
Abstract
RNase H1-dependent phosphorothioate oligonucleotides (PS-ASOs) have been developed to treat various diseases through specific degradation of target RNAs. Although many factors or features of RNA and PS-ASOs have been demonstrated to affect antisense activity of PS-ASOs, little is known regarding the roles of RNase H1-associated proteins in PS-ASO performance. In this study, we report that two nucleolar proteins, NAT10 and DDX21, interact with RNase H1 and affect the potency and safety of PS-ASOs. The interactions of these two proteins with RNase H1 were determined using BioID proximity labeling in cells and confirmed biochemically. Reduction of NAT10 and DDX21 decreased PS-ASO activity in cells, and purified NAT10 and DDX21 proteins enhanced RNase H1 cleavage rates, indicating that these two proteins facilitate RNase H1 endoribonuclease activity. Consistently, reduction of these proteins increased the levels of R-loops, and impaired pre-rRNA processing. In addition, reduction of the two proteins increased the cytotoxicity of toxic PS-ASOs, and treatment of toxic PS-ASOs also altered the localization of these proteins. Together, this study shows for the first time that NAT10 and DDX21 interact with RNase H1 protein and enhance its enzymatic activity, contributing to the potency and safety of PS-ASOs.
Introduction
RNase H1 is an endoribonuclease that cleaves the RNA strand in DNA/RNA heteroduplex, and has been shown to be involved in R-loop solving and mitochondria DNA replication likely through affecting RNA primes [1–3]. Endogenous RNase H1 is utilized by antisense oligonucleotide (ASO)-mediated antisense technology to degrade specific target RNAs complementary to the ASOs [4]. The commonly used ASOs are modified with phosphorothioate (PS) backbones, and 2′ modifications at 3–5 nucleotides in both ends of the ASOs, such as methoxyethyl (MOE) and constrained ethyl (cEt) [5]. These chemical modifications dramatically improve ASO drug performance through enhanced tissue and cell uptake, subcellular distribution, and nuclease resistance in biological systems [6].
The action of PS-ASOs involves at least three components, the PS-ASO and target RNA, which form a heteroduplex through hybridization, and RNase H1 to cleave the RNA strand within the heteroduplex. Thus, factors or features in all three components can affect antisense activity. For example, from the RNA side, it has been shown that RNA structure can affect ASO accessibility; subcellular localization can influence ASO-targeted RNA degradation; and translating ribosomes can reduce ASO activity when targeting mRNA coding regions [4].
However, the copy number and half-life of RNA substrates (except extremely short-lived RNAs) have minor effect on ASO activity. From the PS-ASO side, it has been well documented that chemical modification, conjugation, ASO sequence, and configuration can dramatically affect ASO performance through different mechanisms [6]. In recent years, it has been demonstrated that PS-ASO interactions with cellular (and extracellular) proteins, that is strongly affected by ASO sequence and chemical modifications, play critical roles in PS-ASO activity and safety [4,6,7].
RNase H1, the key enzyme used by PS-ASOs to cleave target RNAs and mediate PS-ASO cytotoxicity [8,9], is also well studied, especially as a holoenzyme. The contributions of individual domains and key amino acids in duplex binding and catalytic activities have been identified [10–13], and the interactions of this enzyme with ASOs and with ASO/RNA duplexes have been evaluated in detail [14–16]. Despite the fact that ample knowledge has been learned previously regarding the enzyme itself, not much is known about the partner proteins that may regulate the activity of RNase H1. Understanding the interactions of RNase H1 with other cellular proteins and their roles in modulating RNase H1 activity and function is important for both biology and antisense technology.
Several proteins have been found to interact with RNase H1. Single-stranded (ss) DNA binding proteins (SSBs), which bind and protect exposed ssDNA, have been shown to be able to complex with RNase H1 and stimulate RNase H1 activities in bacteria [17]. RPA70 protein interacts with RNase H1 and enhances the cleavage activities in vitro [18]. The RPA proteins may regulate R-loop formation and genome stability mediated by RNase H1 [18]. In addition, our group discovered that P32 could bind RNase H1 and alter the enzyme activity [19]. However, the interaction of P32 and RNase H1 can be disrupted by the binding of PS-ASO or ASO/RNA duplex to RNase H1 [15].
Recently, we also found that toxic PS-ASOs can induce the interactions between RNase H1 and P54nrb/PSF proteins [15,20], causing these nucleoplasmic paraspeckle proteins to mislocalize to the nucleoli and contributing to cytotoxicity. These observations suggest that proteins that interact with RNase H1 may regulate RNase H1 function, affecting the performance of PS-ASOs. Identifying such proteins can provide more detailed understanding of antisense mechanisms and facilitates better design of PS-ASO drugs.
RNase H1 is present in the cytosol, mitochondria, and nucleus [21]. It has been shown that RNase H1 is also enriched in the nucleolus and can shuttle between nucleoplasm and nucleolus [22]. The nucleolus is a membrane-less organelle that can form de novo and undergoes dynamic molecule exchange with surrounding environment, especially due to the rapid synthesis of rRNA and ribosomal subunits [23]. Many proteins have been identified in the nucleolus, including NAT10 and DDX21, the two proteins featured in this study. NAT10 is an acetyltransferase and a member of the Gcn5-related N-acetyltransferase (GNAT) family of KATs, which acetylates RNA [24] and proteins, including transcriptional factor p53 [25], transcriptional cofactor Che-1 [26], and (ADP-ribose) polymerase 1 (PARP1) [27]. NAT10 has multiple functions, for example, in ribosome synthesis [28], midbody formation [29], autophagy [26], and cell division [27].
This is consistent with the view that the nucleolus is not only the major site for the biogenesis of ribosomes but also plays important roles in regulating cell stress, cell cycle, telomerase installment, and interaction with other nuclear bodies [30,31]. On the other hand, the RNA helicase DDX21 regulates the activities of both RNA polymerase I and II [32]. DDX21 promotes rRNA transcription, processing, and modifications [32], and has been shown to associate with different types of RNAs, including rRNAs, snoRNAs, 7SK RNA, mRNA [32], and miRNAs [33]. DDX21 can unwind dsRNA and acts as a foldase and contributes to rRNA procession and R-loop formation [34]. DDX21 can be ADP ribosylated by snoRNP-associated PARP-1 [35].
To further identify RNase H1-associated proteins and to understand how these proteins affect RNase H1 activity in cells, in this study, we used BioID system to identify new protein partners of RNase H1. We found that NAT10 and DDX21 interact with RNase H1 in cells. Reduction of these proteins reduced PS-ASO activity and caused R-loop accumulation, suggesting that these proteins can facilitate RNase H1 function. Indeed, RNase H1 cleavage activity could be enhanced with purified NAT10 and DDX21. In addition, toxic PS-ASOs also caused mislocalization of these two proteins and reduction of NAT10 and DDX21 exacerbated cytotoxicity of toxic PS-ASOs. These results indicate that NAT10 and DDX21 serve as protein partners of RNase H1, facilitating RNase H1 activities and PS-ASO performance.
Materials and Methods
Plasmids, reagents, primers, and ASOs
Plasmids (Supplementary Table S1), PS-ASOs (Supplementary Table S2), siRNA, and primer probe sets for quantitative real-time polymerase chain reaction (qRT-PCR) are listed in Supplementary Data .
BioID pull down
Around 1 × 107 Hela cells were transfected with H1-BioID2 (H1) plasmids for 36 h, and then transfected with safe and toxic PS-ASOs at 50 nM. Two hours later, 50 μM biotin was added to the media, and incubated with cells for an additional 16 h. The cells were harvested by trypsin digestion and the nuclear fraction was obtained using Paris Kit (AM1921; Thermo Fisher Scientific).
IP buffer (Thermo Scientific; 87787) was used to prepare the nuclear lysate, and the proteins bound to expressed BioID2-HA-tagged H1 were pulled down with NeutrAvidin resin. Resin was washed stringently by W100 [50 mM Tris–HCl (pH 7.5), 100 mM KCl, 5 mM EDTA, and 0.1% NP-40, 0.05% SDS] five times, moved to new tubes, and washed by W300 (same as W100, but with 300 mM KCl) another five times. The isolated proteins were analyzed on 4%–12% SDS-PAGE, stained by silver staining, and used for mass spec analysis.
Cell culture and transfection
HeLa, HEK293 (FT and CT), and Hepa1–6 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 0.1 μg/mL streptomycin, and 100 U/mL penicillin. Cells were seeded at 60%–70% confluency and grown in 15 cm dishes for 16 h before transfection. The luciferase control, human NAT10, and DDX21 siRNAs were transfected into cells using Lipofectamine RNAiMax (Life Technologies) at 4.0 nM final siRNA concentration to deplete the NAT10 and DDX21 in HeLa and 293CT. For plasmid transfection into 293 cells, 5 million cells in 10 cm dishes were transfected either with empty vector plasmid or plasmids carrying NAT10 or DDX21 expression constructs using Lipofectamine 3000 (Life Technologies), based on the manufacturer's instructions. For PS-ASO transfection, 36 h after siRNA or plasmid transfection, HeLa cells were re-seeded in 96-well plates at ∼70% confluency.
The HeLa cells were incubated overnight in a 37°C incubator with 5% CO2. The following morning, PS-ASOs were transfected at 0–120 nM (1:2 dilution) or 0–20 nM (for Malat1) for 4 h using Lipofectamine 2000 (Life Technologies). The same Lipofectamine 2000 and PS-ASO condition were applied for the reverse transfection of 293CT cells at 48 h after NAT10 and DDX21 knockdown. Next, cells were rinsed with PBS and total RNA was extracted for qRT-PCR analysis, as shown in Fig. 4 and Supplementary Figs. S6–S8.
