotoxicity on Days 1 and 7 in vivo to examine the effect of repeated administration over time (Fig. 2A).
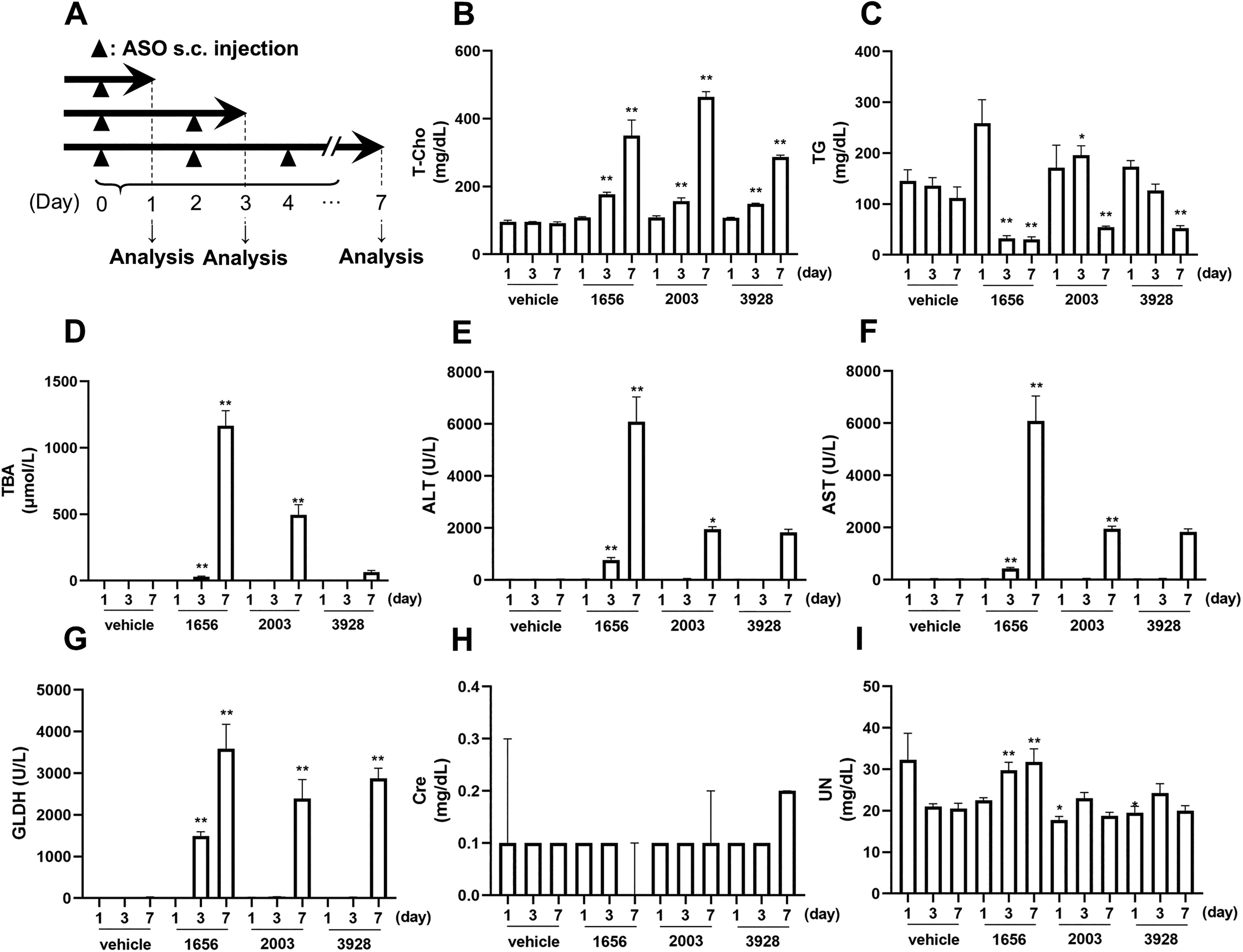
AmNA and scpBNA reduction of hepatotoxicity. For the second experiment, BALB/c mice (n = 4 animals per group) were subcutaneously administered a single dose, twice, and three times of PS-gapmers at 30 mg/kg at indicated time for indicated duration
All modified gapmers showed increase in plasma T-Cho on Days 3 and 7 (Fig. 2B). LX-A1656 showed decrease in plasma TG and increase in plasma TBA, ALT, AST, and GLDH on Days 3 and 7. The elevation of these parameters was relatively reduced in the groups of LX-A2003 and LX-A3928 (Fig. 2C–G). Interestingly, LX-A3928 significantly attenuated the elevation of ALT, AST, and TBA, but only modestly suppressed GLDH. No remarkable change was observed in plasma UN and Cre in all groups (Fig. 2H, I).
