
Abstract
As oligonucleotides (ONs) and similar nucleic acid therapeutic modalities enter development pipelines, there is continual need to develop bioanalytical methodologies addressing unique challenges they pose. Novel ONs back bone chemistries, especially those enabling stereochemical control, and base modifications are being exploited to improve pharmacological properties, potency, and increase half-lives. These changes have strained established methods, oftentimes precluding development of assays sensitive and specific enough to meet the needs of preclinical programs. For stereopure ONs representing a single molecular species, nontrivial presence of chain-shortened metabolites in biological samples necessitate assays with high specificity. To meet these needs, this report presents a toolbox of novel techniques, easy to implement for existing hybridization-ligation enzyme-linked immunosorbent assay formats, which address this challenge and yield significant sensitivity and specificity enhancements. Ligation efficiency was improved up to 61-fold through addition of polyethylene glycol, betaine, or dimethylsulfoxide, mitigating major differences among sequence-matched ONs of varying stereopurity, enabling sensitivities below 0.100 ng/mL for quantitation. These improvements enabled further refinement of capture probe designs engendering sufficient specificity to discriminate N-1 chain-shortened metabolites at both the 5′ and 3′ end of the ONs. These generalizable methods advance the performance of mainstay bioanalytical assays, facilitating research and development of innovative ONs therapeutics.
Introduction
Oligonucleotide (ONs) therapeutics represent a burgeoning drug modality, where most commercial examples were approved recently over the past 5 years [1]. These molecules represent synthetic, nucleic acid polymers with diverse nucleotide and backbone chemistries. At the time of writing, there are over 50 active clinical trials investigating ONs treatments for diseases, including cancer, cardiomyopathies, viral infections, and rare diseases [2]. When also considering those in various stages of preclinical development, the scope broadens considerably, including many companies and academic institutions. While there are still ongoing challenges in effective delivery and widespread application of these potential medicines, it is clear that there are considerable untapped potential and broad research and development activities accelerating this field [3–5].
Introduction of modified backbone chemistries, for example, phosphorothioate (“PS”) linkages, engenders exponential new chemical space, through stereochemical control at each linkage. This has become an important offshoot for the modality, increasingly appreciated as a critical factor in development of potent antisense oligonucleotides (ASOs) with increased metabolic stability and uptake [6–8].
The bioanalytical assays supporting the development of these ONs have evolved considerably over the past decade from the initial hybridization enzyme-linked immunosorbent assay (ELISA) capable of quantifying ONs in the pg/mL range [9–12]. These original reports focused on simple ONs and became the foundation, from which multiple methods have been developed. With recent advancements introducing novel ONs chemistries, conjugates, and manufacturing enabling production of ONs with specific stereoisomers, diverse methods were developed to quantify modified ONs [1,13,14].
More recent methodologies have been reported on platforms, including liquid chromatography-tandem mass spectrometry (LC-MS/MS), hybridization-ligation ELISA (HL-ELISA), and reverse-transcription quantitative polymerase chain reaction (RT-qPCR) to address emerging challenges associated with novel chemistries [15–17]. Oftentimes, there is a trade-off between sensitivity and specificity which cannot be reconciled, due intrinsically to limitations of the method.
Recent advancements have been reported, with liquid chromatography/mass spectrometry (LC/MS)-based methods reaching <1 ng/mL sensitivity and retaining high specificity [15] and RT-qPCR methods achieving <10 fg/mL sensitivity, although without considerable evidence of retaining good specificity [16]. However, it is difficult to ascertain the practical application of these newer methods across the chemically diverse space of ONs molecules, for which hybridization-based assays have been a mainstay for analytical assessments. ELISA-based hybridization methods continue to have broad application across diverse ONs modalities, including siRNA, peptide-conjugated morpholinos, and siRNA-monoclonal antibody conjugates, highlighting the flexibility and ease of implementation for these assay formats [18–20].
This report highlights novel advancements to the HL-ELISA-mediated quantification of “ASOs,” addressing both sensitivity and specificity (Supplementary Fig. S1). Toward this end, a toolbox of additives and probe design schemes were evaluated and shown to yield significant benefits for both stereopure and stereorandom ASOs containing diverse chemical backbones and nucleotide modifications.