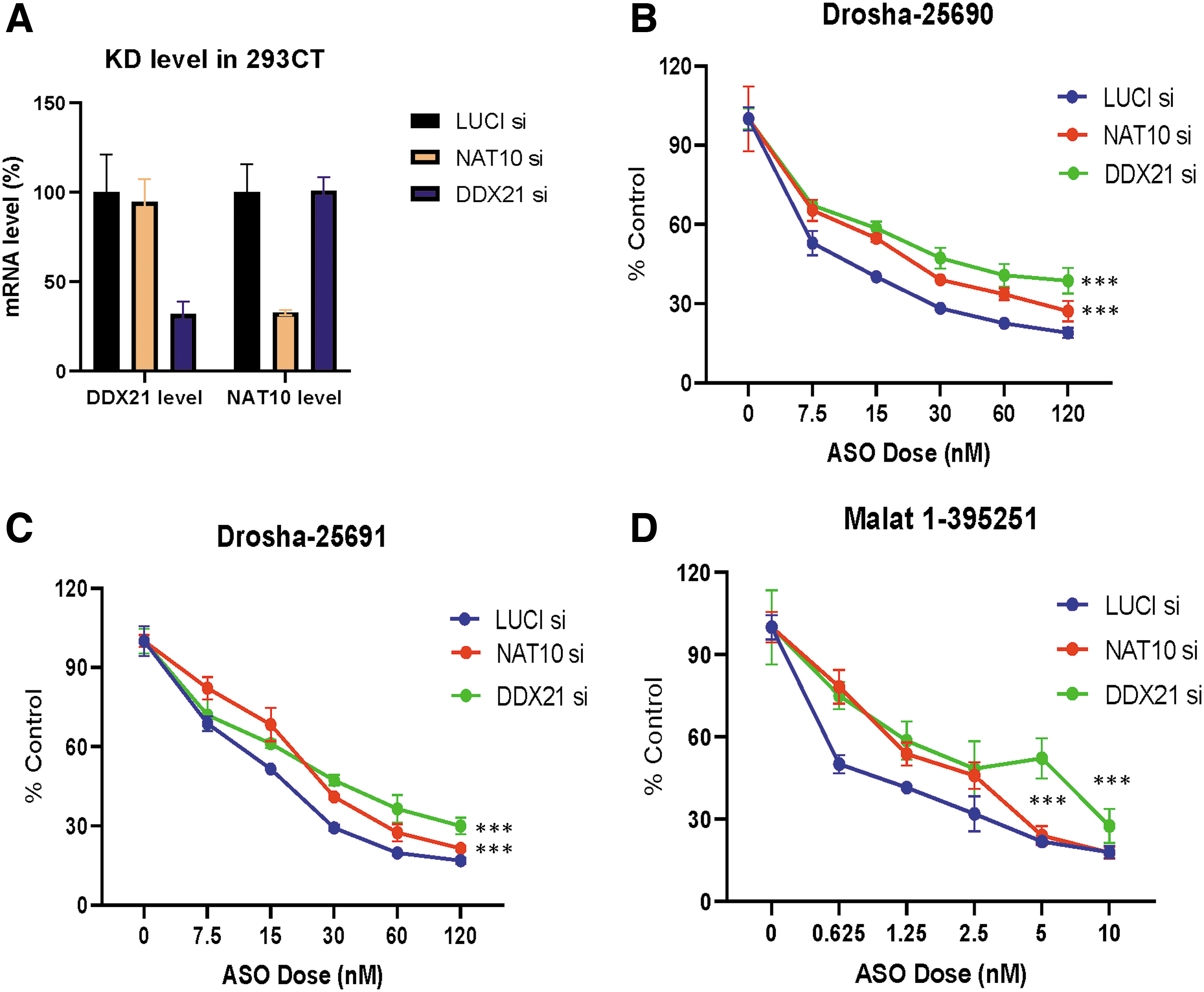
The depletion of NAT10 and DDX21 reduced ASO activities in 293CT cells.
RNA preparation and qRT-PCR analysis
RNA preparation and the qRT-PCR assay using TaqMan primer-probe sets were performed as described previously [36]. Total RNA was prepared using RNeasy mini kit in a 96-well column plate (Qiagen), based on the manufacturer's instructions, and eluted with 120 μL water. qRT-PCR was performed in quadruplicate using the QuantStudio 7 Flex system and TaqMan primer-probe sets as listed in Supplementary Data, using the AgPathID™ One-step RT-PCR kit (Applied Biosystems). qRT-PCR in 20 μL reactions was performed using the following program: 48°C for 10 min, 94°C for 10 min, and 40 cycles of 20 s each at 94°C and 60°C. The qRT-PCR results were quantified using the QuantStudio Software v.1.7.1, calculated, and plotted in Excel.
The tested RNA levels per reaction were normalized to RNA levels measured using RiboGreen (Life Technologies) or GAPDH mRNA level determined by qRT-PCR for aliquot RNA samples. Average values and standard deviations from four experiments were calculated and plotted. Statistics were performed using GraphPad Prism with the F-test for curve comparison based on nonlinear regression (dose–response curves) for XY analyses, using equation “log(agonist) versus normalized response–variable slope.” The y axis (relative level) was used as the normalized response.
Immunoprecipitation
Five million of HEK293FT cells were transfected with various plasmids (2–3 μg/each construct) using Lipofectamine 3000 (Life Technologies), and collected 48 h later. Cell pellets were lysed in lysis buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 1.5 mM MgCl2, 5 mM KCl, 0.1% NP40, 10% glycerol, 1 mM DTT, and protease inhibitor cocktails). The cells were sonicated thrice at 30% strength. Cell lysates were centrifuged at 18,000 g for 30 min at 4°C and 2 mg of supernatants was incubated with 40 μL anti-Flag M2 agarose beads (Sigma) or protein G beads (Sigma) precoated with anti-HA antibody for 3 h at 4°C.
Beads were washed with W100 buffer five times, then transferred to new tubes, and washed with W300 buffer for another five times. Ten microliters of beads and 20 μg input (1% lysate) were boiled with 2 × SDS-sample buffer, and analyzed by SDS-PAGE and Western blot for interested proteins, as shown in Fig. 2 and Supplementary Figs. S2–S4A.
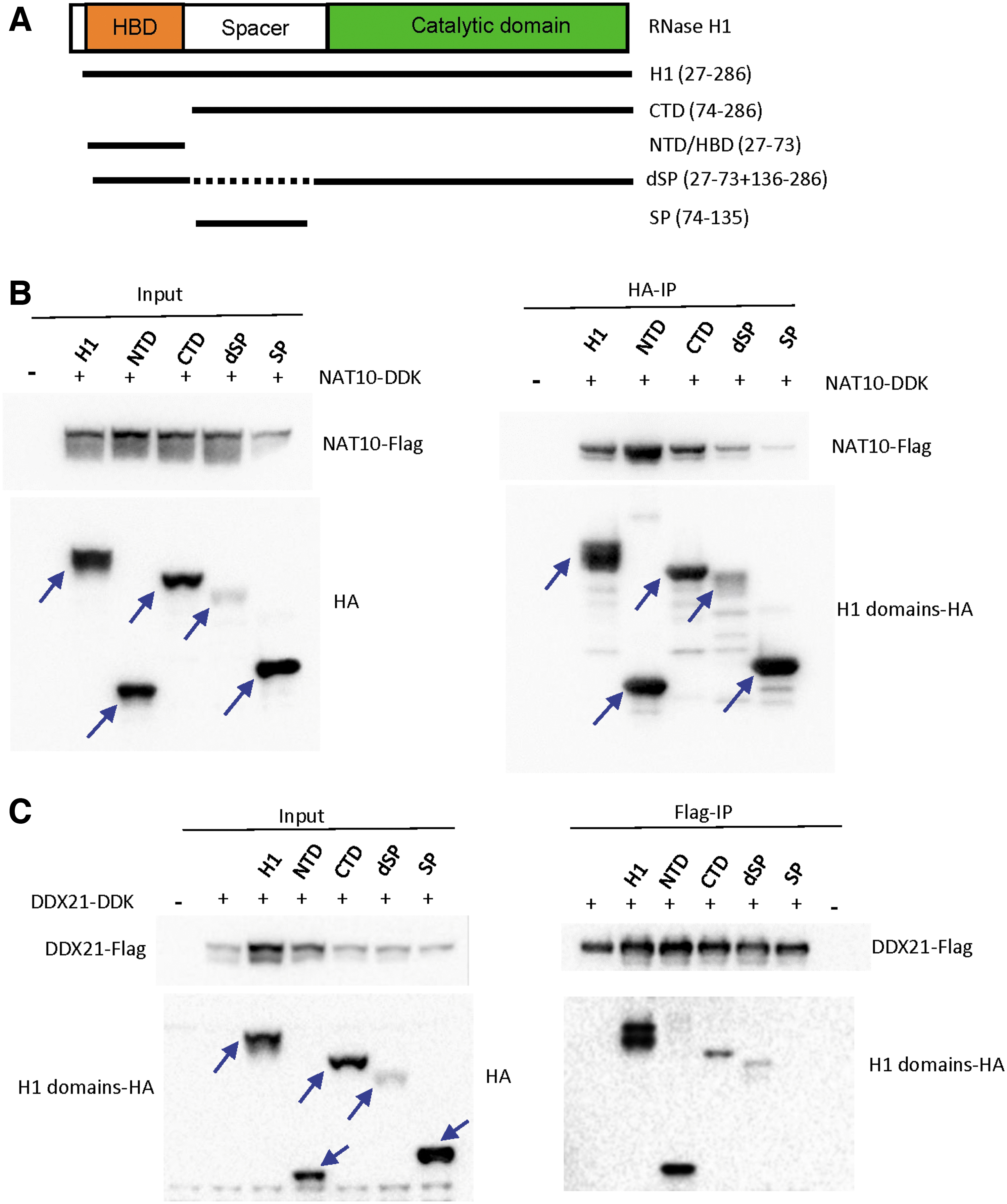
Both the NTD and CTD of RNase H1 contribute to the binding with NAT10 and DDX21.
Western blot analyses
Following 36 h of siRNA transfection, cells were then re-seeded in 10 cm dishes at ∼70% confluency and left to grow overnight in a 37°C incubator with 5% CO2. The following morning, safe and toxic ASOs were transfected at 80 nM into cells for 8 h using Lipofectamine 2000 (Life Technologies). Cells were washed with cold 1 × PBS and collected using a cell scraper. Cell lysate was prepared using RIPA buffer (ThermoFisher) and cleared by centrifugation at 10,000 g for 20 min at 4°C. Proteins (40 μg/lane) were separated by 4%–12% SDS-PAGE and transferred to membrane. Interested proteins were detected with specific antibodies, and visualized using ECL.
The following antibodies were purchased from Abcam: NAT10 (ab194297), DDX21 (ab182156), NPM1 (ab10530), NCL (ab134164), acetylated alpha-tubulin (ab24610), and cleaved PARP1 (ab4830). PSF (sc-374502) and P54 (sc-376865) antibodies were from Santa Cruz Biotechnology. The RNase H1 antibody (15606-I-AP) was obtained from ProteinTech. Quantification of proteins detected in Western blot was performed using ImageJ and normalized to the levels of GAPDH detected from the same blot using the GAPDH antibody (G9545; Sigma Aldrich). Anti-rabbit secondary antibody conjugated to HRP (170-6515) and anti-mouse secondary antibody conjugated to HRP (170-6516) were purchased from Bio-Rad.
Immunofluorescence staining
Thirty-six hours after siRNA transfection, cells were reseeded in 35 mm glass-bottom dishes at ∼75% confluency and left to grow overnight in a 37°C incubator with 5% CO2. The following morning, safe and toxic ASOs were transfected at 100 nM into the siRNA-treated cells for 6 h using Lipofectamine 2000 (Life Technologies). Cells were then washed with PBS, fixed with 4% paraformaldehyde for 30 min, and permeabilized for 5 min with 0.1% Triton X-100 in PBS. After blocking at room temperature for 30 min with block buffer (1 mg/mL BSA in 1 × PBS), cells were incubated for 2 h with primary antibodies in block buffer as listed: PSF (Abcam; sc-3774502), P54 (Abcam; sc-376865), NAT10 (Abcam; ab194297), DDX21 (Abcam; ab182156), Lamin A + Lamin C (Abcam; ab108595), total alpha-tubulin (Abcam; ab7291), acetylated alpha-tubulin (Cell Signaling; D20G3), R-Loop (Kerafast; ENH001), and NCL (ab134164).
Cells were then washed thrice (5 min each) using wash buffer (0.1% NP-40 in 1 × PBS), and incubated for 1 h with anti-Mouse secondary antibody conjugated with AF488 (Abcam ab150113; 1:200) and anti-Rabbit secondary antibody conjugated with AF647 (Abcam ab150075; 1:200). After washing thrice, cells were mounted with anti-fade reagent containing DAPI (Life Technologies). Images were taken using confocal microscope (Olympus FV-1000) and processed using FV-10 ASW 3.0 Viewer (Olympus). Fluorescence quantification was obtained using the Olympus CellSens Dimension software's Count and Measure feature. The experiments were repeated at least thrice.
