
Abstract
Pathogenic variants in ABCA4 are the underlying molecular cause of Stargardt disease (STGD1), an autosomal recessive macular dystrophy characterized by a progressive loss of central vision. Among intronic ABCA4 variants, c.4253+43G>A is frequently detected in STGD1 cases and is classified as a hypomorphic allele, generally associated with late-onset cases. This variant was previously reported to alter splicing regulatory sequences, but the splicing outcome is not fully understood yet. In this study, we attempted to better understand its effect on splicing and to rescue the aberrant splicing via antisense oligonucleotides (AONs). Wild-type and c.4253+43G>A variant-harboring maxigene vectors revealed additional skipping events, which were not previously detected upon transfection in HEK293T cells. To restore exon inclusion, we designed a set of 27 AONs targeting either splicing silencer motifs or the variant region and screened these in maxigene-transfected HEK293T cells. Candidate AONs able to promote exon inclusion were selected for further testing in patient-derived photoreceptor precursor cells. Surprisingly, no robust splicing modulation was observed in this model system. Overall, this research helped to adequately characterize the splicing alteration caused by the c.4253+43G>A variant, although future development of AON-mediated exon inclusion therapy for ABCA4 is needed.
Introduction
Pathogenic variants in ABCA4 are the underlying molecular cause of Stargardt disease (STGD1; OMIM: 248200), a common autosomal recessive macular dystrophy characterized by a progressive central vision loss. STGD1 presents a high variability in phenotype and progression of the symptoms, as well as genotypic heterogeneity, which adds up to the complexity for designing therapies to find a suitable treatment for STGD1 patients [1,2]. More than 2,400 unique variants (www.lovd.nl/ABCA4) have been reported in ABCA4, which encodes the ATP-binding cassette type A4 (ABCA4) protein [3], a flippase localized at the disc membrane of photoreceptor outer segments.
Affected transporter activity can provoke toxic accumulation of visual cycle metabolites in photoreceptor and retinal pigment epithelium (RPE) cells, which eventually leads to oxidative stress and cell death. Changes in protein folding, protein expression, stability, substrate binding, or ATPase activity are the main alterations in the presence of pathogenic ABCA4 variants [4]. Among these, (deep-)intronic variants have been found to affect the splicing at the pre-mRNA level, often leading to the inclusion of a premature termination codon in the final transcript, and thereby reduced ABCA4 protein levels.
One of the most frequent intronic ABCA4 variants is c.4253+43G>A, which is located in intron 28 and is classified as a hypomorphic allele. Interestingly, this variant was shown to be substantially enriched in patients with late-onset STGD1 and has been demonstrated to be clinically expressed only when it is present in trans with a severe ABCA4 allele [5]. The c.4253+43G>A variant can be carried alone or in a complex allele with the c.6006-609T>A variant, both options being reported as equally pathogenic. In these cases, the age of onset can range between the third and seventh decade of life [6]. Based on the late onset of disease and the corresponding phenotypic characteristics, c.4253+43G>A has been classified as a mild ABCA4 allele within the category of rare hypomorphs [7].
The c.4253+43G>A variant is not predicted to create new splice sites or strengthen already existing cryptic splice sites. Instead, this variant has a predicted effect on splicing regulatory motifs. The nucleotide substitution eliminates one exonic splicing silencer (ESS) motif and creates the exonic splicing enhancer (ESE) SF2/ASF motif around this position. As a result, the presence of this intronic variant in a midigene system mostly led to the skipping of exon 28, and at a lesser extent, skipping of both exons 27 and 28 as well. In addition, the wild-type midigene condition also showed skipping of exon 28 at a lower degree, suggesting that this region in ABCA4 is susceptible to natural exon skipping [8]. Attempts to promote exon 28 inclusion into the final transcript have been made by employing antisense oligonucleotides (AONs), small RNA molecules that can modulate pre-mRNA splicing [9]. Importantly, a notable number of intronic ABCA4 variants have been already targeted with AONs, and the respective splicing defect, generally pseudoexon inclusion, was successfully rescued not only in midigene-based assays but also in patient-derived models such as fibroblasts, photoreceptor precursor cells (PPCs), and retinal organoids [8,10–17].
So far, the effect of the c.4253+43G>A variant was based on a midigene containing a relatively short fragment of genomic ABCA4. We hypothesized that other alterations on splicing of the neighboring exons may also arise, but cannot be validated due to the limited context of the vector. In this study, we aim to further analyze the effect of the c.4253+43G>A variant on exon skipping through the broadening of the genomic context used for splice assays. After validation of the effect, we also aim to promote exon inclusion since substantial reduction of skipping events through AONs has not been achieved yet. Ultimately, the screening of multiple AONs in both splice vector systems and relevant retina-like cellular models may contribute to discover the best regions to be targeted to restore exon inclusion.
