
Abstract
Long antisense RNAs (asRNAs) have been observed to repress HIV and other virus expression in a manner that is refractory to viral evolution. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of the coronavirus disease 2019 (COVID-19) disease, has a distinct ability to evolve resistance around antibody targeting, as was evident from the emergence of various SARS-CoV-2 spike antibody variants. Importantly, the effectiveness of current antivirals is waning due to the rapid emergence of new variants of concern, more recently the omicron variant. One means of avoiding the emergence of viral resistance is by using long asRNA to target SARS-CoV-2. Similar work has proven successful with HIV targeting by long asRNA. In this study, we describe a long asRNA targeting SARS-CoV-2 RNA-dependent RNA polymerase gene and the ability to deliver this RNA in extracellular vesicles (EVs) to repress virus expression. The observations presented in this study suggest that EV-delivered asRNAs are one means to targeting SARS-CoV-2 infection, which is both effective and broadly applicable as a means to control viral expression in the absence of mutation. This is the first demonstration of the use of engineered EVs to deliver long asRNA payloads for antiviral therapy.
Introduction
Long antisense RNA (asRNA) targeting is one means to specifically repress transcript expression. Compared to small interfering RNAs (siRNA) and short antisense oligonucleotides (ASO), which range between 18 and 22 base pairs (bp) in length, long asRNAs are generally more than 200 bp long [1]. Although the exact mechanism is not well understood, asRNA-mediated gene repression can occur through transcriptional interference or sequestration [2], through the RNA interference machinery utilizing the ribonuclease III-like enzyme, Dicer [3], or recently, through the endogenous human adenosine deaminase acting on RNA enzyme [4].
Long asRNAs have been used previously to repress virus expression [5,6]. The effectiveness of current antivirals, including antibody-based ones, is waning due to the rapid emergence of new variants of concern (VOC), more recently the omicron variant [7]. Indeed, siRNAs [8–10] and short ASOs [11] have been used to repress severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in vivo. The ability of the virus to evolve resistance around single siRNA targeting may, however, prove eventual, rendering this form of viral repression obsolete [12]. One means of avoiding the emergence of viral resistance to siRNA targeting is by using long asRNA to target SARS-CoV-2.
A modular programmable therapeutic platform that can be developed to rapidly treat SARS-CoV-2 could prove paradigm shifting. In this study, we describe the use of extracellular vesicle (EV)-packaged anti-SARS-CoV-2 asRNAs to repress SARS-CoV-2 infection (Fig. 1). EVs are small nanoparticles (50–150 nm) that are constitutively shed by all cells and taken up by neighboring cells. EVs are the perfect delivery vehicle for various genetic therapeutics, including RNA, as they are relatively inert, nonimmunogenic, and anti-inflammatory [13,14]. We set out to use this system to package an asRNA targeting SARS-CoV-2, as asRNAs have been found to repress gene expression and given extensive target binding, the emergence of mutations against asRNA is rare [15].
CD63-L7Ae and asRdRP-CD (anti-SARS-CoV-2 therapeutic RNA) are transfected as plasmids into A4-NSC producer cells. The CD63-L7Ae fusion protein binds to the RNA-binding C/Dbox domain embedded into the therapeutic RNA (asRdRP-CD) and is packaged into the EVs. The EVs are then purified before exposing the EVs to target cells. asRdRP-CD, anti-SARS-CoV-2 therapeutic RNA targeting RdRP region; EVs, extracellular vesicles; NSC, neural stem cell; RdRP, RNA-dependent RNA polymerase; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Materials and Methods
Vectors
EXOsomal transfer into cell (EXOtic) device plasmids, pDB68 (Connexin43 S368A), pDB30 (CD63-nLuc), pSA465 (CD63-L7Ae), and pSA462 (nanoluc-C/Dbox), were also kind gifts from Dr. Martin Fussenegger [16]. The C/Dbox was introduced in the 3′’UTR of asRNAs through standard oligo cloning methods. asRNAs were all cloned into a pcDNA3.1(+) mammalian expression vector.