Chromatin immunoprecipitation-quantitative PCR
After siRNA knockdown for 48 h, Hela cells (1 × 107) were crosslinked by treatment with 1% formaldehyde for 10 min. Reactions were quenched by addition of glycine to 125 mM and incubation for 5 min at room temperature. Cells were lysed, and chromatin fragmented by sonication to an average length of 250–500 bp. Chromatin immunoprecipitation (ChIP) assays were performed using SimpleChIP® Enzymatic Chromatin IP Kit (Cell Signaling), according to the manufacturer's instructions and the R-CHIP protocol [37]. Antibodies used for ChIP is anti-R-loop (Kerafast ENH001). The immunoprecipitated and input DNAs were isolated and used as template for TaqMan real-time PCR assay. Primer and probe sequences used in this study are listed in Supplementary Data and detailed information for q-PCR. Briefly, purified DNAs from immunoprecipitated or input samples were dissolved in 20 μL nuclease-free water.
Real-time PCR was set up in a 10 μL reaction by mixing primer probe sets containing forward and reverse primers (10 μM of each) and fluorescently labeled probe (3 μM) in 7.5 μL 2 × TaqMan® Gene Expression Master Mix (Thermo Fisher Scientific), and 2.5 μL DNAs (DNA was diluted 50 times from input, and 10 times from immunoprecipitated samples). The reactions were denatured at 95°C for 10 min and 40 cycles of PCRs were then conducted at 95°C for 15 s and 60°C for 40 s within each cycle, using QuantStudio 7 Flex system. The immunoprecipitation recovery rate was calculated as follows: 100 × (quantity [immunoprecipitated by specific antibody] – quantity [immunoprecipitated by IgG control])/quantity [total input]. With this method, signals obtained from each immunoprecipitation are expressed as a percentage of the total input [38]. Average values and standard deviations from three experiments were calculated and plotted.
Caspase activity assay
Hepa1-6 cells (2 × 105) were seeded in six-well plates at 60%–70% confluency for 16 h before transfection. Fifty nM mouse NAT10 and DDX21 siRNAs were transfected into Hepa1-6 cells using Hepa1-6 Cell Avalanche™ Transfection Reagent (EZT-HEPA-1) from EZ BIOSYSTEMS, according to manufacturer's instruction. After 48 h of siRNA transfection, Hepa1-6 cells were then re-seeded in triplicate in 96-well plates with 2.5 × 104 cells/well. The cells were electroporated with different ASOs (0–20 μM, 1:2 serial dilution) and incubated at 37°C for 24 h. For each time point, the treated 96-well plates were removed from incubator and equilibrated to room temperature before processing with the Caspase-Glo® 3/7 assay kit (Promega Biosciences; G8090). One hundred microliters of Caspase-Glo 3/7 reagent was added to each well containing 100 μL of media, and the plates were left to incubate at room temperature for 20 min. The luminescence of each sample was measured using the Tecan Spark plate reader, and the average values and standard deviations from triplicate were calculated in Excel and plotted in Prism.
RNase H1 cleavage assay
Five nM GST-RNase H1 or 2 nM mitochondria localization signal (MLS)-deleted H1 proteins purified in-house [15] were incubated with 300 nM preannealed PS-ASO/Cy3-RNA duplex and reacted at different times (0–10 min). The buffer condition was described previously [15], except that NAT10 and DDX21 were added at 60 nM in the reactions.
Pulse-chase labeling
HeLa cells were treated with DDX21 and NAT10 siRNA for 36 h, washed twice with PBS, and incubated with prewarmed medium lacking methionine (RPMI 1640; Invitrogen) at 37°C for 40 min. Cells were then pulse labeled at 37°C with 35 μCi/mL S35-methionine in the RPMI 1640 medium for 15 min, and chased for 40 min with 2 mM unlabeled methionine. Cells were washed with PBS containing 100 μg/mL cycloheximide and collected using trypsin. Cells were incubated on ice for 20 min with 500 μL lysis buffer (Thermo Scientific; 87787), and cell lysate was cleared by centrifugation. Immunoprecipitation was performed with 2 mg of lysate proteins and 2 μg P54nrb antibody prebound protein A/G magnetic beads (Sigma-Aldrich, Inc.; LSKMAGAG10). After stringent wash, 20 μg cell lysate and immunoprecipitated proteins were separated by SDS-PAGE, transferred to the membrane, and visualized by autoradiography.
Northern blot assays
Following 48 h of siRNA transfection, HeLa cells were then re-seeded in 15 cm dishes at ∼70% confluency and left to grow overnight in a 37°C incubator with 5% CO2. ASOs were transfected at 80 nM into the siRNA-treated cells for 8 h using Lipofectamine 2000 (Life Technologies). Cells were washed with cold 1 × PBS and collected using a cell scraper. Total RNA was extracted using TRIzol™ Reagent (Invitrogen; 15596026), according to manufacturer's protocol. Briefly, cells were washed with PBS, and 1 mL TRIzol reagent was directly added to the cells in plates. Whole lysates were collected and transferred into Eppendorf tubes, mixed, and incubated on ice for 5 min. Next, 200 μL chloroform was added and incubated at room temperature for 5 min. The mixture was then centrifuged at 12,000 g for 15 min.
The supernatant was transferred to clean tubes and equal volume of isopropanol was added and incubated at room temperature for 5 min. Total RNA was precipitated by centrifugation at 12,000 g for 20 min at 4°C. RNA pellet was washed with ice-cold 75% ethanol and then with 100% ice-cold ethanol, and dissolved in water. Ten micrograms total RNA was mixed with formaldehyde loading dye, and separated on a 1.2% denaturing agarose gel following the NorthernMax™ Kit protocol (Thermo Fisher Scientific; AM1940) in 1 × MOPS buffer. The RNA was transferred onto a membrane overnight using paper blot, and probed using P32-labeled probes specific to different regions of pre-rRNA. The results were visualized by autoradiography using the Storm 860 imager.
Results
NAT10 and DDX21 can interact with RNase H1
To identify cellular proteins that interact with RNase H1, the human RNase H1 protein without the MLS peptide (1-26AA) was fused at its C-terminus with BioID2, a second-generation biotin ligase [39]. The fusion protein also contains a C-terminal HA tag (Supplementary Table S1). The BioID2-HA tagged H1 expression plasmid was transfected into HeLa cells, and the expression of the tagged protein was confirmed by Western analysis using three different antibodies (Fig. 1A).
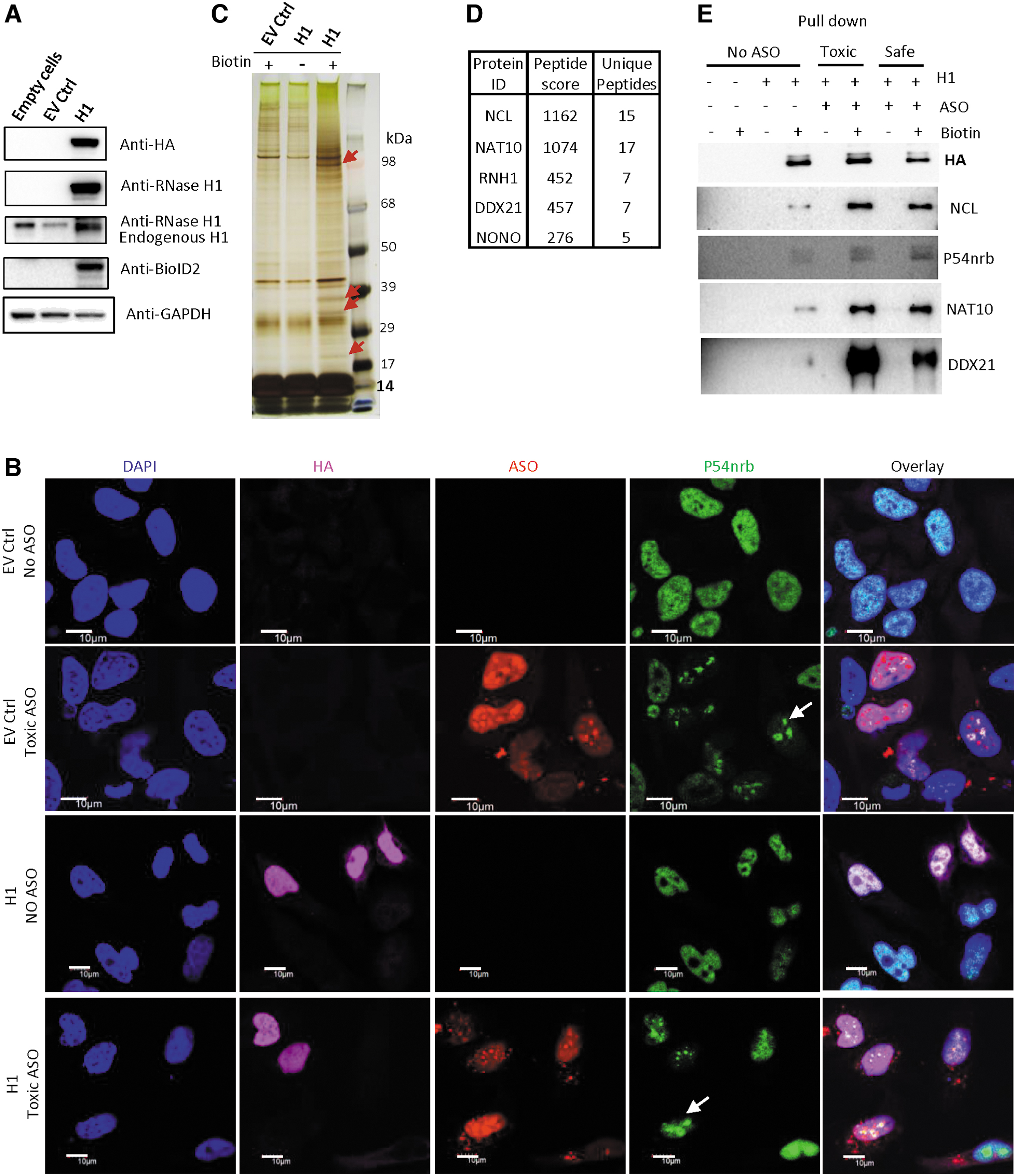
NAT10 and DDX21 can interact with RNase H1.
We note that the empty vector does not express the BioID2-HA peptide due to the lack of an in-frame start codon, as shown by Western blot (Supplementary Fig. S1A and data not shown) and immunofluorescence staining (Fig. 1B). The localization of the H1-BioID2 protein in cells was determined by immunofluorescence staining. As expected, the tagged RNase H1 without MLS localized in the nucleus, consistent with previous studies [18,40]. Upon toxic PS-ASO transfection, the tagged H1-BioID2 did not substantially alter the localization pattern, yet the paraspeckle protein P54nrb mislocalized to the nucleoli (Fig. 1B), in agreement with our previous observations [7].
Cells were then incubated with biotin to label RNase H1 interacting proteins. To eliminate biotinylated proteins from mitochondria, cell nuclei were prepared (Supplementary Fig. S1B). The biotinylated nuclear proteins were isolated using NeutrAvidin resin, separated on SDS-PAGE, and visualized by silver staining (Fig. 1C). Multiple proteins were isolated specifically in biotin-treated cells expressing the H1-BioID2 protein compared with cells containing the empty vector (lane 3 vs. lane 1 in Fig. 1C). Several proteins were identified through mass spectrometry, including not only RNase H1 but also NAT10, DDX21, NCL, and previously reported RNase H1-interacting proteins P54nrb/NONO (Fig. 1D) [20]. The unique peptides of identified proteins are listed in Fig. 1D.