Materials and Methods
Hybridization-ligation enzyme-linked immunosorbent assay
The assay protocol was performed as described previously [10,21], with the following modifications. Unless otherwise noted, all solutions were equilibrated at room temperature (“RT”) for at least 60 min before use. In addition, all wash steps were performed over 4 cycles (350 μL each) with tris-buffered saline with 0.05% tween (TBS-T), repeated once again after rotation of the plate by 180°. Reagent manufacturer and catalog numbers are provided in Supplementary Table S1.
Day 1
The capture probe working solution was prepared at probe concentrations previously optimized over a range of concentrations (2–200 μM). After 95.0°C heat denaturation of this solution, an equivalent volume of the ASO-containing samples was added, briefly vortexed, and incubated for 90 min at 42.0°C, followed by 4°C for 5 min. Samples were then incubated for 60 min (RT, 22°C ± 2°C) in a NeutrAvidin™-Coated High-Capacity Plate, previously blocked with TBS SuperBlock blocking buffer for a minimum of 1 h at RT.
The detection probe working solution (1 × 96-well plate volume) was prepared by addition of the following reagents to an appropriate volume of detection buffer (total volume of 20 mL): (1) 1.0 mL 200 mM dithiothreitol (DTT) (prepared in detection buffer and stored in single use aliquots at −80°C), (2) 710 μL T4 DNA ligase buffer, (3) 5.0–10.0 mL polyethylene glycol (PEG) solution (varying w/v percentages), (4) 4 μL 10 μM detection probe (for HTT-170 assays, concentration previously determined to optimize for signal:noise and dynamic range), and (5) 5 μL T4 DNA ligase (100 U/mL). Plates were incubated in this solution at 2–8°C overnight, for at least 16 h, on an orbital plate shaker set at 400 RPM.
Day 2
S1 nuclease working solution was prepared by dilution of 2 mL S1 nuclease reaction buffer to 18 mL 100 mM NaCl. Before use, 80.0 μL S1 nuclease (100 U/μL) was diluted into this working solution. After washing, samples were incubated with this solution for 60 min at 37°C. Anti-digoxigenin-AP working solution was prepared by dilution of 8.0 μL anti-digoxigenin-AP into 20 mL SuperBlock TBS blocking buffer.
AttoPhos working solution was prepared according to manufacturer's instructions and allowed to equilibrate at RT for 90 min before use. After 150 μL of this working solution was added to each well, plate was incubated at RT for ∼20 min and read on a Molecular Devices, USA, SpectraMax M5E plate reader (440 nm excitation, 555 nm emission, 550 nm cutoff). After initial read, plate was quenched with 75.0 μL 20% (w/v) ethylenediaminetetraacetic acid (EDTA) solution and a final read was performed.
HL-ELISA solutions preparation
For all solutions, Milli-Q purified water was used for preparation unless noted otherwise. Tris-EDTA-bovine serum albumin (BSA) (0.5% w/v BSA, “TE-BSA”) buffer solution was prepared by dissolving 2.5 g BSA powder completely in a 500 mL bottle of tris-EDTA, pH 7.6, and kept at 2–8°C. Capture probe buffer (60 mM Na2HPO4, 0.9 M NaCl, 5.0 mM EDTA, 0.24% Tween-20, pH 7.6) was prepared by dissolving 8.52 g Na2HPO4, 180 mL 5 M NaCl, and 10 mL 0.5 M EDTA in 500 mL water and adjusting pH to 7.6 with HCl or NaOH. The solution was 0.2 μm filtered, 2.4 mL Tween-20 was added, and the volume was adjusted to 1.0 L with water.
Aliquots of 12 mL were stored at 2–8°C. “PEG” solutions of varying weight percentages (w/v) were prepared by dissolving the appropriate mass of PEG into a minimal volume of detection buffer. Once fully dissolved, the volume was adjusted (eg, for 40% w/v PEG-6000, 100 g PEG-6000 was completely dissolved in ∼100 mL detection buffer and thereafter adjusted to a final volume of 250 mL). Capture probe and detection probe stocks (Integrated DNA Technologies; see Supplementary Table S2) were prepared at a concentration of 200 μM with TE buffer, pH 7.6, whose 10.0 μL aliquots were stored at −20°C.