Cell lines
HEK293T cells, Vero E6 cells, immortalized mesenchymal stem cells (MSC), lung derived-induced pluripotent stem cells, and neural stem cells (NSCs) (A4 lineage) were cultured in Dulbecco's modified Eagle's medium (DMEM) (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS) and incubated at 37°C and 5% CO2. Monocyte-derived macrophages (MDMs) were cultured in RPMI1640 under similar conditions.
asRNA design
Long asRNA sequences were designed toward regions of the SARS-CoV-2 viral genome, which were functionally important and have a high degree of sequence conservation between the SARS-CoV-1 and SARS-CoV-2 genomes, including current circulating SARS-CoV-2 VOCs. These regions include the SARS-CoV-2: (1) Nucleoprotein (N) genomic region that plays a critical role in virion assembly; (2) 5′ region of the RNA-dependent RNA polymerase (RdRP) (nsp12) gene that includes the frameshift stimulating pseudoknot, and (3) catalytic region (Cat) of the RdRP (nsp12) gene, which is critical for the replication of the viral genome inside othe host cell. For each of these regions, the length varied between 600 and 800 bp. As a control, an asRNA designed toward enhanced green fluorescent protein (eGFP) was used. Each of the sequences used is listed in Supplementary Table S1.
asRNA cloning and transfection in vitro
Long asRNAs used in this study were cloned in a manner to be expressed from the cytomegalovirus promoter. asRNAs were cloned into a pcDNA3.1(+) plasmid. Nontargeting asRNAs are against eGFP. Cells were seeded overnight to 70%–80% confluency before transfecting asRNA plasmids with FuGENE 6 (Promega) in OptiMEM (Gibco-Invitrogen), as per the manufacturer's protocol.
Virus
SARS-CoV-2 Wuhan (Ancestral—VIC1) strain was obtained from the Peter Doherty Institute for Infection and Immunity and Melbourne Health, Victoria, Australia, and cultured in Vero E6 cells.
Viral plaque and immunoplaque assays
For viral plaque assays, Vero E6 cells were infected with SARS-CoV-2 for 1 h before overlaying with 1% methylcellulose viscosity (4,000 centipoises) (Sigma-Aldrich). Cells were incubated for 4 days at 37°C before fixing in 8% formaldehyde and stained with 1% crystal violet to visualize plaques. Viral immunoplaque assays for SARS-CoV-2 were performed on Vero E6 cells, as described previously [9], using recombinant monoclonal antibodies that recognize SARS-CoV-2.
Viral copy number determination
To determine viral copy numbers in infected cells, digital polymerase chain reaction (PCR) against the N gene of SARS-CoV-2 (CDC primers from IDT—SARS-CoV-2 N1) was performed in Quant-Studio 3D Digital PCR 20K chips (Thermo Scientific) on a ProFlex 2 × Flat Block Thermal Cycler (Thermo Scientific). Results are analyzed on the QuantStudio 3D Analysis Suite software (Thermo Scientific) and expressed as viral copies per microliter of template RNA.
EV production, asRNA packaging, and characterization
Control and asRNA-packaged EVs in this study were produced using the following plasmids from the EXOtic packaging system (Table 1):
EXOtic Packaging System Plasmids
Producer cells were transfected using Lipofectamine 3000 (Thermo Fisher Scientific), according to the manufacturer's instructions, over 24 h before washing cells with DMEM and replacing media with DMEM +10% EV-depleted FBS (Thermo Fisher Scientific). Supernatant was collected at 48 and 96 h later and centrifuged at 300 g for 10 min at 4°C. The viability of cells was determined at the time of EV collection, which was greater than 90%. Supernatant was transferred to a new 50-mL conical tube and further centrifuged at 2,000 g for 20 min at 4°C before passing the supernatant through a 0.45-mm filter (Millipore).
The filtered supernatant was then centrifuged at 100,000 g for 120 min at 4°C to pellet the EVs, which were resuspended in phosphate-buffered saline (PBS). The final precipitate was resuspended in sterile PBS and passed through 0.22-micron Ultrafree® Centrifugal Filter Units (Millipore Sigma). EVs were then quantified using Nanoparticle Tracking Analysis on a NanoSight (Malvern Panalytical) and further confirmed that these are CD63-positive EVs by determining luciferase activity using the Nluc tagged to CD63 relative to ultracentrifuged supernatant from untransfected cells (ie, no EVs) (Fig. 2B). For all in vitro experiments, exosome to cell ratio of 104 exosomes per cell was maintained.