Materials and Methods
AON design
Two different approaches were followed to design the oligonucleotides used in this study, the oligo-walk targeting the region around variant c.4253+43G>A and the blocking of splicing silencer motifs predicted in the region (all reported in Supplementary Table S1). Oligonucleotide sequences and their properties are shown in Supplementary Table S2. Further details on AON design are provided in the Supplementary Data S1.
Maxigene transfection and AON rescue studies in HEK293T cells
To analyze the effect of the c.4253+43G>A variant on splicing, the wild-type and mutant maxigenes harboring a broader genomic ABCA4 region were generated and transfected in human embryonic kidney (HEK293T; ATCC No. CRL-3216™) cells. The used cloning primers are listed in Supplementary Tables S3 and S4. These maxigene vectors were employed to test the designed AONs. The detailed cloning, transfection, and rescue procedures can be found in the Supplementary Data. Experiments were performed in two independent replicates (n = 2).
AON rescue studies in patient-derived PPCs
Control and patient-derived induced pluripotent stem cells (iPSCs) were reprogrammed from fibroblasts by the Stem Cell Technology Center (Radboudumc, Nijmegen, The Netherland) as previously described [12,15,16,18]. The patient-derived iPSC line (RMCGENi020-A) was previously fully characterized [19]. To derive PPCs from iPSCs, a 30-day differentiation protocol was adapted from previous research on two-dimensional to three-dimensional (3D) retinal organoid differentiation [20,21]. Differentiation markers were analyzed by quantitative polymerase chain reaction (qPCR; primers listed in Supplementary Table S5). All the iPSC culture conditions, PPC differentiation, characterization, and rescue procedure are further detailed in the Supplementary Data. All differentiation experiments were performed in two independent replicates (n = 2).
Analysis of ABCA4 splicing
HEK293T cells and PPCs were harvested for further RNA isolation and real time-PCR (RT-PCR) analysis of ABCA4 transcripts. Amplification of ABCA4, ACTB as a loading control, and RHO as transfection control was performed for these transcript analyses (all primers sequences are provided in Supplementary Table S6). All PCR products were resolved on 2% (w/v) agarose gels and validated by Sanger sequencing. Semiquantitative analysis of correct and skipping transcripts was performed with the Fiji software (Supplementary Tables S7 and S8) [22]. Statistical significance between conditions was determined for p < 0.05, presented with one or multiple asterisks (*) in the figures. Detailed methods on splicing analyses by RT-PCR are shown in the Supplementary Data.
Results
A larger genomic context reveals new skipping events induced by the c.4253+43G>A variant
The predicted effect of the c.4253+43G>A variant is not directly related to the creation or strengthening of a particular splice site, although GeneSplice predicts a 0.8 weakening of intron 28 splice donor site (Fig. 1A). However, no differences in scores were found by other prediction tools. Instead, c.4253+43G>A changes the splicing regulatory pattern around the region. In the wild-type sequence, three ESS motifs and two ESE motifs are detected by the Human Splicing Finder Pro tool. Meanwhile, the nucleotide change breaks two of the three ESS motifs and creates two new ESE motifs. This is translated in a modification of the exonic splicing regulatory profile around the intronic region harboring the variant, showing a higher ESE/ESS ratio in the mutant sequence.