Next, we observed pathological and hepatotoxic changes in these modified gapmer-treated livers on Day 7 (Fig. 3a, b). Single cell necrosis of hepatocyte with mild degree was observed in the LX-A1656 group and the change with minimal degree was observed in the LX-2003 and 3928 groups. Moderate eosinophilic change in centrilobular hepatocyte was observed in the LX-A1656 and LX-2003 groups and minimal but diffuse eosinophilic change of hepatocyte was observed in the LX-A3928 group. Mild basophilic change/vacuolation in the periportal hepatocyte was observed in the LX-1656 group and the change with minimal degree was observed in the LX-A2003 group. It was not observed in the LX-A3928 group. Additionally, mild macrophage infiltration in the periportal area was observed in the LX-A1656 group. Similar tendency in terms of comparison among LX-1656, 2003, and 3928 was observed on Day 3 (Supplementary Fig. S4B, C), and no toxicologically significant change was observed on Day 1 (Supplementary Fig. S4A).
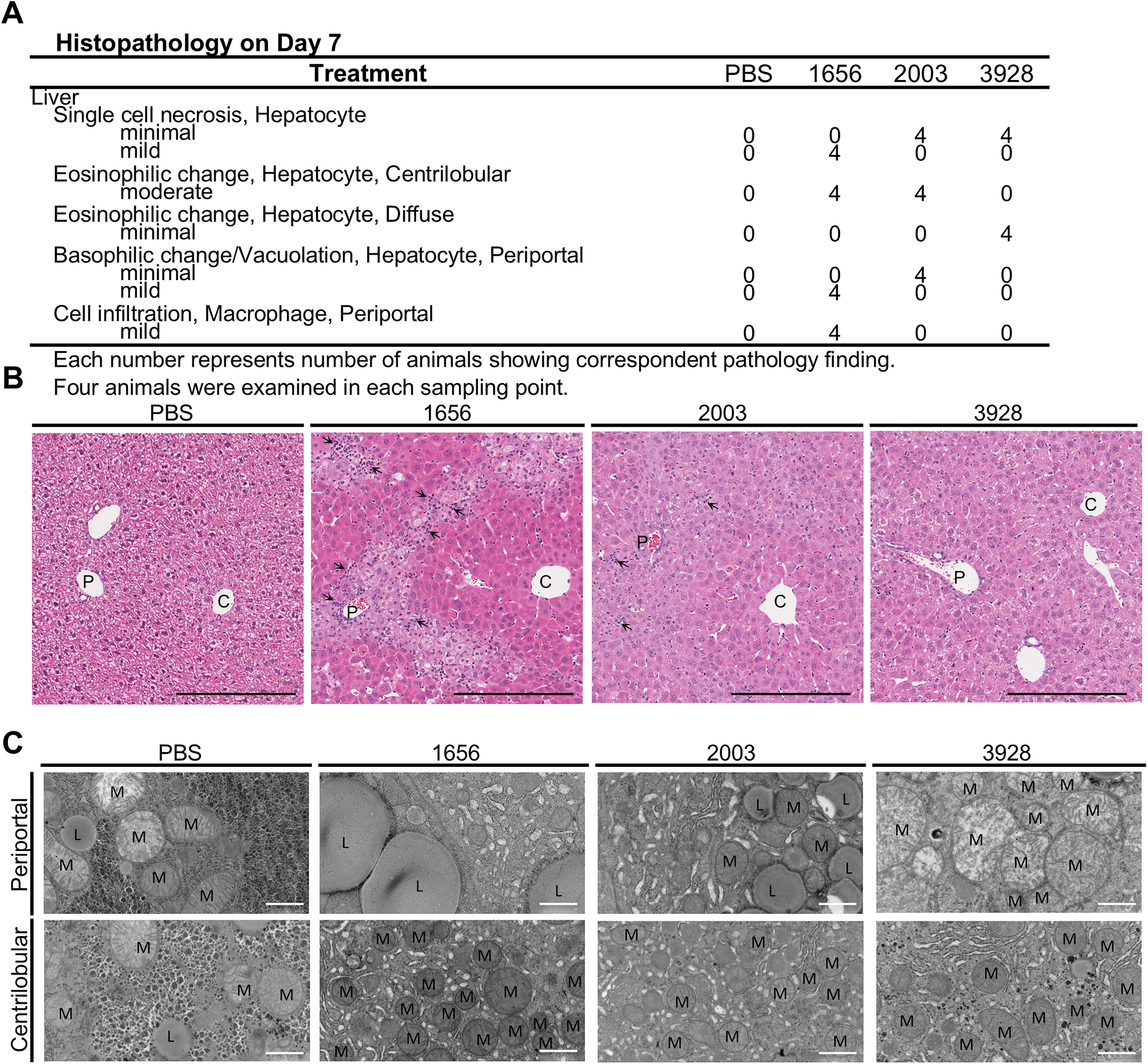
From these results, it was concluded that the overall pathological changes were consistent with changes in blood parameters such as AST and ALT. Furthermore, it was found that the level of lesions was not uniform within the liver.
Ultrastructural hepatocellular findings induced by modified gapmer
Because of the differences within the liver lobules were observed in HE, we performed electron microscopy for further analysis in the samples from Day 7. An increase in mitochondria in the centrilobular area was observed in all treated groups (Fig. 3C). It was also observed in the periportal area in the LX-A3928 group (Fig. 3C). This change was thought