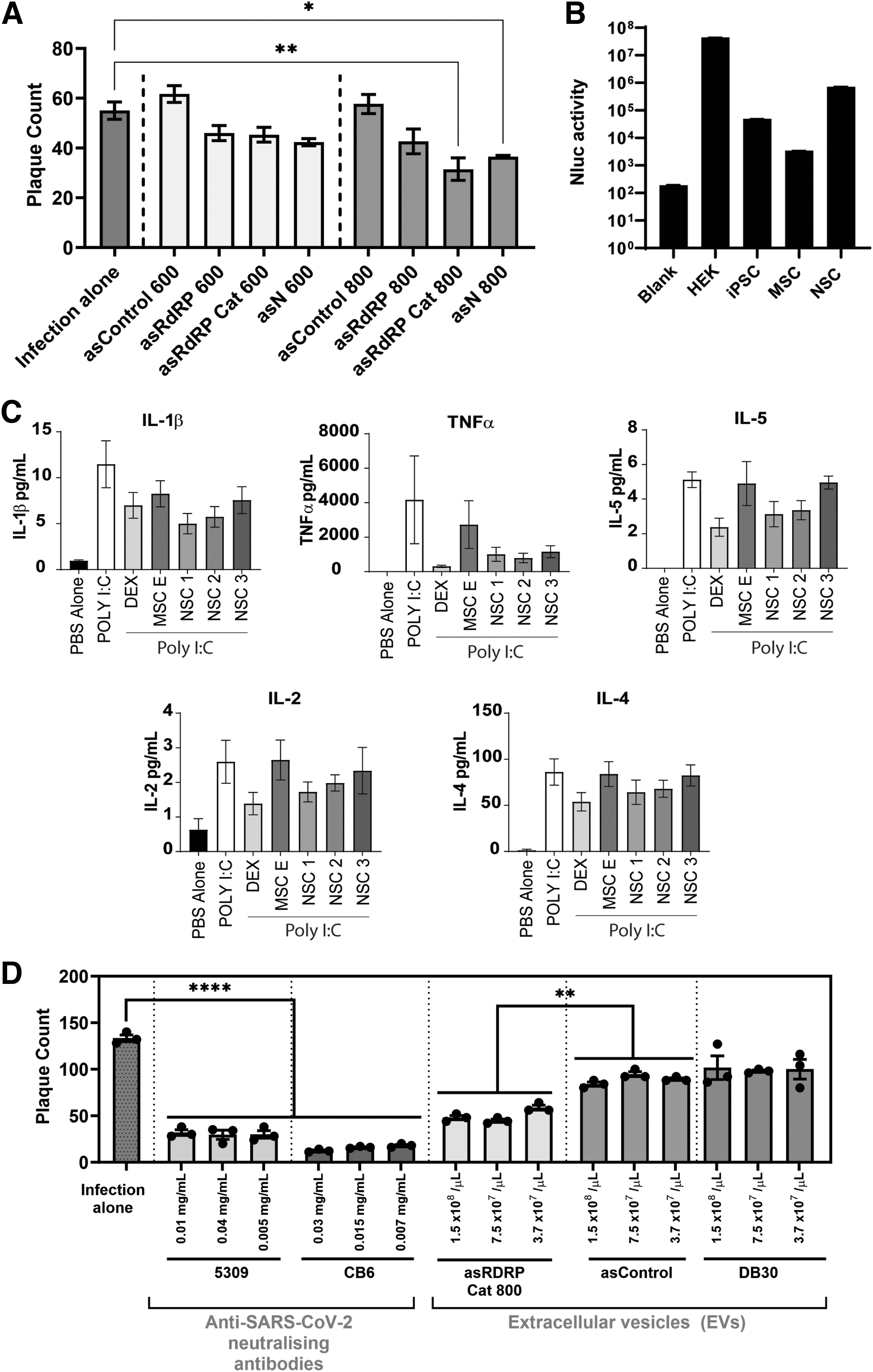
Delivery of asRNAs targeting the SARS-CoV-2 RdRP gene packaged in NSC EVs represses SARS-CoV-2 infection in vitro.
Determining luciferase activity in EVs
Nano-Glo® Luciferase assay system (Promega) was used to determine the luciferase activity in purified nanoluc (Nluc) exosomes as per the manufacturer's instructions before measuring luciferase activity on the Promega GloMax Discover Microplate Reader Detection System with GM3000 Software (Promega).