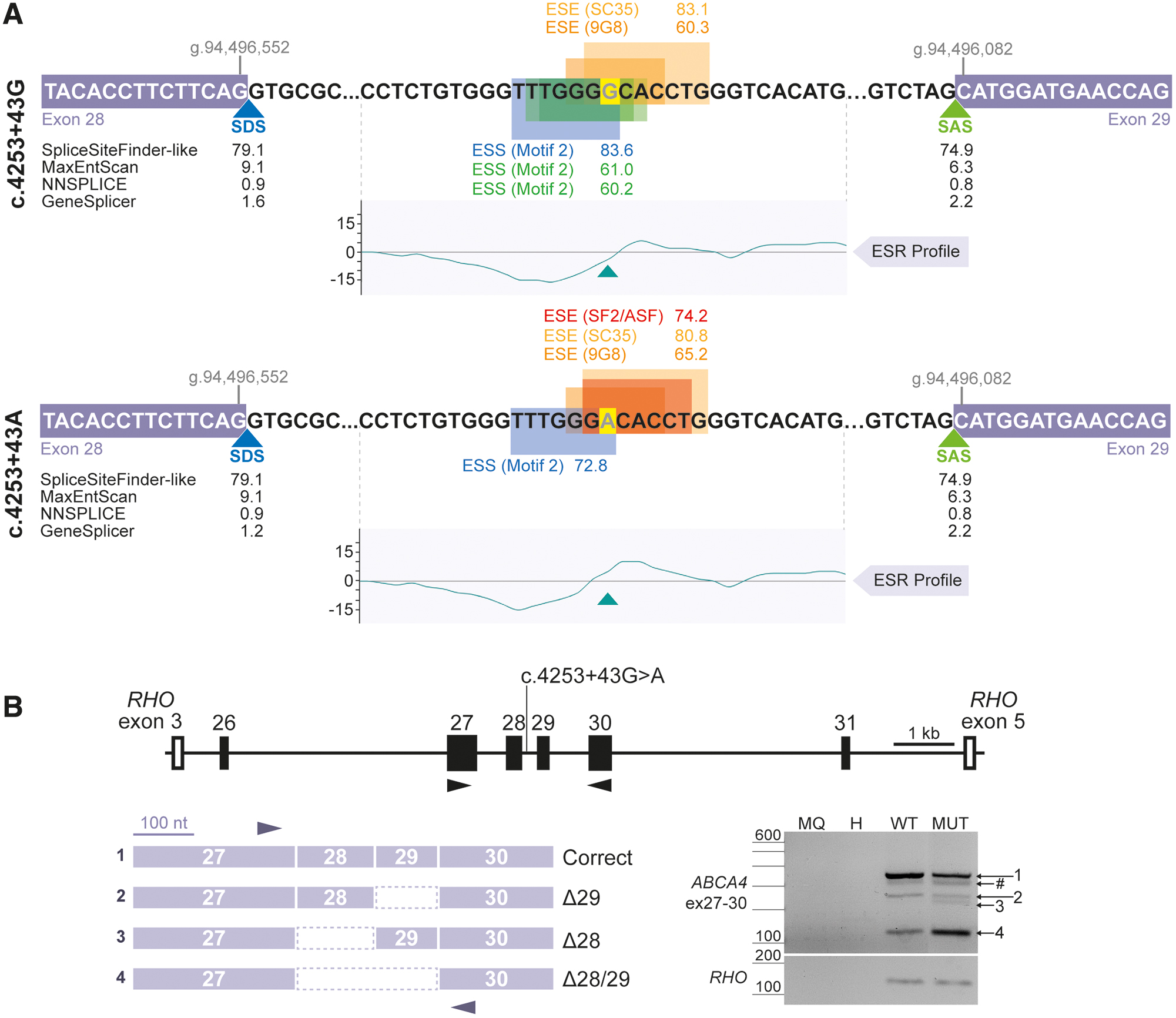
To evaluate the effect of c.4253+43G>A variant in a broader genomic context, this study was based on a new vector, which included a ∼13-kb region from intron 25 until intron 31, from now on referred as maxigene vector since it is derived from a combination of two contiguous midigene vectors [23]. Upon transfection of the wild-type maxigene vector in HEK293T cells, the presence of correct ABCA4 transcript, skipping of exon 29 (Δ29), and co-skipping of both exons 28 and 29 (Δ28/29) were detected by RT-PCR analysis (Fig. 1B). Interestingly, transfection with the c.4253+43G>A variant-harboring maxigene showed a similar splicing pattern except for one additional transcript corresponding to the skipping of exon 28 (Δ28), although the ratio between correct and skipped transcripts notably differs when the intronic variant is present, mainly because of the increase of the Δ28/29 event.
In brief, increasing the genomic context around c.4253+43G>A position contributed to obtain a wider overview of a variety of exon-skipping transcripts (Supplementary Fig. S1) in the wild-type and mutant conditions, including new ones that were not detected in the previous midigene-based assay, which were also increased by the presence of the variant.
ESS-blocking approach did not lead to increased exon inclusion in maxigene-based splice assays
To restore correct transcript levels, the highest-scored ESS motifs (shown in Supplementary Table S1 and Supplementary Fig. S2) within the region of interest were targeted with AONs to block the recognition of sequences that might be playing a role in favoring the skipping of exons 28 and/or 29. The regions where ESS motifs were predicted are indicated as R1, R2, R4, and R5 in Fig. 2A.