The association of NAT10, DDX21, P54nrb, and NCL with H1-BioID2 was also confirmed using Western analysis for proteins isolated using BioID2 system from cells transfected or not transfected with PS-ASOs (Fig. 1E). Consistent with our previous observations that the presence of toxic PS-ASOs induced P54nrb/PSF interaction with RNase H1 [15,20], the interactions of endogenous DDX21 and NAT10 with H1-BioID2 were also substantially enhanced by PS-ASOs. Furthermore, NAT10 and DDX21 could be directly co-immunoprecipitated using anti-HA beads from lysates of cells expressing H1-BioID2 in the absence of PS-ASOs (Supplementary Fig. S1C). These results together indicate that these proteins interact with RNase H1, and the interactions can be enhanced by PS-ASOs.
Both N-terminal domain and C-terminal domain of RNase H1 contribute to the bindings with NAT10 and DDX21
To identify the domains of RNase H1 that participate in the interactions with NAT10 or DDX21, we constructed different RNase H1 domains on the BioID2-HA vector (Fig. 2A). The various RNase H1 plasmids were co-transfected with plasmids expressing Flag-tagged NAT10 or DDX21 into 293CT cells. The expression of different HA-tagged RNase H1 domains and the Flag-tagged NAT10 was confirmed by Western analyses (Fig. 2B, left panel). Immunoprecipitation was performed using anti-HA antibody to pull down different RNase H1 domains, and co-precipitated NAT10 was detected by Western analyses using anti-Flag antibody (Fig. 2B, right panel). The results demonstrated that the NTD, C-terminal domain (CTD), the full-length RNase H1 protein, and the spacer-domain-deleted (dsp) RNase H1, can bind to NAT10 (Fig. 2B), yet the spacer domain has much weaker interaction. We note that both NTD and CTD of H1 appear to have tighter interaction with NAT10 than the full-length H1.
Similar results were also observed when immunoprecipitation was performed using anti-Flag antibody to pull down NAT10 protein, and the associated H1 domains were detected by Western analyses using anti-HA antibody (Supplementary Fig. S2A). This is likely due to different conformations of individual domains compared with that present in the full-length protein. As a control, no NAT10 was co-isolated with anti-HA beads from cells without expression of HA-tagged RNase H1 (Supplementary Fig. S2B), or from cells expressing HA-tagged Hsp90 protein (Supplementary Fig. S2C), suggesting that the co-isolation of NAT10 with HA-tagged RNase H1 domains was not due to nonspecific interaction with the anti-HA beads.
A similar strategy was applied to detect the interactions between HA-tagged RNase H1 and Flag-tagged DDX21 co-expressed in cells (Fig. 2C, left panel). Anti-Flag antibody conjugated beads were used to immunoprecipitate the tagged DDX21 and the associated RNase H1 proteins. Western blot analyses showed that the NTD of RNase H1 contributes the major binding with DDX21, whereas CTD can also weakly interact with DDX21 protein (Fig. 2C, right panel). The expression of the spacer-domain-deleted RNase H1 protein was poor, which might contribute to the low level of co-immunoprecipitated protein. Together, these results indicate that the NTD of H1 can interact with both NAT10 and DDX21, and the CTD of H1 showed strong binding to NAT10, but much weaker binding to DDX21.
The CTD of DDX21 binds to RNase H1
Next, we sought to determine what domains in DDX21 and NAT10 interact with RNase H1. Different domains or full-length DDX21 were constructed with GST tag (Fig. 3A), expressed, and purified from Escherichia Coli. GST-tagged DDX21 proteins were immobilized on the glutathione resin (Fig. 3B, lower panel) and incubated with or without a toxic PS-ASO, since PS-ASO binding may affect protein conformation and protein-protein interactions [15]. Unbound PS-ASOs were removed by washing. Next, the resins preloaded with different domains of DDX21 were incubated with the same amount of purified MBP-RNase H1, and resin-retained proteins were co-isolated and analyzed by Western analyses.
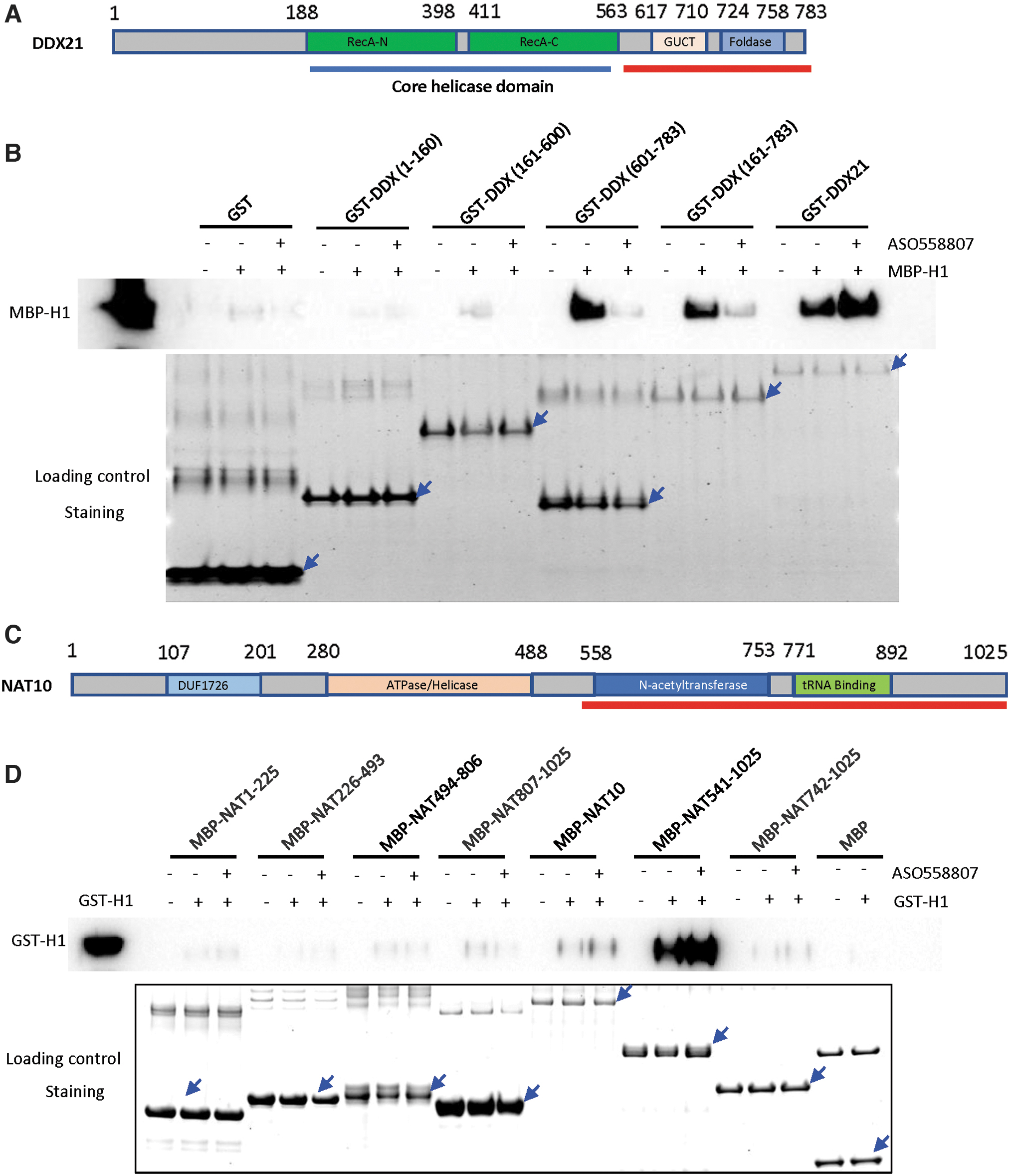
The C-terminal domains of purified DDX21 and NAT10 can bind RNase H1.
The results showed that the C-terminus (601–783) of DDX21 was associated with purified RNase H1 protein (Fig. 3B, upper panel). Interestingly, the toxic PS-ASO stimulated the binding between full-length DDX21 and RNase H1, but reduced the association of RNase H1 with DDX21 domains lacking the NTD (1–160) (Fig. 3B, upper panel). Similar results were observed in a repeat experiment (Data not shown). Currently, it is unclear how the NTD of DDX21 affects the interaction with RNase H1 in the presence of toxic PS-ASOs. The NTD may contribute to the folding of purified DDX21 domains, which may affect the interaction with toxic ASO, leading to altered interactions of DDX21 with purified RNase H1 protein. Understanding the detailed causes of these observations awaits further evaluation.
When using purified proteins containing the different domains of GST-DDX21 to pull down the RNase H1 from lysates of cells expressing the H1-BioID2, without PS-ASO or with safe or toxic PS-ASOs spiked in the lysates, the DDX21 domains containing 601-783AA interacted with RNase H1 (Supplementary Fig. S3A), similar to the observations using purified RNase H1 protein. However, the addition of PS-ASOs had no substantial effect on the interactions of DDX21 protein domains with RNase H1 present in cell lysate.
This experiment was repeated and similar results were observed (data not shown). The cause of different effects of PS-ASOs on the interactions of DDX21 with purified RNase H1 and with RNase H1 in cell lysate is currently unknown, but may be explained by the fact that in cell lysates, many other proteins exist that can bind competitively to PS-ASOs or DDX21 or RNase H1, reducing the effects of PS-ASOs on the interactions of DDX21 with RNase H1.
To further confirm that the CTD of DDX21 interacts with RNase H1, constructs were established to express Flag-tagged DDX21 domains in human cells. These plasmids were co-transfected with either H1-BioID2 plasmid or the empty vector into 293FT cells. Immunoprecipitation was performed using anti-Flag beads and co-precipitated RNase H1 protein was analyzed by Western blot (Supplementary Fig. S3B). The results showed that again the CTD of DDX21 (aa601-783) interacts with RNase H1, consistent with the observations using purified proteins (Fig. 3B). These results further confirmed that RNase H1 can interact with the CTD of DDX21 protein. The interaction between RNase H1 and DDX21 proteins was also confirmed in live cells using Nano-BiT assay (Supplementary Fig. S3C).
The NTD of NAT10 can interact with RNase H1 in cells
To evaluate the domains of NAT10 that interact with RNase H1, NAT10 was fused at the N-terminus with a MBP tag. To increase the solubility of different domains of NAT10, the dissections of all the NAT10 domains are instructed from the combination of the secondary structure prediction and the known crystal structure of homologous protein Kre33 [28] (Fig. 3C). The MBP-tagged NAT10 domains expressed well in E. coli, but the full-length NAT10 protein still showed limited expression (Fig. 3D, lower panel). We incubated the same amount of amylose resin carrying different domains of purified NAT10 proteins with or without toxic PS-ASOs, and then detected the association with purified, GST-tagged full-length RNase H1 protein.
Western blot analyses using RNase H1 antibody showed that, using the purified protein system, the CTD (541-1025 AA) of NAT10 associated with RNase H1. Since neither aa494-806 nor aa807-1025 domain showed robust binding to RNase H1, the entire CTD (541–1025) may be needed to form the binding pocket for RNase H1. The full-length NAT10 had a low expression level, yet still showed the association with RNase H1 (Fig. 3D, upper panel). Similar results were observed in a repeat experiment (data not shown). Consistent with the above observations, NAT10 can bind RNase H1 in the absence or presence of a toxic PS-ASO (Fig. 3D, upper panel).