to be correlated with eosinophilic change of hepatocytes on HE. Further, a decrease in glycogen granules was observed in all treated groups both in the centrilobular area and periportal area compared with the control (PBS) group (Fig. 3C). In addition to the above abnormalities, an increase in the rough endoplasmic reticulum was observed both in the centrilobular and periportal area in the LX-A1656 and LX-A2003 groups (Fig. 3C), and it was thought to be correlated with basophilic change of hepatocytes on HE. An increase in lipid droplets in the periportal area (Fig. 3C) was observed in the LX-A1656 and LX-A2003 groups and it was thought to be correlated with vacuolation of hepatocytes on HE.
Modified gapmer distribution in the liver
To assess the relationship between lesions and modified gapmer localization in the liver, modified gapmer distribution was examined by modified gapmer-IHC. Strong positive reaction of modified gapmer in the Kupffer cells was observed in all modified gapmer groups at every time points (Fig. 4A–C, Supplementary Fig. S5A–D). On the other hand, positive reaction of modified gapmer in the hepatocytes for all modified gapmer groups was observed only on Day 7. The positive intensity on Day 3 and/or Day 7 was the following order: LX-A1656 > LX-A2003 > LX-A3928. In addition, positive signals were observed in macrophages that infiltrated in the periportal lesion were observed in the LX-A1656 group on Day 7. The modified gapmer signals in the centrilobular and periportal hepatocytes were compatible in each group. Therefore, no direct correlation between the modified gapmer lobular localization and the lobular lesion distribution in the hepatocytes was found.