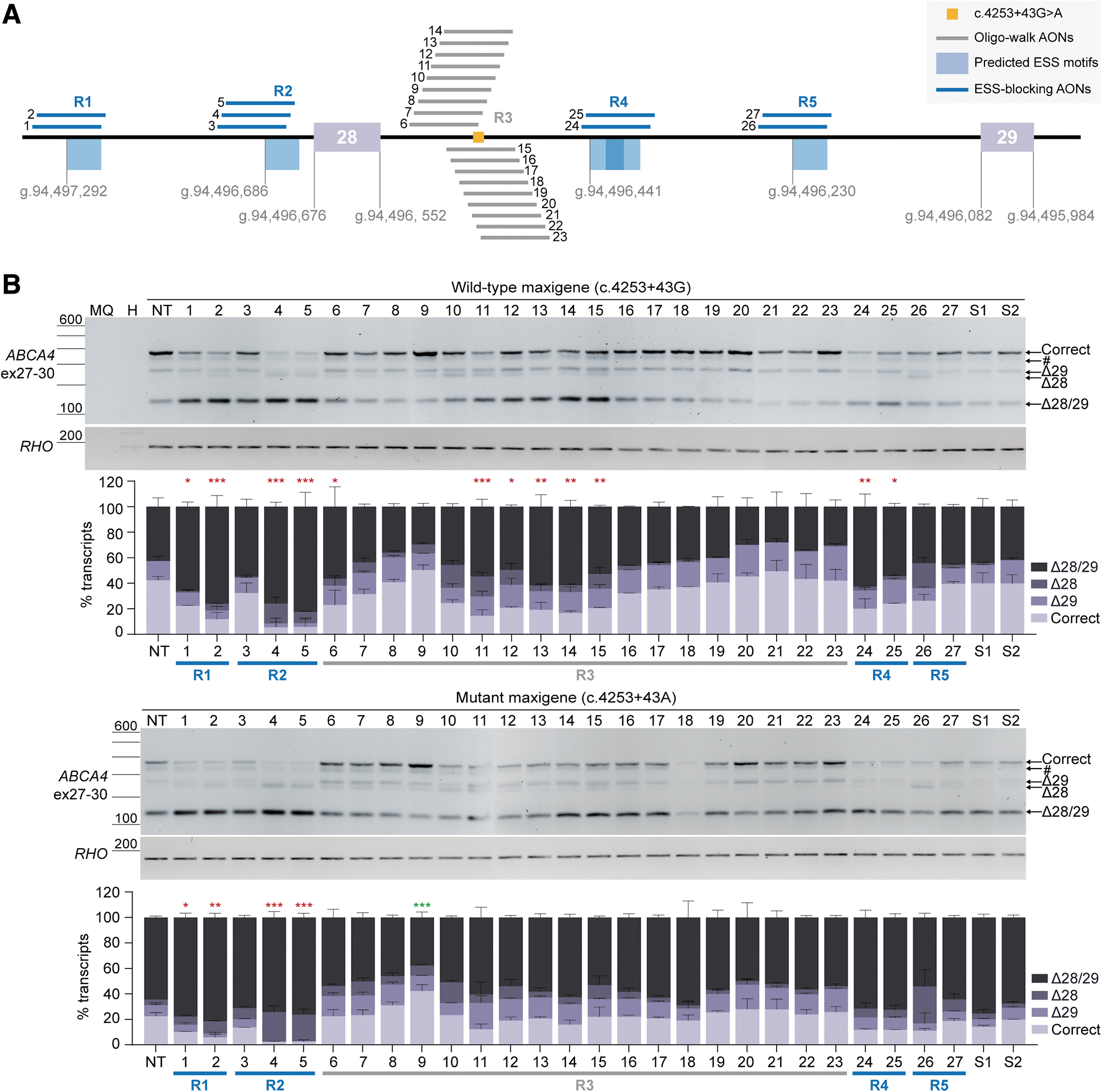
The nontreated mutant maxigene-transfected cells revealed the appearance of Δ28, a decrease of correct transcript from 42% to 22% and an increase of the Δ28/29 transcript from 43% to 65% relative to the total transcript levels (Fig. 2B and Supplementary Table S7). Overall, none of the AONs designed to block ESS motifs promoted exon inclusion when delivered to the cells transfected with the mutant maxigene. AONs targeting regions R1 and R2 significantly reduced the levels of correct transcript, except for AON #3. AONs targeting the ESS motif located within R4 slightly increased Δ28/29 transcript levels, whereas those blocking R5 motifs either enhanced the presence of Δ28 transcript levels (AON #26) or kept a similar pattern compared with the respective nontreated mutant condition (AON #27).
Similar effects were observed in the wild-type maxigene condition since AONs targeting R1 and R2 induced a significant reduction in correct transcript levels, as well as those blocking R4. In addition, sense oligonucleotides (SONs) #1 and #2 did not produce apparent changes in splicing pattern, although it is important to notice a slight decrease in correct transcript induced by SON #1. In conclusion, blocking the predicted high-scored ESS motifs could not help restoring correct transcript levels, and instead, it enhanced the skipping events in this intronic region.