Next, we evaluated if the interactions of NAT10 with RNase H1 observed with purified proteins also apply in cells. New constructs were established to express Flag-tagged NAT10 domains in human cells. These plasmids were co-transfected with either H1-BioID2 plasmid or an empty vector into 293FT cells, and co-immunoprecipitation was performed using the whole cell lysate and anti-Flag beads. The co-isolated H1 protein was analyzed by Western analysis (Supplementary Fig. S4A). Interestingly, the NTDs of NAT10 (especially aa1-226 and aa226–493), and not the CTD (aa 541–1025), were found to be the primary domains interacting with RNase H1 in cell lysate. The experiment was repeated, and a similar trend was observed (data not shown). These results suggest that in cells, the NTDs of NAT10 contact RNase H1.
Although it is unclear how the RNase H1 interaction domains of NAT10 differ between the purified proteins and cells, this is not unexpected, since in cells where many other cellular components exist, NAT10 and/or RNase H1 may adapt conformations different from purified proteins that affect their interactions. Despite this uncertainty, the above results clearly showed that NAT10 can interact with RNase H1 protein. The interaction of RNase H1 and NAT10 was also detected in live cells using the Nano-Bit assay (Supplementary Fig. S4B).
DDX21 and NAT10 enhance the antisense activity of PS-ASOs in cells
Since both DDX21 and NAT10 are known to be able to bind RNAs [35,41,42], these proteins may also bind PS-ASOs, which can bind many DNA/RNA binding proteins [43]. To evaluate this possibility, the NanoBRET assay was applied to examine the protein binding affinity to PS-ASOs with different chemistries (Supplementary Fig. S5). The results indicated that indeed, both proteins can bind PS-ASOs with decent affinity (Kd at 25–550 nM range), and the 2′-fluro-modified PS-ASO had the tightest binding, followed by 2′-cEt- and 2′-MOE-modified PS-ASOs, consistent with previous observations regarding the effects of 2′ modifications on protein binding [15,20].
To determine whether DDX21 and NAT10, which can bind both RNase H1 and PS-ASOs, impact the antisense activities of PS-ASOs, 293CT cells were treated with siRNAs targeting DDX21, NAT10, or Luciferase as a control for 48 h, followed by transfection of 2′-MOE-modified PS-ASOs targeting cytoplasmic Drosha mRNA, or nuclear Malat1 RNA. The effects of reduction of NAT10 or DDX21 proteins on the antisense activity were determined by analyzing the levels of PS-ASO target RNAs.
The levels of NAT10 and DDX21 mRNA were specifically reduced by their corresponding siRNAs, as analyzed by qRT-PCR (Fig. 4A). The siRNA-treated cells were subsequently transfected with two different PS-ASOs targeting Drosha mRNA (Fig. 4B, C), or an ASO targeting Malat1 RNA (Fig. 4D). The levels of Malat1 or Drosha RNAs were significantly reduced in a dose-dependent manner by their corresponding PS-ASOs, with IC50s of ∼0.6, 8.0, and ∼16 nM for Malat1 ASO and Drosha ASOs 25690 and 25691, respectively. However, less reductions were observed in cells depleted of NAT10 or DDX21 proteins, compared to that in control cells (Fig. 4B–D). Upon NAT10 reduction, the IC50s for Malat1 ASO, and Drosha 25690 and 25691 ASOs were ∼1.8, 19, and 25 nM, respectively. Similarly, the IC50s of these ASOs in DDX21-reduced cells were ∼2.1, 25, and 25 nM, respectively.
As a control, the levels of an untargeted NCL mRNA were not affected by the PS-ASOs or by the reduction of these proteins (Supplementary Fig. S6), suggesting the specificity of the PS-ASOs and the effects of reduction of these proteins on antisense activity. Similar effects were observed in HeLa cells, which had more obvious decrease in the activities of PS-ASOs than in 293CT cells. The IC50s of Malat1 ASO, and Drosha ASOs 25690 and 25691 in control HeLa cells were ∼2, 12, and 60 nM, respectively, whereas upon reduction of NAT10, the IC50s of these ASOs were above 20, ∼90, and 120 nM, respectively. A similar effect was also found in DDX21-reduced cells (Supplementary Fig. S7).
We note that the different activities of ASOs in different cells may be due to the different transfection efficiency in different cells as in 293CT cells, reverse transfection was used. Moreover, overexpression of DDX21 or NAT10 in 293CT cells can also modestly increase the activities of PS-ASOs, without substantially affecting the levels of RNase H1 protein (Supplementary Fig. S8). All the results indicate that NAT10 and DDX21 proteins can enhance PS-ASO activity in cells.
DDX21 and NAT10 can enhance the cleavage activity of RNase H1 and affect the R-loop levels
To further determine the mechanism of how NAT10 and DDX21 enhance the PS-ASO activities, we investigated whether these proteins stimulate RNase H1 cleavage activities using purified proteins. RNase H1 was purified from E. coli, and NAT10 and DDX21 were generated from mammalian cells and also purchased from Origene since it has low solubility in bacteria. The heteroduplexes were annealed with PS-ASO 558807 and its length matched complementary RNA, and RNase H1 cleavage assay was performed with GST-tagged full-length H1, as described previously [15]. NAT10 or DDX21 proteins enhanced the cleavage activities, especially at early time points of cleavage reactions (Fig. 5A), as clearly seen from the quantification results (Fig. 5B).
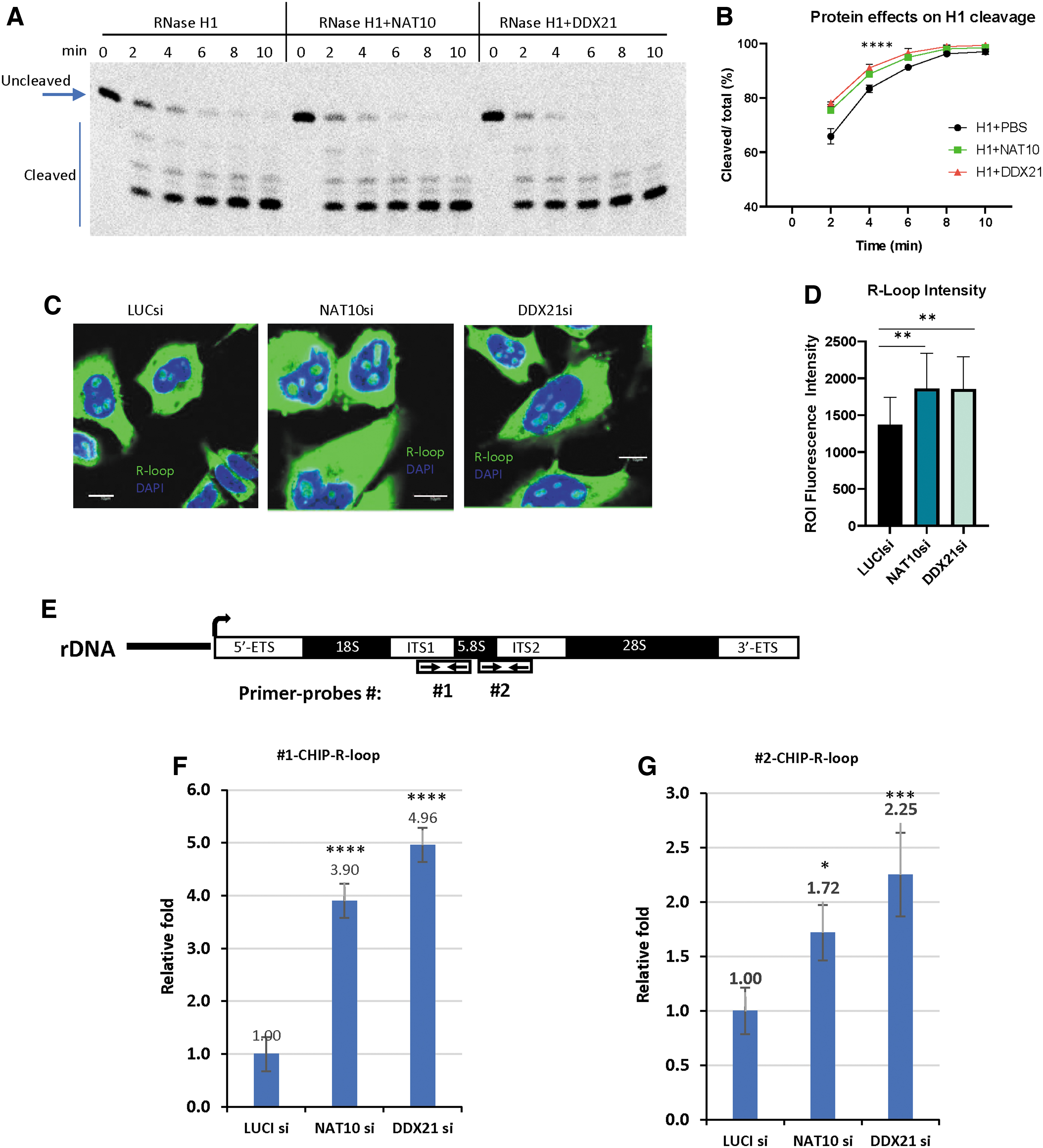
DDX21 and NAT10 can enhance the cleavage activity of RNase H1 and affect the R-loop levels.
These experiments were repeated using an untagged RNase H1 and similar trends were observed (Supplementary Fig. S9). These results indicated that NAT10 and DDX21 proteins can enhance the cleavage activity of RNase H1, which explains why reduction of NAT10 or DDX21 decreased (Fig. 4), whereas overexpression of NAT10 or DDX21 enhanced the PS-ASO activities in cells (Supplementary Fig. S8).
Since DDX21 and NAT10 enhance RNase H1 cleavage activity, and RNase H1 plays important roles in the regulation of R-loop levels and genome stabilities, next, we evaluated whether NAT10 and DDX21 affect R-loop levels. HeLa cells were treated with specific siRNAs for 48 h, and the levels of R-loop were first evaluated by immunofluorescence staining with an R-loop antibody (S9.6). Although this antibody can nonspecifically stain RNA in the cytoplasm, nucleolar staining was readily detected where pre-rRNA transcription occurs, which leads to the formation of R-loops [38,44]. Modestly greater nucleolar signal intensity was observed in cells depleted of DDX21 or NAT10, compared with that in control cells (Fig. 5C). Quantification results of nucleolar signals also showed a modest, but statistically significant increase in the levels of nucleolar staining signal upon DDX21 and NAT10 depletion (Fig. 5D).
Since the S9.6 antibody has been shown to nonspecifically stain other RNAs in immunofluorescence staining assay [44], to further confirm the R-loop accumulation in the NAT10- or DDX21-depleted cells, CHIP was performed to analyze the effect of reduction of NAT10 or DDX21 on R-loop levels in HeLa cells after a 48-hr siRNA treatment. The R-loop antibody was used to enrich R-loops from fragmented genome DNA, and quantitative PCR was employed to detect the R-loop levels using the probes targeting the boundary between ITS1 and 5.8S rRNA (#1), or between 5.8S rRNA and ITS2 of the rRNA gene locus (#2), which has been shown to form R-loops [22].