Efficiency of asRNA packaging into EVs
The packaging efficiency of 800 bp long asRNA overlapping the RdRP gene (asRdRP 800) in CD63-L7ae and CD63 EVs from HEK293T cells was determined by co-transfecting 50 ng CD63-L7ae with 50 ng asRdRP. Twenty four hours later, the supernatant was collected and passed through a filter column for RNA extraction detected asRdRP 800 RNA by real-time PCR (RT-PCR) relative to U6sRNA (housekeeping gene).
RT-PCR detection of asRNAs in EVs
RNA was isolated from exosomes using the Maxwell® automated simplyRNA isolation kit according to the manufacturer's instructions (Promega). Equal amounts of RNA were used for Luna® Universal One-Step RT-qPCR Kit (NEB) according to the manufacturer's instruction. Roche® LightCycler 96 was used to perform the RT-qPCR and results were analyzed using the LightCycler 96 software (Roche).
EV biodistribution in mice
NSC EVs diluted in sterile PBS were complexed with DiIC18(7);1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DIR), a lipophilic, near-infrared fluorescent cyanine dye at a final concentration of 1.55 ng/μL DIR. A total of 108 NSC EVs were administered to BALB/c mice intravenously (IV) by tail vein injection (20 μL). Fluorescence was detected using a single photon animal imager at various indicated time points post-administration.
Cytokine profiling and liver enzyme analysis of mouse administered with EVs
A total of 100 μL of 100 billion HEK derived EVs per mouse (C57BL/6) were injected through IV, or 100 μL of 30 billion NSC-derived EVs were injected per mouse through IV. PBS was used as control. Mice bled at 4 and 72 h postinjection and serum was collected to measure cytokine expression by performing RT-PCR of mouse interferon (IFN)α/β mRNA. Two hundred microliters of blood was taken from mice for VetScan Mammalian Liver Profile analysis (Abaxis) 72 h post-EV administration. Liver enzymes analyzed were alkaline phosphatase, aspartate aminotransferase, and blood urea nitrogen. For SARS-CoV-2-infected mice lungs, tissues were homogenized in trizol before extracting RNA for RT-PCR analysis to determine mouse IFNβ mRNA expression on the Rotor-Gene (Thermo Fisher Scientific) using the Rotor-Gene SYBR Green RT-PCR kit (Qiagen, Hilden, Germany).
Cytokine detection from human macrophages
Blood from consented and deidentified donors was used in this study under an approved IRB 19582 (City of Hope, Duarte, CA). Human monocyte isolation and macrophage differentiation were performed as previously described [17]. Granulocyte-macrophage colony-stimulating factor (GM-CSF) primed human macrophages were either untreated (sterile PBS) or treated with polyinosinic:polycytidylic (poly I:C; Sigma Aldrich), dexamethasone (Sigma Aldrich), MSC EVs, or increasing concentrations of NSC EVs, and supernatant collected 24 h later. Supernatants collected from cells were subjected to cytokine detection by Luminex using a Cytokine 10-Plex Human Panels (LHC0001M; Thermo Fisher Scientific) on a Bio-Plex® 200 (Bio-Rad) by the Analytical Pharmacology Core (City of Hope).
SARS-CoV-2 in vivo work
K18-hACE2 mice (3–4 months old) were purchased from the Jackson Laboratory (Bar Harbor, ME) and bred in-house at the Griffith University Animal Resource Center. Mice were intranasally (IN) infected with 104 plaque-forming unit (PFU) (20 μL total volume) of live SARS-CoV-2, while under isoflurane anesthesia. Mice were subsequently treated with EVs retro-orbitally (IV) (100 μL total volume), while under isoflurane anesthesia. Mice were monitored daily for weighing and clinical scoring.
Results and Discussion
To determine if SARS-CoV-2 is susceptible to asRNA-directed silencing, we generated and tested various asRNAs of various lengths to highly conserved regions within the RdRP and nucleoprotein (N) genes of SARS-CoV-2 (Supplementary Fig. S1). The N protein plays a critical role in virion assembly, whereas RdRP (nsp12 gene) is critical for the replication of the viral genome inside the host cell. We found that an 800 bp long asRNA overlapping the RdRP catalytic (Cat) region (asRdRP Cat 800) reproducibly repressed SARS-CoV-2 infection with two independent asRNA controls (Fig. 2A and Supplementary Fig. S2A) and confirmed by knockdown of SARS-CoV-2 viral copy numbers (Supplementary Fig. S2B).