Increased exon inclusion levels achieved by oligo-walk approach in maxigene-based splice assays
In parallel, a large set of AONs was designed following an oligo-walk approach directly targeting the region where the variant is located, referred as R3 in Fig. 2A. The binding of these AONs did not have such a negative impact as the silencer-blocking AONs. However, some significantly induced more Δ28/29 transcript in the wild-type maxigene condition, which is the case of AONs #6 and #11 to #15. Despite not being a drastic change, it is important to mention the formation of Δ28 transcript at different levels upon delivery of AONs #6 to #15, a splicing event that is not visible in the absence of the intronic variant. In contrast, AONs #16 to #23 did not hamper the levels of correct transcript in the wild-type situation. When delivered after mutant maxigene transfection, most of the AONs did not further increase or decrease the levels of correct transcript compared with the nontreated lane, only sequences #11 to #14 and #18 tended to promote more co-skipping events (Fig. 2B).
Importantly, AONs #8, #9, and #19 to #23 managed to restore, to some extent, the presence of the correct transcript. AON #9 consistently outperformed the rest of oligonucleotides in both wild-type and mutant conditions since it could significantly increase the levels of correct transcript from 22.4% to 42.2% compared with the respective nontreated condition, which was accompanied by a decrease of the co-skipping event but not a decrease in Δ28 and Δ29 transcripts individually (Fig. 2B and Supplementary Table S7). AON #8 could also reach a modest increase of correct transcript without reducing exon 28 and 29 skipping bands. Notably, skipping of exon 28 alone was barely visible after administration of AONs #19 to #23, but there was a generalized increased skipping of exon 29 by ∼10% compared with the mutant maxigene condition without any treatment.
Overall, these results showed the capacity of AONs to promote exon inclusion in a maxigene-based system in HEK293T by implementing an oligo-walk design. In contrast with the designed ESS-blocking AONs, this approach was able to induce the desired effect on splicing when targeting the intronic c.4253+43G>A variant directly.
Skipping rescue analysis in patient-derived PPCs
The best-performing AONs in HEK293T were selected to be further assessed on a more retina-like cell model with the full ABCA4 context. This model was also employed to confirm the presence of exon 28 and 29 skipping in a context more similar to the retinal environment. The iPSC line derived from the patient harboring the c.4253+43G>A variant (present in cis with c.6006-609T>A and in compound heterozygosity with c.1822T>A) [19] was differentiated for 30 days toward the formation of PPC culture, together with the control iPSC line. Subsequently, the obtained culture was characterized by qPCR analysis of a series of retinal markers to confirm a correct differentiation (Supplementary Fig. S3). In brief, both lines showed a clear increase in expression of ABCA4, indicating the suitability of these cultures to analyze splicing at the RNA level. The characterization of these PPC cultures was in line with previously reported studies [10,12,15,16,18] and suggested the early retinal differentiation stage with a heterogeneous cell population producing higher levels of ABCA4 transcript.
At day 20 of differentiation of both healthy control and patient-derived PPCs, seven selected AONs and one of the SONs (#2) were delivered gymnotically at two different concentrations (1 and 5 μM) to evaluate their exon inclusion efficacy in this cell model. Furthermore, cycloheximide (CHX) treatment was conducted 24 h before harvesting the cells to block nonsense-mediated decay (NMD) and be able to adequately assess the effect of the AONs. Overall, these data notably differed from the splicing assays using the maxigene vector. Clear differences in the splicing pattern between the control and patient-derived cells were detected, even though the treatment with CHX did not considerably increase the accumulation of aberrant transcripts in either of the two PPC lines (Fig. 3).