The results indicate that the R-loop levels detected with the two different primer sets were significantly higher in NAT10 or DDX21 depletion cells than that in control cells (Fig. 5E–G). Together, these results suggest that NAT10 and DDX21 proteins may facilitate RNase H1 function both in R-loop solving in cells and in facilitating PS-ASO-induced RNase H1 cleavage of target RNAs.
Reduction of NAT10 and DDX21 did not reduce the level of RNase H1 protein
To determine if reduction of DDX21 and NAT10 affects the expression of RNase H1, leading to reduced PS-ASO activity, HeLa cells were treated with corresponding siRNAs for 48 h, followed by transfection with or without toxic PS-ASOs for 8 h. The RNase H1 protein levels did not substantially change upon NAT10 or DDX21 siRNA treatment (Fig. 6A), suggesting that the reduced PS-ASO activity in DDX21- and NAT10-depleted cells was not due to reduced RNase H1 protein levels. Reduction of these two proteins also did not alter the levels of PSF (Fig. 6A), La, and NPM1 (Fig. 6B), proteins known to affect PS-ASO activities [6,43,45].
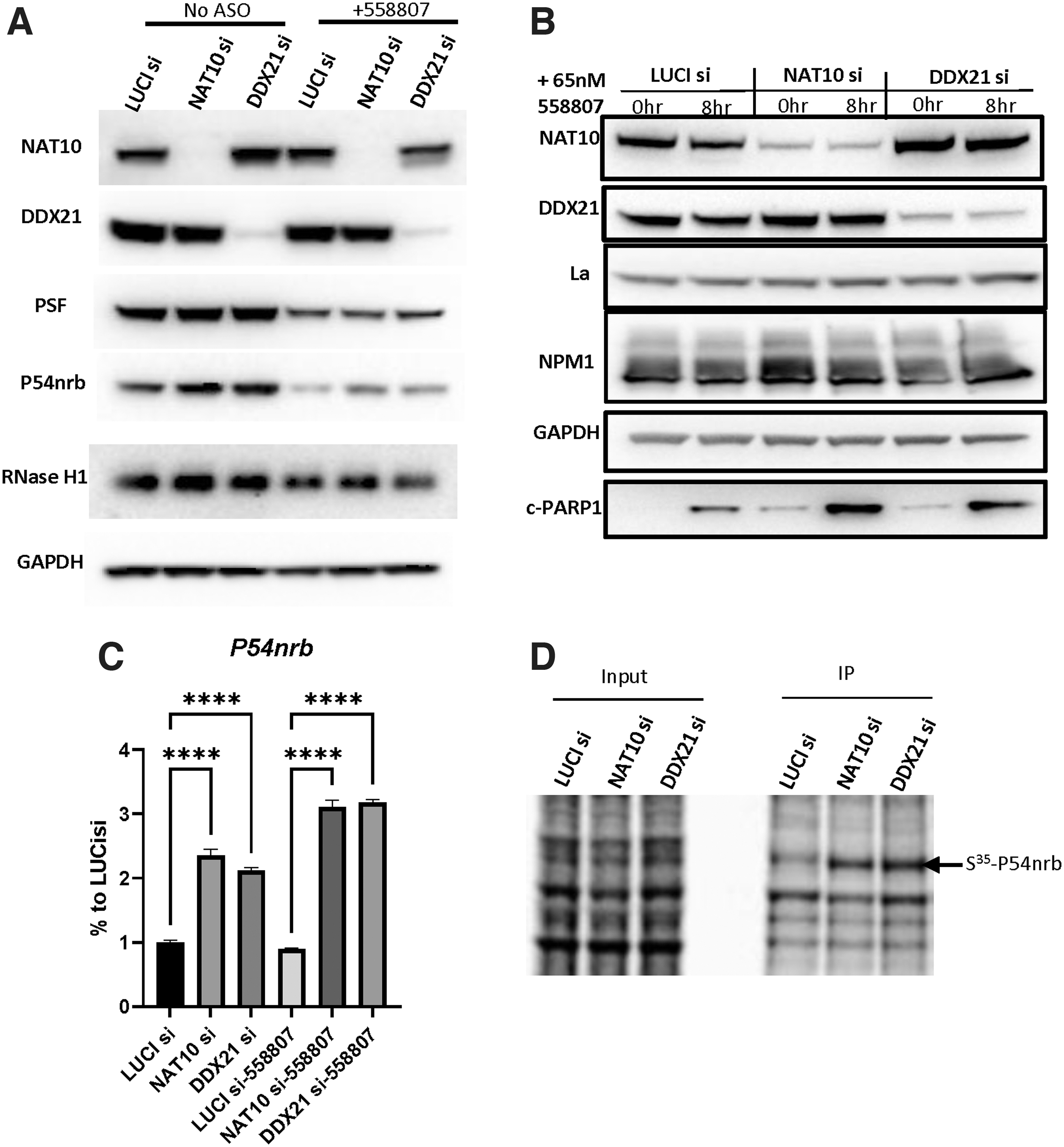
NAT10 and DDX21 reduction did not reduce the protein level of RNase H1.
However, reduction of DDX21 and NAT10 caused modestly increased levels of P54nrb protein (Fig. 6A), a paraspeckle protein that inhibits PS-ASO activity by competition with RNase H1 for binding to PS-ASO/RNA duplexes [43]. The P54nrb mRNA levels were also increased upon depletion of these two proteins, as examined by qRT-PCR (Fig. 6C). In addition, depletion of DDX21 or NAT10 enhanced translation of P54nrb protein, as determined for nascent P54nrb protein levels using pulse-chase labeling with S35-methionine, followed by P54nrb co-immunoprecipitation and autoradiography (Fig. 6D). These results together suggest that P54nrb expression was increased upon reduction of these two proteins, which may contribute to the reduced PS-ASO activity since P54nrb inhibits PS-ASO activity [45]. Consistent with our previous observations, transfection of toxic PS-ASOs caused reduction of P54nrb, PSF, and RNase H1 proteins (Fig. 6A).
Reduction of NAT10 and DDX21 increases the cytotoxicity induced by toxic PS-ASOs
Since reduction of NAT10 and DDX21 caused the increase in levels of cleaved PARP, and greater increase in PARP levels in the presence of toxic PS-ASOs compared with that in control cells (Fig. 6B), it is possible that the reduction of DDX21 and NAT10 may exacerbate the toxicity of toxic PS-ASOs. To evaluate this possibility, the effects of reduction of NAT10 or DDX21 on PS-ASO and P54nrb mislocalization were determined, since toxic PS-ASOs tend to localize to the nucleolus and trigger P54nrb nucleolar mislocalization [7,46]. At 48 h after siRNA treatment, we transfected cells with 300 nM Cy3-labeled safe PS-ASOs (446654) containing 2′-MOE modification, and imaged the live cells at different times from 3 h to 24 h.
Safe PS-MOE-ASOs usually accumulate in the nucleoplasm, but not nucleolus, and form nuclear PS bodies containing TCP1β protein [47], yet toxic PS-ASOs tend to also localize in the nucleolus and trigger P54nrb nucleolar mislocalization [20]. Interestingly, we found the safe PS-MOE-ASOs mislocalized into nucleolar structures upon depletion of NAT10 and DDX21 5 h after transfection, although at 3 h, the safe PS-ASO showed normal localization pattern similar to that in control cells (Fig. 7A). In contrast, a toxic PS-ASO is already localized to the nucleolus at this time (Fig. 7B). Similar observation was also made when the safe PS-ASO was transfected at low concentration (20 nM) (Supplementary Fig. S10). Together, these results indicated that reduction of NAT10 and DDX21 sensitized the toxicities of PS-ASOs.
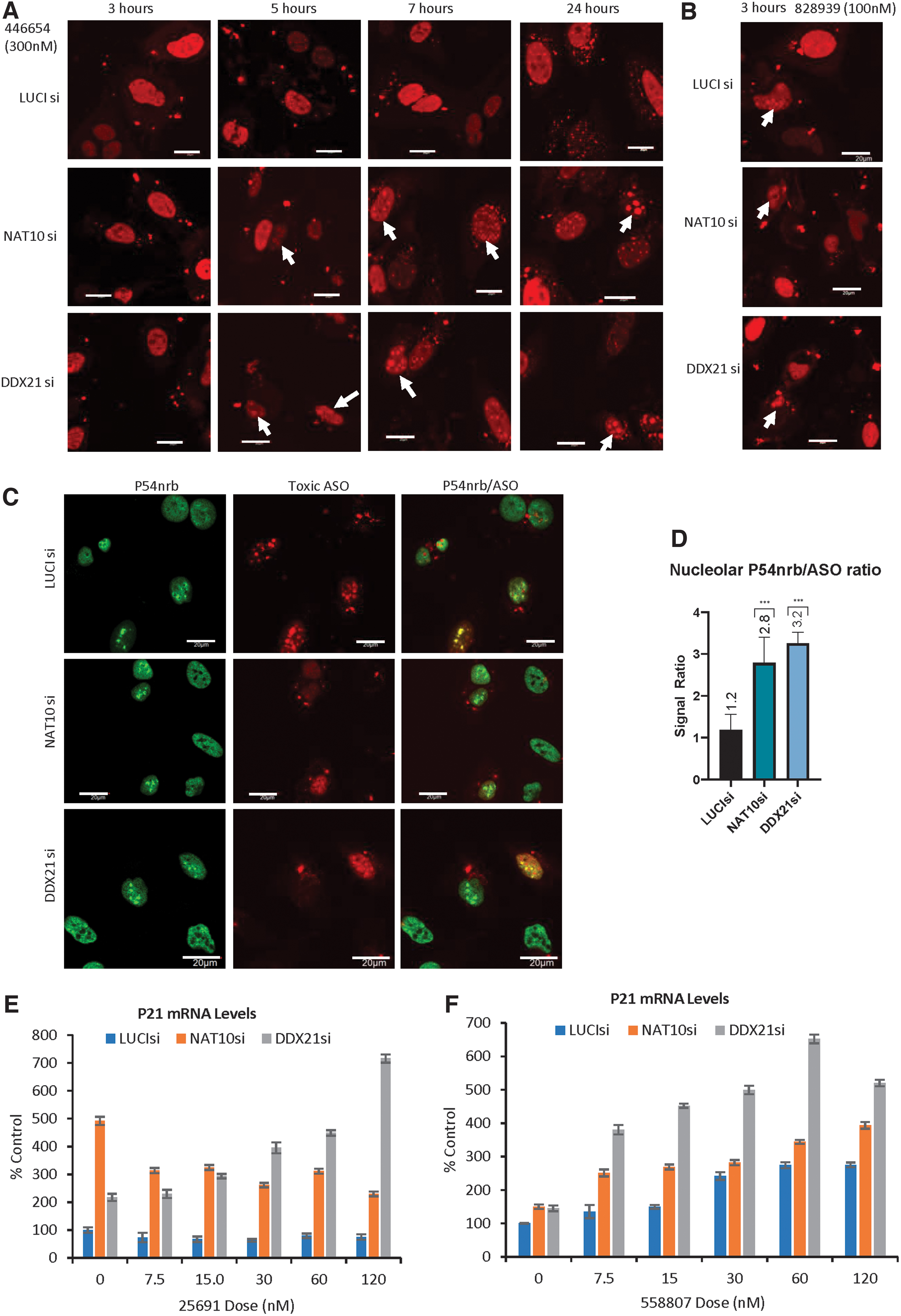
Reduction of NAT10 and DDX21 increases cytotoxicities induced by toxic PS-ASOs.