Next, we designed the top candidate asRNA, asRdRP Cat 800, to be packaged with the EXOtic system into EVs. The EXOtic EV packaging system allows packaging of virtually any RNA into any cell type [16]. As we have previously generated EVs from HEK293T cells [18,19], we wanted to compare the EXOtic packaging efficiency of nanoluciferase (Nluc) in EVs purified from other cell types, in particular, stem cells relative to HEK293T cells. We found that NSCs efficiently package Nluc in EVs (Fig. 2B). Using the EXOtic system, we then proceeded to package asRdRP Cat 800 into NSC EVs.
In this system, the ubiquitous exosome marker protein CD63, a tetraspanin protein, is fused to an archaebacterial derived L7Ae peptide, which allows for the recruitment and encapsulation of those RNA containing C/Dbox to budding exosomes. The DNA sequence for C/Dbox was cloned at the 3′end of asRdRP Cat 800 to express mRNA containing the C/Dbox RNA domain. Importantly, we found that the addition of a C/Dbox RNA domain into asRdRP Cat 800 allowed more effective asRdRP Cat 800 packaging into EVs (Supplementary Fig. S3). These data suggest that asRdRP Cat 800 is a viable RNA capable of repressing SARS-CoV-2 infection in vitro and that this antisense transcript can be packaged into EVs, similar to observations with protein coding mRNAs [19,20].
An added benefit from using NSCs to produce our antiviral EVs is that NSCs have been observed to impart anti-inflammatory effects [21], suggesting the use of these cells to make EVs may provide an approach that not only delivers antiviral RNAs to virus infected cells but may also diminish SARS-CoV-2-mediated immune dysregulation and severe acute respiratory distress. To determine if the NSCs used to develop the asRdRP Cat 800 containing EVs are imbued with anti-inflammatory properties, we treated polyinosinic:polycytidylic acid (poly I:C)-exposed GM-CSF primed human MDMs with NSC- and MSC-derived EVs, which have been reported to have anti-inflammatory properties [22,23].
We found that NSCs reduced key cytokines [interleukin (IL)-1β, tumor necrosis factor (TNF)-α, IL-2, IL-5, and IL-4] that drive coronavirus disease 2019 (COVID-19) severe acute respiratory distress complications [24] to similar levels in poly I:C-exposed cells treated with a known anti-inflammatory agent, dexamethasone (Fig. 2C). We also demonstrated that NSC-derived EVs comparably had better anti-inflammatory effects than MSC-derived EVs. Overall, this shows that NSC-derived EVs can offer an added anti-inflammatory effect when used to deliver the antiviral RNA payloads to target cells. We then tested the ability of asRdRP Cat 800 packaged in NSC EVs to dampen SARS-CoV-2 infection in vitro. Compared to a nontargeting control asRNA, asRdRP Cat 800 EVs significantly repressed SARS-CoV-2 replication (Fig. 2D).
To explore the ability of these EVs to functionally inhibit CoV-2 in vivo, we explored the biodistribution of NSC EVs in vivo following IV administration (IV). We find that the Nluc containing NSC EVs disseminate to liver, spleen, and lungs when IV delivered (Fig. 3A). IV delivery may prove advantageous in localizing delivery to the lung, an organ afflicted by SARS-CoV-2. To assess this notion, we tested the EVs in a SARS-CoV-2 mouse model by testing the therapeutic effect of delivering five daily doses of asRdRP Cat 800 EVs by IV administration (Fig. 3B).