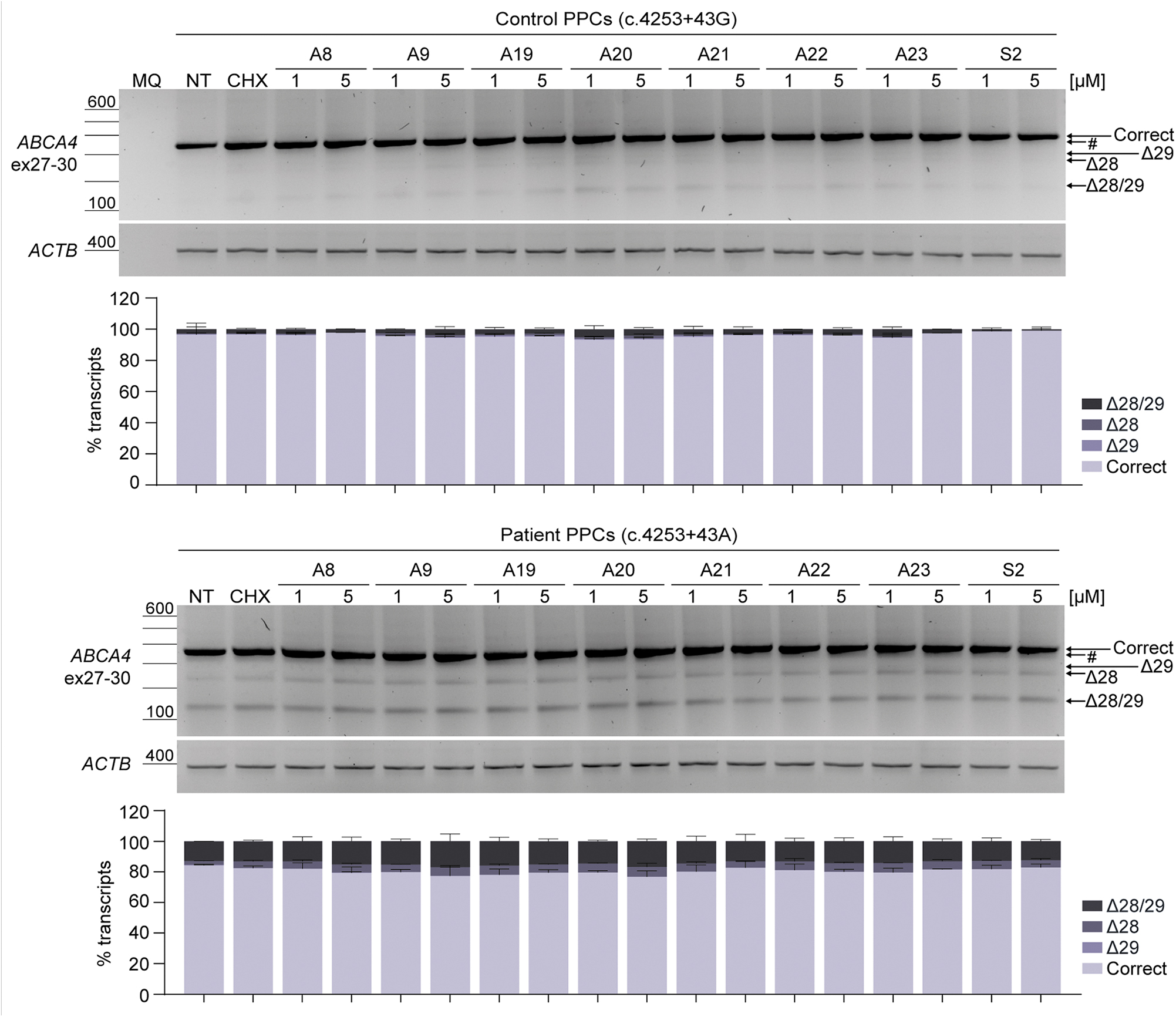
The presence of higher levels of skipping events was observed in the patient-derived PPCs compared with the healthy control-derived PPCs after CHX treatment. Correct and skipped transcripts (Δ28, Δ29, and Δ28/29) in PPCs could be confirmed by Sanger sequencing (Supplementary Fig. S4), except for the Δ29 transcript in patient-derived PPCs. In contrast with the noticeable increase of the Δ28/29 transcript in mutant maxigene conditions, an increase of the Δ28/29 transcript from ∼3% to 13% of the total transcript levels was detected when evaluating CHX-treated healthy control and patient conditions, respectively, and skipping of exon 28 only represented around 4% of the total transcript levels (Supplementary Table S8). Despite producing fewer amounts of skipped transcripts, none of the delivered AONs were able to promote the inclusion of exons 28 and/or 29 in patient-derived PPCs for both tested concentrations. In conclusion, the decrease of exon skipping levels observed in maxigene-based splice assays was not recapitulated in the PPC model following previously validated protocols [10,12,15,16,18].
Discussion
Most of the intronic ABCA4 variants affect the splicing process by inducing the recognition of cryptic exons or pseudoexons, while a smaller proportion causes exon elongation or exon skipping [1]. Variant c.4253+43G>A was originally described to alter the pre-mRNA splicing process by disrupting splicing motifs around this position, and as previously mentioned, led to exon 28 skipping that could not be rescued by AON delivery on midigene-based assays [8]. This in vitro model may not always show the natural splicing profile due to limitations in genomic and cellular context [24]. For that reason, we generated a maxigene vector able to harbor a larger genomic content to recapitulate a more precise splicing pattern, which was reproduced in a retina-like cell model. The current study has helped to elucidate other skipping events that were not yet revealed, and demonstrates how crucial genomic and cellular context are to adequately characterize exon skipping events.