To further confirm the increased toxicity caused by depletion of NAT10 and DDX21, we transfected toxic PS-ASOs (558807) and co-stained the cells with a paraspeckle protein P54nrb after fixing the cells. P54nrb normally localizes to the nuclear paraspeckles with or without safe PS-ASOs [7,45]. Consistent with our previous findings [7], upon toxic ASO transfection, P54nrb protein mislocalized into the nucleoli in control and DDX21- or NAT10-depleted cells (Fig. 7C). Quantification results indicate that the ratio between nucleolar P54nrb and PS-ASO signal significantly increased in NAT10 or DDX21 siRNA treatment cells (P < 0.001) (Fig. 7D). The results together suggest that reduction of NAT10 and DDX21 further increased the toxicity of toxic PS-ASOs.
The enhanced toxicities caused by NAT10 or DDX21 depletion were also determined using qRT-PCR quantification for the levels of P21 mRNA, a toxic marker for apoptosis. Our results showed that depletion of NAT10 significantly increased the P21 mRNA level in cells even without PS-ASO transfection, and the elevated P21 mRNA level remained similar upon a nontoxic ASO treatment at different doses compared with that in control cells transfected with the same concentrations of nontoxic PS-ASOs (Fig. 7E). Increased P21 mRNA levels were also observed in DDX21-depleted cells compared with that in control cells, although the P21 level increased in a dose-dependent manner upon the nontoxic ASO treatment. Similarly, transfection of a toxic PS-ASO caused dose-dependent increase in P21 mRNA levels, and reduction of NAT10, especially reduction of DDX21, further increased P21 mRNA levels (Fig. 7F).
To further determine if reduction of DDX21 and NAT10 enhances toxicity of toxic ASOs, caspase activity was evaluated in murine Hepa1–6 cells, which showed robust caspase activation upon toxic PS-ASO treatment [7]. The results indicate that again, reduction of NAT10 and DDX21 further increased caspase activities in Hepa1–6 cells transfected with either a nontoxic or a toxic PS-ASO (Supplementary Fig. S11A, B). The knockdown level was confirmed by qRT-PCR (Supplementary Fig. S11C), and the enhanced toxicity upon depletion of NAT10 and DDX21 was also indicated by the elevated P21 mRNA levels (Supplementary Fig. S11D), consistent with the above observations (Fig. 7E, F). Together, these data indicate that reduction of NAT10 or DDX21 protein exacerbates toxicity of PS-ASOs, and suggest that these two proteins can affect the safety of PS-ASOs.
Toxic PS-ASOs can alter nuclear membrane structure similarly to reduction of NAT10
It has been shown that under certain disease or stress conditions, the nuclear envelop structure can be disrupted [48,49]. We thus evaluated if toxic PS-ASOs affect the architecture of nuclear envelop. A GFP-tagged Lamin A, a nuclear membrane-associated protein, was expressed in HeLa cells. The GFP-Lamin A localized normally to the nuclear membrane, and the nuclear membrane appears round and smooth in cells without PS-ASO or in cells transfected with a nontoxic PS-ASO (Fig. 8A). However, upon toxic PS-ASO transfection, the nuclear envelope appears disrupted with wrinkled morphology (Fig. 8A, right panel).
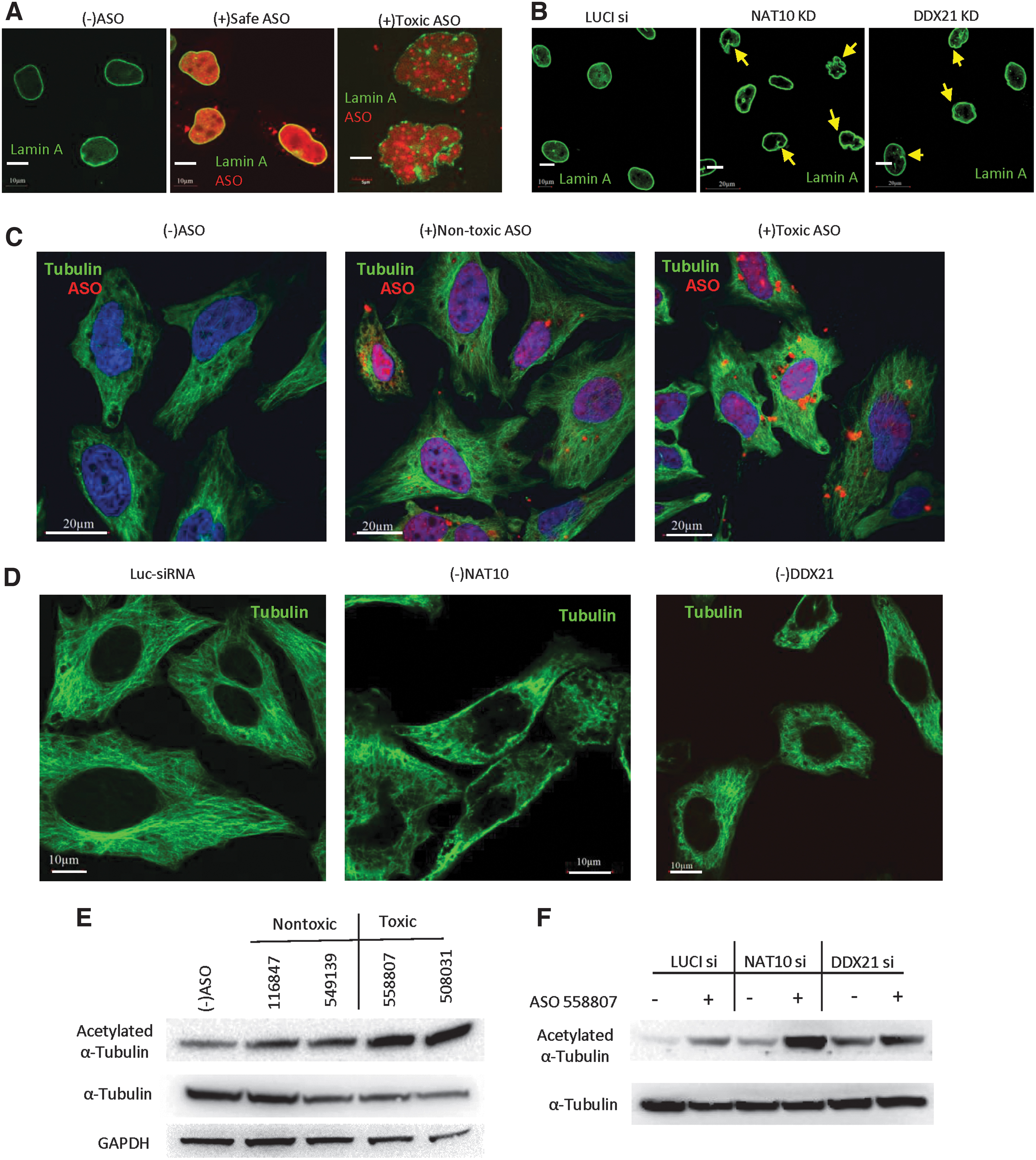
Toxic PS-ASOs or reduction of NAT10 and DDX21 can alter nuclear membrane structure.
Interestingly, deletion of NAT10 also caused a similarly wrinkled nuclear envelope (Fig. 8B, middle panel). Altered nuclear envelope was also observed in cells depleted of DDX21, although to a lesser extent (Fig. 8B, right panel). These data indicate the depletion of NAT10 and DDX21 can cause toxic effects on nuclear membrane structure similar to the outcome of toxic PS-ASO treatment.
Since the nuclear envelop can be affected by the microtubule network [50], the altered morphology of the nuclear envelope observed above may suggest altered microtubule systems. Indeed, in cells transfected with a toxic PS-ASO, and not a nontoxic PS-ASO, the microtubule network marked with alpha-tubulin appeared to be substantially distorted, compared with that in control cells (Fig. 8C). Interesting, reduction of NAT10 and DDX21 also caused a distorted microtubule network (Fig. 8D). These results are consistent with the altered nuclear envelop structures under these conditions.
It has been known that alpha-tubulin is acetylated by a TAT1 and is also a main cytoskeletal protein that supports the cell structure and maintains normal function of cells [51]. We thus evaluated if PS-ASOs can affect the acetylation of alpha-tubulin. HeLa cells were transfected with different toxic PS-ASOs and safe PS-ASOs, and the elevated levels of acetylated alpha-tubulin were indeed found for toxic PS-ASOs compared with control cells or safe PS-ASO-treated cells (Fig. 8E). The reduction of NAT10 and DDX21 slightly increased the acetylated tubulin compared to the control cells in the absence of PS-ASOs. However, the acetylation of alpha-tubulin was substantially increased in NAT10- or DDX21-depleted cells upon toxic PS-ASO transfection (Fig. 8F). These results further indicated that the reduction of NAT10 and DDX21 increased the toxicity and that toxic PS-ASOs can cause more acetylation of alpha-tubulin.
NAT10 and DDX21 are involved in pre-rRNA processing, and toxic PS-ASOs can alter the nucleolar localization of these proteins
Since NAT10 and DDX21 normally localize to the nucleolus where pre-rRNA transcription and processing occur, these two proteins may affect pre-rRNA synthesis. Indeed, reduction of DDX21 or NAT10 caused accumulation of pre-rRNA processing intermediates, as determined by northern analysis (Fig. 9A) and qRT-PCR quantification of pre-rRNA levels (Fig. 9B). These observations are consistent with previous observations that RNA helicase DDX21 contacts with rRNA and snoRNAs and promotes rRNA transcription, processing, and modification [32], and that NAT10 is an 18S rRNA cytosine acetyltransferase and is essential for pre-rRNA processing during small subunit biogenesis [52]. Transfection of toxic PS-ASO substantially reduced the levels of pre-rRNA species in both control cells and cells depleted of these two proteins (Fig. 9A), in agreement with our previous observations that toxic PS-ASO, and not nontoxic PS-ASO, can affect pre-rRNA transcription [7,22].