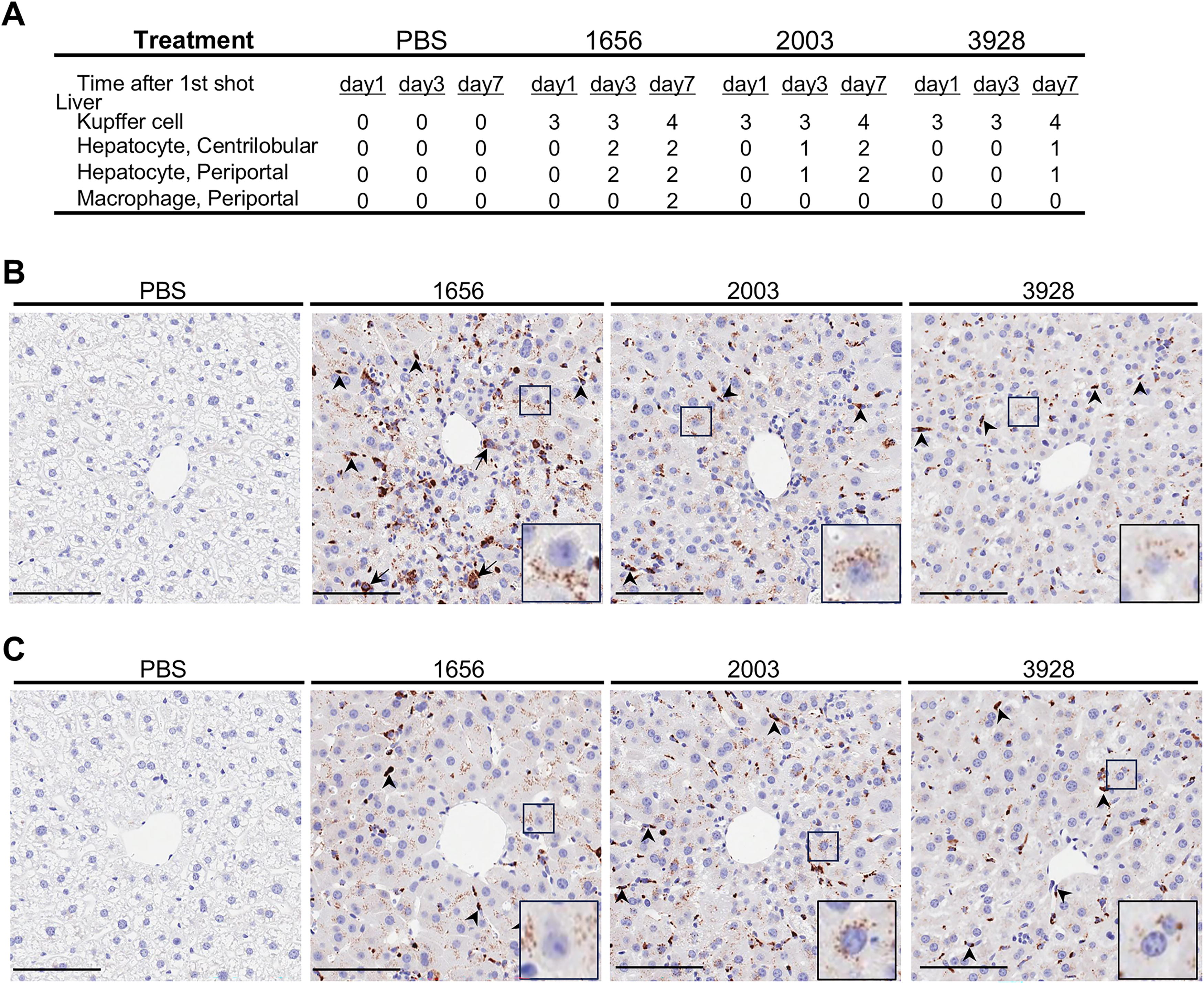
Implications on mitochondria-related genes
We performed transcriptome analysis of modified gapmer or PBS treated mouse livers to investigate the genome wide gene expression change induced by modified gapmers. All three modified gapmers decreased Scarb1 gene expression compared with PBS at 1, 3, and 7 days after first administration, indicating modified gapmer target were consistently downregulated (Fig. 5A). Scarb1 gene expression of all three modified gapmers treated group were at the same level at 3 and 7 days after first administration, but Scarb1 expression level of LX-A3928 treated group at 1 day was higher than that of LX-A1656 and LX-A2003 treated groups at 1 day. In principal component analysis, the expression profile of LX-A1656 treated group was different from that of PBS treated group from 3 days, and the expression profiles of LX-A2003 and LX-A3928 treated groups were different from that of PBS treated group at 7 days, but were similar to LX-A1656 treated group at 3 days (Fig. 5B). The identified DEGs were overlapped among two or three modified gapmer groups, suggesting that the transcriptional effects of these modified gapmers are similar and the effect strength was different (Supplementary Fig. S6A, Supplementary Table S1). Functional enrichment analysis revealed DEGs of all modified gapmers at 3 days after first administration were enriched in cholesterol biosynthesis (Supplementary Fig. S6B). Gene co-expression network analysis revealed five co-expression modules (paleturquoise, darkgreen, darkmagenta, brown, blue) whose gene expressions were changed only in three modified gapmer groups (Fig. 5C). The GO terms related to the electron transport were significantly enriched to the genes in the dark orange module (Fig. 5D). Differential expression in mitochondria-associated genes were common to the three modified gapmers and appeared earlier in LX-A1656. For example, the expression of genes ATP5o, Ndufv1, Ndufv1, and Sdhb was suppressed (Fig. 5E, F). In addition, the expression levels of genes such as Gdf15 increased in the modified gapmers-administered group, and the change was earliest in LX-A1656-administered group (Fig. 5H).