Prediction of splicing regulatory motifs showed a more pronounced presence of ESS motifs than ESE motifs within the sequence, but the nucleotide change increases the ESE/ESS ratio around this region by creating and strengthening SF2/ASF and 9G8 motifs, respectively. These ESE motifs are recognized by the serine/arginine (SR)-rich proteins, which are described to increase exon inclusion by facilitating the recruitment of the spliceosome factors within exonic regions [25]. For example, intronic MTRR gene mutations creating a new SF2/ASF protein-binding motif induced pseudoexon inclusion in the final transcript [26], and disruption of an SF2/ASF-dependent exonic enhancer motif in SMN2 led to suboptimal exon inclusion [27]. Altogether, these studies suggested that SF2/ASF motifs have the capacity to preserve exon inclusion.
Nonetheless, potential binding sites for SR proteins have been found within intronic regions and consequently could produce a negative impact on splicing by interfering with the aforementioned recruitment of splicing factors [28]. Previous research has already suggested the duality of SR proteins within the splicing process, also acting as negative regulator of splicing [29–31].
Furthermore, three previously described ESS motifs [32] around this position are predicted to be recognized by the heterogeneous nuclear ribonucleoproteins (hnRNP) H and F, which have been associated with promoting exon skipping via interaction with splicing repressors [25,33,34]. Interestingly, these were shown to have a role in splicing regulation through participation in both splicing enhancer and silencer complexes [35]. Considering the duality of these trans-acting elements on splicing, it may suggest that the combination of all three ESE motifs together with the abruption of two ESS motifs are disbalancing the outcome toward splicing repression rather than activation, to some extent explaining the skipping of exons 28 and 29 in ABCA4.
In relation to this, dependency on maintaining the correct balance of splicing regulatory sequences was already reported to be essential for those exons vulnerable to nucleotide changes affecting exonic motifs [36], and perhaps might be translated to those located within intronic regions. Another aspect to consider is that these splicing enhancers and silencer motifs often present overlapping sequences, and prediction tools can identify various regulatory elements within similar sequences in both exonic and intronic regions as they correspond to highly degenerate motifs [37]. Therefore, the prediction whether a certain motif is actually acting as a positive or as a negative regulator of splicing is challenging, adding complexity to understanding their exact contribution to the (dys)regulation of splicing.
To restore correct ABCA4 splicing, two different strategies to promote exon 28 and 29 inclusion using AONs have been followed in this study. A common design to reduce skipping of the target region is to block potential splicing silencer motifs to increase the recognition of the exon [38,39]. However, our maxigene-based assay demonstrated a poor exon inclusion efficacy by blocking high-scored ESS motifs. Contrary to these findings, targeting enhancer motifs found in intronic regions with AONs successfully achieved efficient exon 8 inclusion in the SLC26A4 gene associated with sensorineural hearing loss [40], although targeting those motifs within the exonic areas was also shown to promote exon inclusion [41,42]. It is also important to note that the predicted high-score ESSs were not always located in regions with the lowest ESE/ESS ratio, which could be one explanation for the low efficacy observed for ESS-blocking AONs, as they could be disrupting the splicing regulators balance and thereby resulting in a repressor effect.
The second strategy followed was an oligo-walk approach to specifically cover the region around the c.4253+43G>A variant, a strategy that has been recently used to increase exon inclusion [17] for ABCA4. In our maxigene-based splice assay, the approach contributed to the identification of AONs able to increase inclusion of exons 28 and 29, although in some cases, this was due to the decrease of the co-skipping event but not a decrease of individual skipping events. This may suggest that these AONs could be targeting essential motifs that contribute to the inclusion of these exons. In addition, the ABCA4 region harboring exons 28 and 29 seems to be relatively susceptible to be skipped even in absence of the c.4253+43G>A variant and maintaining the balance of splicing regulatory elements could be crucial to avoid excessive skipping of the exons.
Nonetheless, steric blockage by AONs might be either helping to or interfering with restoring the balance toward the wild-type situation due to induced variation in regulatory protein binding. Moreover, the secondary structure of the ABCA4 pre-mRNA may also play a role in differential splicing of exons 28 and 29. It is known that the formation of RNA secondary structures can influence splicing by masking or exposing regulatory motifs through hairpin, G-quadruplexes, or long-distance interactions, and mutations around these regions can completely change the stability of these structures [43–45]. In a similar way, binding of AONs might also alter the formation of these structures and influence exon inclusion.
Despite observing an increase of exon 28 and 29 skipping, patient-derived PPCs carrying the c.4253+43G>A variant showed a less prominent splicing defect Δ28 compared with experiments using the maxigene system in HEK293T cells, and healthy control-derived PPCs were also observed to have only very few skipping events. Curiously, the patient line presented the Δ28 and Δ28/29 transcripts, but not the Δ29 transcript, which differs from the outcome in the maxigene assay. The more retina-like cellular context still showed the defect produced by the intronic variant, although perhaps a more mature 3D model such as retinal organoids [46,47] would be needed to further corroborate that the skipping events are indeed induced by the variant and the molecular context, and it is not an artifact of the maxigene system.