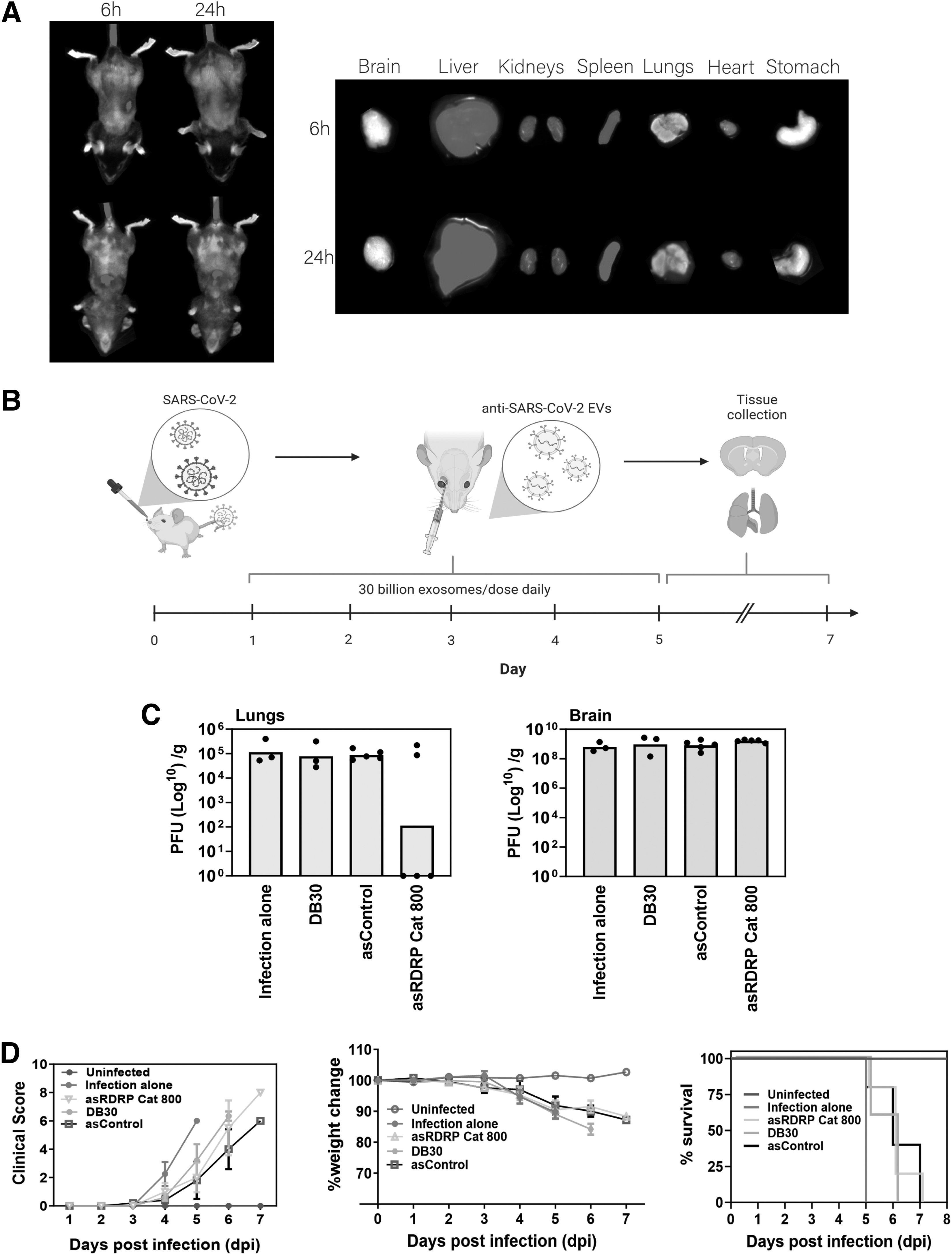
Delivery of NSC EVs containing anti-SARS-CoV-2 asRNA reduces viral lung infection in mice.
We also found that 30 billion NSC EVs per IV injection was well tolerated with no observable change in liver enzyme function (Supplementary Fig. S4) and was not immunostimulatory (Supplementary Fig. S5), suggesting that these EVs are safe to be delivered into mice. Hence, we used this amount of NSC EVs for the subsequent in vivo study. Lungs from three out of five mice treated with asRdRP Cat 800 EVs had no detectable SARS-CoV-2 infection when compared to its control, asGFP packaged in EVs (asControl) (Fig. 3C), suggesting that asRdRP Cat 800 EVs are able to exert a notable antiviral effect in the lungs of mice, although not in all the treated mice. We reasoned that this incomplete antiviral activity could either be attributed to poor EV biodistribution in the lungs (Fig. 3A) or be limited by the amount of daily EVs administered (ie, 30 billion EVs).