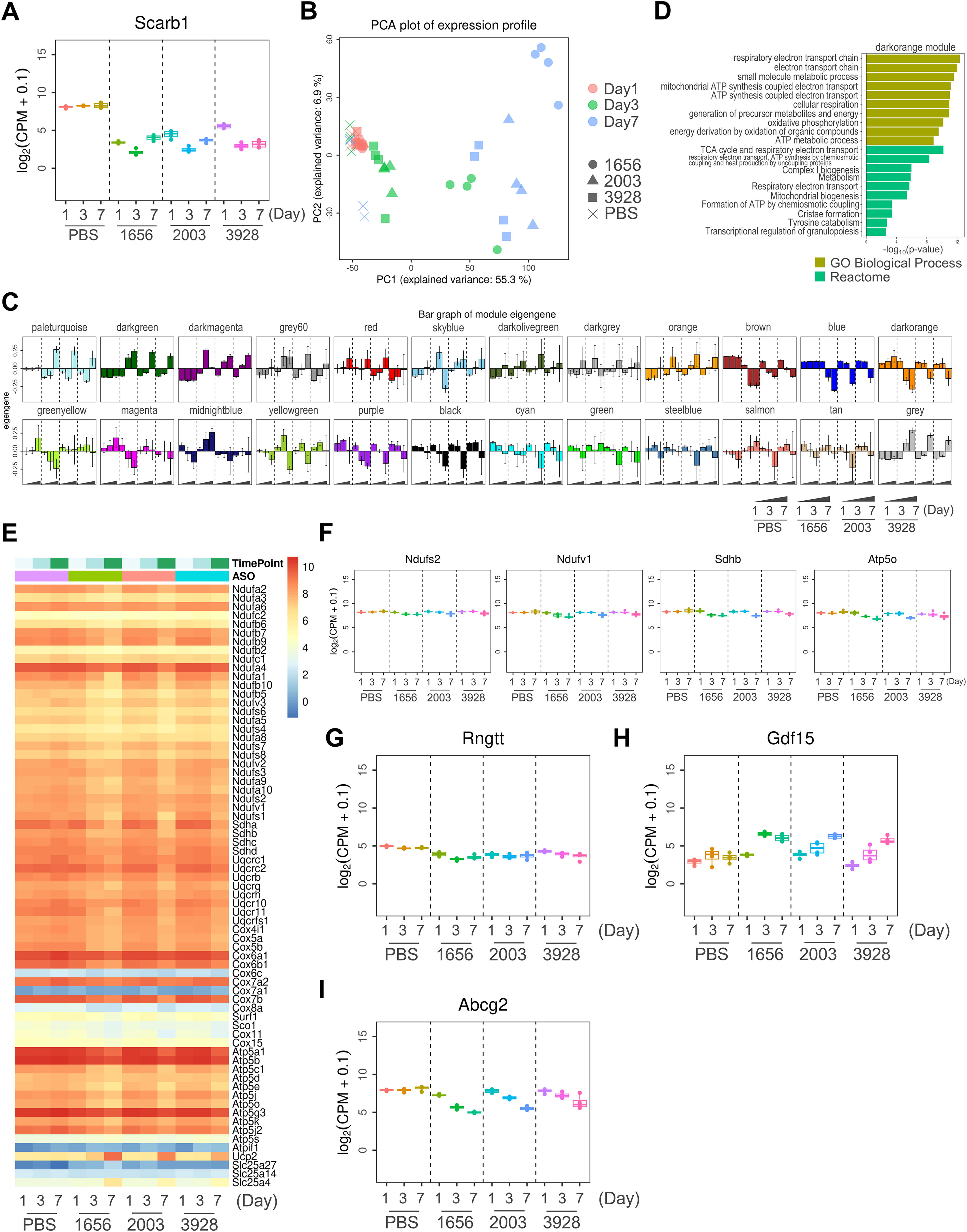
AmpliSeq transcriptome analysis revealed the implications on mitochondria-related genes.