Even with a less noticeable splicing defect, the exon inclusion guided by AON delivery in HEK293T cells could not be translated into the PPC model. This might be the result of, for instance, suboptimal delivery to the target cells, either by an unadjusted concentration of the tested AONs or by a reduced uptake of the molecule by the PPCs. The significant differences in the structures of the ABCA4 pre-mRNA produced by the maxigene and the PPCs may influence the binding of the AONs and their efficacy, as well as the potential impact of different splicing factors that are specific to each cell types.
In addition, CHX treatment did not dramatically increase the presence of skipped transcript in either the control or patient PPCs, hinting that these transcripts might not be substantially subjected to NMD in this model. The only observed difference was a slight increase of skipping of exons 28 and 28/29 upon CHX treatment, while it was not detected for exon 29. This might be explained by the fact that exon skipping of exon 29 results in the production of in-frame transcripts, while Δ28 and Δ28/29 lead to a frameshift, and consequently, create a premature stop codon that may trigger NMD [48].
Despite the low exon inclusion efficacy by either the ESS-blocking or the oligo-walk AONs in maxigene-based splice assay or PPCs, it has been previously shown that both strategies could successfully restore correct splicing. The most recent example is the rescue of exon 39 and 40 skipping in ABCA4 [17,49] caused by the severe variant c.5461-10T>C [50,51]. In this case, the best-performing AON binds in close proximity of the donor site of intron 39, similarly to AON #9 in this study, and the region is not predicted to have a remarkable larger number of silencer motifs compared with the analyzed sequence in intron 28. The Food and Drug Administration (FDA) and European Medicines Agency (EMA)-approved Spinraza (Nusinersen) is a clear example of a successful exon inclusion through the blocking of an intronic splicing silencer, being the very first treatment for spinal muscular atrophy [9].
This AON disrupts the function of a silencer located downstream exon 7 of the SMN2 gene and facilitates exon inclusion, leading to restoration of the protein levels with promising outcomes in clinical trials [52]. A potential alternative for exon 28 and 29 inclusion might be the use of bifunctional AONs, which have a domain complementary to the target region and a tail domain able to recruit splicing regulatory proteins, helping to increase the number of positive signals in a specific region [38,53].
Conclusions
Altogether, this study contributed to obtain a better picture of the splicing defects induced by the intronic ABCA4 variant c.4253+43G>A by implementing the use of a maxigene vector in HEK293T cells, which also helped to narrow down the number of efficacious AONs after comprehensive screening. However, these commonly implemented strategies to promote exon inclusion were not as effective as previously demonstrated, since in this specific case, the beneficial effect could not be translated into a patient-derived retinal-like cell model. In conclusion, further optimization of the AON design is needed to rescue the skipping of exons 28 and/or 29 in ABCA4.
Footnotes
Acknowledgments
The authors would like to thank the healthy individual and the patient for donating the cells and contributing to the search for new therapeutic approaches. We also thank the Radboudumc Stem Cell Technology Centre for generating the healthy individual- and patient-derived iPSCs, and Saskia van der Velde-Visser for cell culture technical assistance. We acknowledge Dr. Jim Swildens and Melita Kaltak for manufacturing AONs and providing helpful feedback, as well as Prof. Frans P.M. Cremers' research group for the engineered ABCA4 minigene constructs used for the generation of the maxigene constructs.
Data Availability Statement
The data supporting the findings of this research are available within the article and supplemental materials. Raw data are available upon reasonable request from the corresponding author.
Disclaimer
The funding organizations provided unrestricted grants and had no role in the design or conduct of this research.
Author Disclosure Statement
R.W.J.C is the Chief Scientific Officer of Astherna B.V. R.W.J.C. and A.G. are inventors on several filed patents describing the use of antisense oligonucleotides (WO2013036105A1, WO2018109011A1, WO2020015959A1, WO2020115106A1, WO2021023863A1) for the treatment of Stargardt disease.
Funding Information
This research was financially supported by European Union's Horizon 2020 research and innovation program Marie Sklodowska-Curie Innovative Training Network (ITN) StarT (Grant No. 813490 to R.W.J.C.), the Foundation Fighting Blindness USA Project Program Award (PPA-0517–0717-RAD to R.W.J.C. and A.G.), and the Translational Research Acceleration Program (TRAP) of the Foundation Fighting Blindness USA (TA-GT-0521-0799-RAD-TRAP to R.W.J.C.).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.