Indeed, work by Shrivastava et al. [19] showed that therapeutic daily IV delivery of 100 billion EVs repressed HIV-I viral load in organs as far as the brain. Future work will focus on delivering higher doses of EVs either IN or intratracheally as EVs will directly reach the lungs in higher amounts compared to IV delivered EVs [25]. Importantly, intranasal administration of NSC [26]- and astrocyte cell [27]-derived EVs has been shown to deliver EVs to the brain. Indeed, we failed to see any antiviral activity with asRdRP Cat 800 EVs in the brains of mice when delivered by IV (Fig. 3A). Within the context of clinical long COVID caused by SARS-CoV-2 [28], a therapeutic EV approach that can deliver anti-SARS-CoV-2 asRNAs into the brain would be clinically beneficial.
Although not statistically significant, we observed that all EV-treated SARS-CoV-2-infected mice have improved clinical well-being and survival (Fig. 3D). This clinical improvement could possibly be attributed to the anti-inflammatory capacity of these NSC EVs (Fig. 2C). Although we showed that these NSC EVs did not activate type I IFNs in either serum from uninfected mice (Supplementary Fig. S5) or lungs from SARS-CoV-2-infected mice (Supplementary Fig. S6), further work is still needed to conclusively determine that these EVs are indeed exerting an anti-inflammatory effect in infected mice by performing a detailed cytokine panel analysis in all organs.
There are several other limitations in this study. SARS-CoV-2 variant used in this study is the ancestral variant, not the most recent VOC. However, asRdRP Cat 800 was designed within a highly conserved region of the RdRP gene, where these is >90% conservation between SARS-CoV-1 and SARS-CoV-2, including current SARS-CoV-2 VOCs (Supplementary Fig. S1). Indeed, the RdRP region (nsp12) that asRdRP Cat 800 targets is highly conserved across all SARS-CoV-2 VOCs [29], making this gene highly druggable for antiviral targeting. Therefore, the antiviral activity of our asRNAs is expected to be equipotent across all SARS-CoV-2 VOCs.
Future work aims to demonstrate this conclusively in other SARS-CoV-2 VOCs, both in vitro and in vivo. To our knowledge, this is the first demonstration of the use of engineered EVs to deliver long asRNA payloads for antiviral therapy. Overall, our pre-clinical data demonstrate that anti-SARS-CoV-2 asRNAs delivered in EVs can ameliorate lung viral infection. Collectively, these data suggest that EV delivery of RNA therapeutics may prove to be a viable approach as an antiviral agent. Our data provide a proof-of-concept approach that this scalable and cost-effective platform technology can be easily amended to target novel pandemic RNA viruses beyond SARS-CoV-2.
Ethics statement
All animal experiments were performed in compliance with relevant laws and institutional guidelines and in accordance with the ethical standards of the Declaration of Helsinki. All animal care and procedures have been performed according to protocols reviewed and approved by the City of Hope Institutional Animal Care and Use Committee (IACUC) held by the principal investigator for this application (Kevin Morris, IACUC 16095). All SARS-CoV-2 animal work was conducted in a BSL3-approved animal facility at Griffith University (Animal ethics approval: MHIQ/09/20/AEC). Blood from consented and deidentified donors was used in this study under an approved IRB 19582 (City of Hope).
Data Availability Statement
All data generated or analyzed during this study are included in this article.
Footnotes
Acknowledgments
We would like to thank Dr Martin Fussenegger from Department of Biosystems Science and Engineering, Faculty of Life Science, University of Basel, Switzerland, for the EXOtic device plasmids. We would like to thank Dr Naphak Modhiran and Associate Professor Dan Watterson (School of Chemistry and Molecular Biosciences, The University of Queensland, QLD, Australia) for providing us with SARS-CoV-2 recombinant monoclonal antibodies. We would also like to thank the Peter Doherty Institute for Infection and Immunity and Melbourne Health, Victoria, Australia, for providing us with SARS-CoV-2 live virus. Research reported in this publication includes work performed by the Analytical Pharmacology core facilities at City of Hope, Duarte, CA.
Author Disclosure Statement
N.A.J.M. and K.V.M. are both consultants for Prorenata Biotech. K.V.M. is the chief scientific officer (CSO) for The Gene Company.
Funding Information
This work was supported by MRFF 2001931 to NAJM, NHMRC 2027569 to AI, and NIH/NIMH 1R01MH134389–01 to KVM.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.