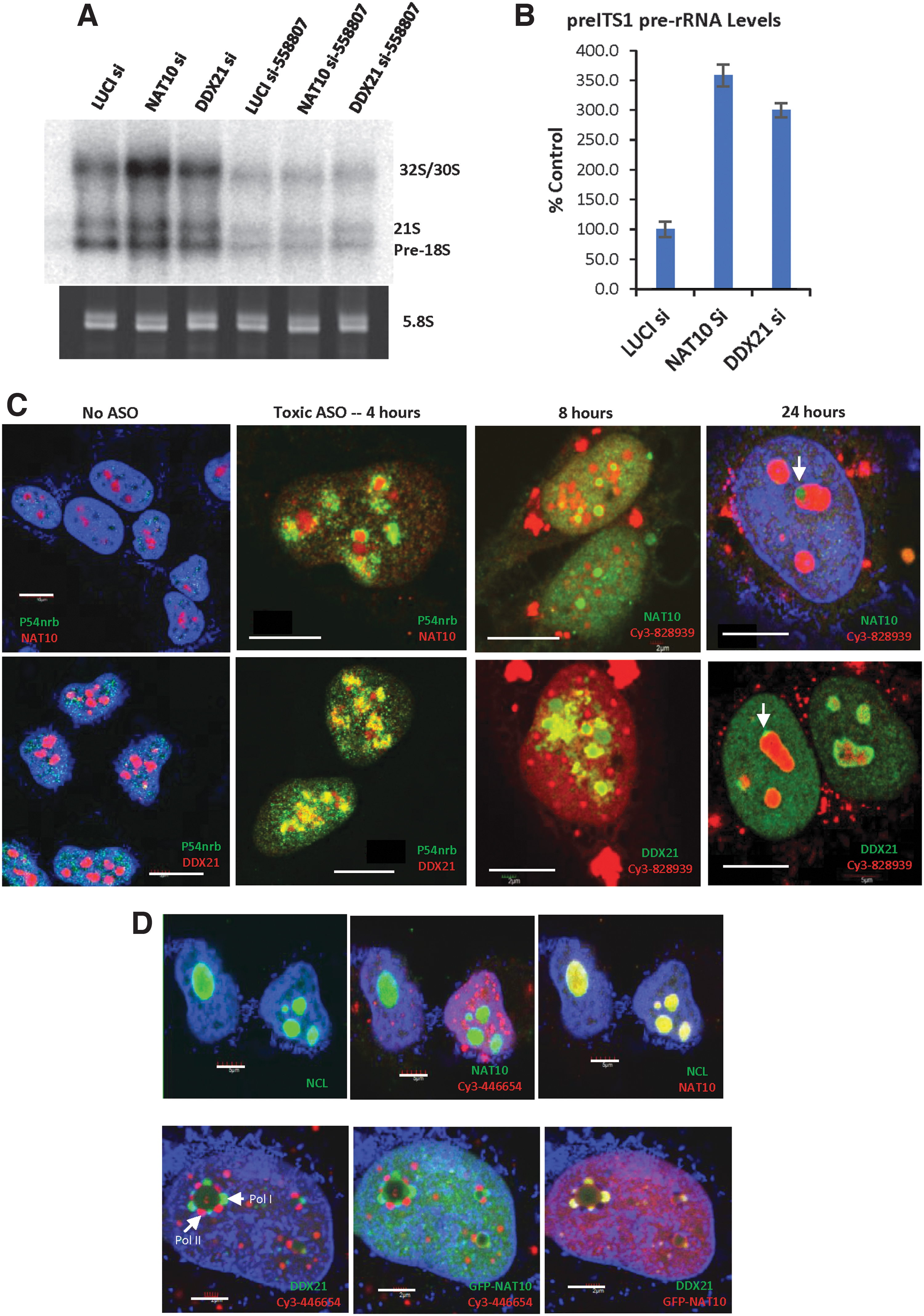
NAT10 and DDX21 are involved in pre-rRNA processing, and toxic PS-ASOs can alter the localization of these proteins.
Since toxic PS-ASOs can cause mislocalization of P54nrb to the nucleolus and nucleolar protein NCL to the nucleoplasm [7,53], we examined how toxic PS-ASOs affect the localization of DDX21 and NAT10 proteins. Immunofluorescence staining results showed that, although transfection of a safe PS-ASO did not affect the localization of these proteins (Supplementary Fig. S12C), toxic PS-ASOs affected the protein distribution (Fig. 9C). Without PS-ASO transfection, NAT10 and DDX21 localized to nucleoli, without colocalization with the paraspeckle protein P54nrb (Fig. 9C and Supplementary Fig. S12A, B). At 4 h after toxic PS-ASO transfection, P54nrb already moved into nucleolar structure and a fraction of NAT10 or DDX21 proteins moved out to nucleoplasm. P54nrb showed partial overlap with NAT10 and DDX21. By 8 h, NAT10 formed smaller foci, surrounded by PS-ASOs, and most proteins were dispersed into the nucleoplasm. DDX21 also appeared dispersed around nucleolar areas.
Twenty-four hours later, the toxic PS-ASOs accumulated in the nucleoli and formed solid-like structures as we recently reported [53], but NAT10 or DDX21 still existed around the PS-ASO containing nucleolus-like structure as small concentrated foci, which are most likely the active polymerase I transcription sites, as we demonstrated recently [53]. The partial colocalization of PS-ASOs with NAT10 and DDX21 around nucleolar area was observed in cells at the early stage of toxic PS-ASO treatment, suggesting dynamic and opposite movements of the PS-ASOs and these nucleolar proteins. These results indicate that the toxic PS-ASOs can alter the localization of NAT10 and DDX21, which might affect their functions and contribute to cytotoxicities.
When transcription was inhibited in HeLa cells using actinomycin D for 4 h, NAT10 and DDX21 were found to colocalize at perinucleolar cap structures (Fig. 9D). Previously, we have shown that upon transcription inhibition, PS-ASOs can also relocate to Pol II perinucleolar caps where they colocalize with P54nrb [45]. The NAT10 and DDX21 containing perinucleolar caps do not contain PS-ASOs, suggesting that these two proteins relocate to Pol I perinucleolar caps, similarly to the case of fibrillarin [45]. Together, these results suggest that toxic PS-ASOs can alter the localization of NAT10 and DDX21 proteins, in a way different from transcription inhibition, which may alter their functions such as in pre-rRNA processing.
Discussion
In this study, we identified two novel RNase H1 binding proteins, NAT10 and DDX21, using BioID proximity labeling. The interactions between NAT10 and RNase H1, and DDX21 and RNase H1 were confirmed by co-immunoprecipitation from cell lysate and by using purified recombinant proteins. These two RNase H1 interacting proteins affect RNase H1 function, since depletion of NAT10 or DDX21 reduced, and overexpression of NAT10 or DDX21 enhanced, the ASO activities in cells. NAT10 and DDX21 can bind to PS-ASOs and facilitate the PS-ASO activity by enhancing the cleavage of RNase H1. In addition, reduction of NAT10 and DDX21 increased the R-loop levels and cytotoxicities induced by toxic PS-ASOs, which are mediated by RNase H1 protein [8]. Our results demonstrated that NAT10 and DDX21 can regulate the activity of RNase H1 and PS-ASO performance.
The interactions of NAT10 and DDX21 with RNase H1 identified in the BioID system were confirmed by coIP with different tags not only in mammalian cells but also with purified proteins, which further indicated the direct binding of these proteins with RNase H1. Using different truncations, we found that the C-terminus of NAT10, including acetyltransferase and tRNA binding domains, is involved in the interaction with RNase H1 in pure protein system, while the N-terminus of, including the putative helicase domain, interacts with RNase H1 in mammalian cells. The C-terminus (601-783 aa) of DDX21 can bind RNase H1 consistently in various systems. The C-terminal tail of DDX21 is critical for trimer formation of DDX21 through intermolecular contacts and carries a low complexity region (LCR)-like sequence, which might bind to RNA or DNA promoter regions like the case of the LCR domains in Drd1p and DDX3X proteins [42].
However, both the NTD and CTD of RNase H1 are involved in the binding to NAT10 or DDX21, indicating that the spatial structure of RNase H1 is important for the interactions. It has been demonstrated that the spacer domain of RNase H1 can bind P54nrb and the Hybrid Binding Domain (NTD) of RNase H1 binds to RPA70 and P32 [18,19]. The direct interaction of NAT10 and DDX21 with RNase H1 can be enhanced by the PS-ASOs. This is different from the toxic PS-ASO-induced interactions between P54nrb and RNase H1 [15,46], and PS-ASO disruptable interactions between RNase H1 and P32 [15]. Since different binding partners can associate with the same domains of RNase H1, it is possible that H1 interactions with other proteins may be dynamic and spatiotemporally regulated.
Importantly, NAT10 and DDX21 can enhance the activities of different PS-ASOs in different cell types. Reduction of NAT10 and DDX21 decreased and overexpression of NAT10 and DDX21 increased PS-ASO activities, suggesting that these proteins facilitate RNase H1 function. This is also supported by the observations that reduction of NAT10 and DDX21 did not reduce the levels of RNase H1 or other nuclear proteins such as NPM and La known to enhance PS-ASO activity [43], but both NAT10 and DDX21 proteins can increase the cleavage rates of RNase H1. In addition, NAT10 and DDX21 also facilitate R-loop solving in cells, a known function of RNase H1, and depletion of these proteins increased R-loop levels.
Currently, it is unclear how these proteins enhance RNase H1 activity. It is possible that these proteins bind to RNase H1 and shape the conformation of RNase H1 for better catalytic activity. It is also possible that these proteins may facilitate ASO and RNA interaction or dissociation after cleavage, and/or facilitate H1 interaction with ASO/RNA duplex, since both proteins have potential helicase domains [32,42,48]. Moreover, since P54nrb protein level increased upon reduction of these proteins, and P54nrb can inhibit PS-ASO activity [45], this may also partially contribute to the reduced PS-ASO activity observed in cells upon depletion of DDX21 and NAT10.
On the other hand, the reduction of NAT10 and DDX21 also increased toxicities of toxic PS-ASOs, as shown by increased PARP cleavage, elevated levels of P21 mRNA, and caspase activity. Reduction of these proteins induced weak, but detectable PARP cleavage, and caused nucleolar localization of a nontoxic PS-ASO that normally does not localize to the nucleoli. These observations suggest that reduction of these proteins can cause detrimental effects that can sensitize cells to PS-ASO stimuli. Currently, it is unclear how these proteins affect ASO toxicity. Since RNase H1 has been shown to be required for the cytotoxicity of toxic PS-ASOs [8,54], it is possible that these proteins may affect the cytotoxicity of PS-ASOs through RNase H1. However, it is also possible that loss of these proteins impaired certain biological processes. Understanding the detailed mechanisms requires further investigation.
The regulation of R-loops is critical for normal cell growth and genome stability. R-loops are involved in gene expression regulation mediated by transcription. The defects in RNase H, RNA-DNA helicases, or some RNA binding proteins can cause R-loop stabilization [55]. As a helicase, DDX21 has been reported to unwind R-loops, and acetylation of DDX21 by CBP inhibited its unwinding activity, but deacetylation of DDX21 by SIRT7 increased the R-loop-unwinding activity [34]. In our study, knockdown of DDX21 increased R-loop levels and impaired rRNA processing. Similarly, reduction of NAT10 also increased R-loop levels and impaired pre-rRNA processing. The involvement of NAT10 in pre-rRNA processing has been reported [24,56]. It is involved in rRNA acetylation [24,52]. It is possible that reduction of these proteins may affect R-loop unwinding or pre-rRNA modification, or reduce RNase H1 function in R-loop solving, leading to impaired pre-rRNA processing. Unveiling the detailed mechanisms of the involvement of these proteins in R-loop and pre-rRNA processing requires further investigation.
Together, we showed that NAT10 and DDX21 can interact with RNase H1 protein and facilitate the function of RNase H1, enhancing the antisense activity of PS-ASOs. These proteins may also contribute to the cytotoxicity of toxic PS-ASOs. As an essential enzyme that can cleave RNA/DNA duplexes, which constantly appear in actively transcribing cells, the function of RNase H1 must be tightly regulated to ensure normal expression of genes or even cell fate. Our results provide examples regarding how the activity of RNase H1 can be regulated by its interacting proteins in cells. In addition, better understanding protein factors that affect PS-ASO performance and the underlying mechanisms will facilitate the design of better drugs. Thus, further unveiling such regulatory proteins of RNase H1 will be important for both biology and for antisense field.
Footnotes
Acknowledgment
We wish to thank all members of Crooke group and Punit Seth for stimulating discussions, and Cheryl Li De Hoyos for technical assistance in Caspase assay.
Funding Information
Internal funding was from Ionis Pharmaceuticals. Funding for open access charge: Ionis Pharmaceuticals internal funding.
Author Disclosure Statement
All authors are employees of Ionis Pharmaceuticals.
Data Availability
No data related to this study was deposited to databases. Research data are available upon request.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.