Next, we evaluated the hybridization-dependent off-target effects of modified gapmer. In silico analysis using GGGenome search engine showed that the modified gapmer sequences were completely matched with Rngtt pre-mRNA, and Rngtt gene expression of three modified gapmers treated groups were decreased compared with PBS group (Fig. 5G). The total of 334 genes were selected by GGGenome as the off-target candidates. We identified the off-target genes that were coincide the in silico off-target candidates and downregulated DEGs. The effects of modified gapmers on mitochondria were considered based on histological abnormalities and changes in the expression levels of mitochondria-related genes. Indeed, in these off-target genes, Abcg2 gene was identified as the LX-A1656 off-target genes at 3 and 7 days after administration and as the LX-A2003 and LX-A3928 off-target genes at 7 days (Supplementary Table S2, Fig. 5I). The above-mentioned suppression of gene expression was also demonstrated by individual qRT-PCR analysis (Supplementary Fig. S3A). These results suggest that LX-A1656 may have caused hepatotoxicity at least by affecting mitochondria and AmNA and scpBNA reduced it.
Discussion
Recently, strategy for enhancing the therapeutic profile of modified gapmers with modified nucleic acids has been reported. For instance, LNA and cEt can enhance modified gapmer potency. However, some of the LNA and cEt gapmers have shown hepatotoxicity. In this study, we demonstrated that replacing LNA with AmNA-scpBNA in modified gapmers can significantly reduce hepatotoxicity, which was verified by the hepatotoxicity markers; plasma AST, ALT, and GLDH. We further evaluated plasma TGs and cholesterol as liver markers. A decrease in plasma TGs was observed, whereas cholesterol showed the opposite behavior. As for cholesterol, it was reported that knocking out of Scarb1 inhibited the absorption of cholesteryl ester from HDL, leading to an increase in plasma cholesterol levels. 23 In terms of gene expression, genes associated with cholesterol synthesis were elevated in all modified gapmer administration groups. It was speculated that this result may reflect the change in blood cholesterol concentration.
Compared to LNA-modified gapmers, AmNA-scpBNA-modified gapmers showed strong potency in the liver, which suggests that AmNA-scpBNA can reduce toxicity without altering modified gapmer activity. On the other hand, immunohistochemistry with antimodified gapmer antibody showed slightly different tissue distribution in the liver between LNA and AmNA-scpBNA gapmers. AmNA-scpBNA and LNA gapmers showed significant accumulation in Kupffer cells, however AmNA-scpBNA gapmer was distributed less into hepatocytes than LNA gapmer. It was reported that modified gapmer is preferably accumulated in nonparenchymal cells than hepatocytes. 24 Furthermore, introduction of a lipophilic moiety in an oligonucleotide such as through lipid conjugation is known to increase accumulation of siRNA and PS-DNA in the tissues.25,26 Such conjugation of fatty acids and/or cholesterol also showed that efficient and selective uptake of conjugates depends on interaction with lipoprotein particles, lipoprotein receptors, and other cell-surface receptors.27,28 ScpBNA has a hydrophobic cyclopropyl group in its structure, which is expected to interact with lipoprotein as well as lipid. Indeed, dual hybridization assay showed that the accumulated amount of modified gapmer in the liver would increase as the amount of scpBNA modification increases. The results suggest that it is due to the hydrophobicity of scpBNA.
Pathological analysis for modified gapmer suggested LNA, AmNA-scpBNA and PO would potentially distribute in the different area in the liver. Because the histopathological change was more prominent in the periportal area in the LNA group and this phenomenon was less remarkably or not observed in the AmNA-scpBNA and PO modification groups. However, modified gapmer-IHC showed no apparently different signals in the hepatocytes between periportal area and centrilobular area in any group. The reason for the histopathological lobulation for LNA remained unknown, but considering that lipid metabolism is generally carried out in the periportal area, 29 the periportal histopathology might be related to oxidative stress caused by excess lipid metabolism, such as β-oxidation, which might happen in ATP depletion due to paraspeckle disruption. 30 Additionally, macrophages were present in the periportal area in the LNA-treated group at 7 days, and these cells showed strong positive for IHC. It has been reported that macrophages are more likely to take up modified gapmer than hepatocytes. 31 Therefore, it was suggested that macrophages that had taken up modified gapmer infiltrated in the area following the hepatocellular toxicity. On the other hand, AmNA-scpBNA gapmer did not show hepatocellular pathological findings in the periportal area compared with LNA gapmer and PO modification, but it showed only diffuse and weak lesions throughout the lobules. In the above, further investigation was required into the relationship between changes in distribution and hepatotoxicity.
The Scarb1 gapmer used in this study is known to cause aberrant localization of P54nrb. 13 Electron microscopy showed decreased number of glycogen granules in livers treated with these modified gapmers. P54nrb-deficient mice exhibit impaired glucose tolerance and lower hepatic glycogen. 32 Therefore, the normal localization of P54nrb could potentially be disrupted by the modified gapmer, leading to a decrease in the amount of glycogen in the liver. Increased number of mitochondria was also observed in livers. Mitochondria were increased in the central lobule with LNA and AmNA-scpBNA, and in the central and peripheral lobules with PO modification. Mitochondrial fission facilitates uncoupled respiration, resulting to reduced ATP synthesis. 33 Therefore, it was speculated that the increase in mitochondria might indicate a state of decreased energy production.
Our Ampliseq analysis showed that there was a difference in the number of DEGs among LX-A1656, LX-A2003, and LX-A3928. Since the gene expression level of LX-A1656 changed the most, off-target gene knockdown was assumed to be one of the causes of hepatotoxicity. 11 Interestingly, it revealed significant changes in multiple mitochondrial function-related genes including Gdf15, Slc25a family, and Abcg2. Among these genes, the decrease in the expression level of respiratory chain-related genes was considered to be related to ATP depletion. Interestingly, Abcg2 was an off-target gene of Scarb1 gapmers and expression of Abcg2 was decreased in the modified gapmer dosing groups. Abcg2-deficient hepatocytes express Drp-1 by increasing protoporphyrin IX leading to upregulation of mitochondrial fission via mitochondrial movement and impaired function have been reported. 34 In addition, slc25a47-deficient mice have decreased glycogen production due to decreased mitochondrial function. 35 Both of these inferred events and increased expression of Gdf15 were consistent with mitochondrial disorders. AmNA-scpBNA possibly slowed down or suppressed these changes, suggesting that AmNA-scpBNA could possibly reduce hepatoxicity by mitochondrial alteration. Although further examination should be necessary to confirm the hypotheses described above, reducing mitochondrial toxicity of ASO, nevertheless, is essential to the development of effective oligonucleotide therapies. Thus, the appropriate combination(s) of AmNA, scpBNA, and PO will lead to the creation of ASOs with more therapeutic potency.
Footnotes
Acknowledgments
The authors thank all the members of Luxna Biotech and Axcelead Drug Discovery Partners for discussions. The authors also thank Dr Shuuichi Miyakawa, Dr Masahiko Hattori, Dr Akio Uchida, and Dr Kenichi Miyata of Takeda Pharmaceutical Company Limited for supplying the anti-PS-ASO antibody.
Data Availability Statement
Transcriptome data were deposited at Gene Expression Omnibus with the accession code GSE289964. Research data are available upon request.
Author Disclosure Statement
T.K., A.R.S., K.S., and T.U. are employees of Luxna Biotech. S.A., H.K., M.A., K.Y., and R.F. are employees of Axcelead Drug Discovery Partners.
Funding Information
This research was conducted with a research fund from Luxna Biotech and Axcelead Drug Discovery Partners, which the authors belong to.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.