Intermediate detection probe stocks were prepared fresh in TE buffer and stored at 2–8°C for up to 3 months; intermediate capture probe stocks were stored at −20°C for up to 1 year. Detection buffer (66 mM tris-HCl, 10 mM MgCl2, pH 7.6) was formulated at Boston BioProducts (Cat. No. C-7823W).
DTT solution (200 mM) was prepared by dissolving 1.389 g DTT in 45 mL detection probe buffer, vortexed until complete, whose 1.0 mL aliquots were stored at −80°C for up to 6 months. S1 nuclease reaction buffer (200 mM sodium acetate, 1.5 M NaCl, 10 mM ZnSO4, pH 4.5) was prepared by diluting 16.7 mL 3 M sodium acetate, 75 mL 5 M NaCl, and 1.25 mL 2 M ZnSO4 into 100 mL water. The pH was adjusted to 4.5 with HCl or NaOH, volume adjusted to 250 mL, 0.2 μm filtered, and stored at 2–8°C for up to 6 months.
Standard preparation
ASOs standards were formulated at 1 mg/mL in phosphate-buffered saline, assessed for purity, and used for the preparation of purity-corrected diluted standards in TE-BSA buffer. These intermediate standards from 1.00 × 105 to 1.00 × 102 ng/mL were used to prepare 50 × working standards from 10.0 × 103 to 2.50 ng/mL in TE-BSA buffer, stored at 2–8°C for less than 3 months. On the day of analysis, these working standards were diluted 50-fold into TE-BSA buffer, yielding standard curves ranging from 2.00 × 102 to 5.0 × 10−2 ng/mL, unless otherwise noted and remaining at RT. Quality control (“QC”) samples were prepared analogously at the noted levels. Negative controls, unless otherwise noted, are nonspiked TE-BSA buffer samples used for background subtraction.
Antisense ONs
ASOs used in this study were synthesized by Wave Life Sciences USA, Inc. (Cambridge, MA, USA), previously described with minor modifications [22]. Characterization of identity and purity was assessed by LC/MS and ultra performance liquid chromatography, respectively. Chemistry and purity for all ASOs used in this study are shown in Supplementary Table S3.
Data analysis
Raw plate data were captured with SoftMax Pro 7.0.2 software from a Molecular Devices, USA. This software was used for evaluation of cross-reactivity and interference parameters, as well as other select standard curve titrations, fitting standard curves with a 5-parameter logistic regression model and 1/Y2 weighting. Data were further plotted in GraphPad Prism 8, version 8.1.0. Statistical analyses were performed using 1-way analysis of variance (ANOVA) followed by Tukey's multiple comparisons test, unless otherwise noted for each figure.
Results
Sensitivity
A key challenge in the development of bioanalytical assays to quantitatively measure ASOs is in reaching a balance of sensitivity and specificity. For stereopure ASOs that contain diverse chemical modifications, this endeavor has shown that long-standing hybridization ELISA analytical techniques were insufficient in meeting the needs of a rapidly evolving platform. Changes in stereochemistry, backbone chemistry, and nucleotide modifications can contribute to meaningful changes to the sensitivity of these assays; in some cases, major changes to the methodology are required to meet even the most basic requirements required by most programs [∼1 ng/mL lower limits of quantification (LLOQ)]. To address prohibitively low responses of select molecules, we sought to investigate the impact of chemical additives that were long used to facilitate ligation-based PCR methodologies [23–25].
Early evaluations of PEG-6000 on the performance of ATXN3-766 were found to markedly improve mean responses across a calibration range from 0.0500 to 50.0 ng/mL (Fig. 1A). Further characterization of dimethylsulfoxide (DMSO), betaine, and “PEG” (of varying average molecular weights) revealed that all additives significantly increase the responses of a representative batch of ASOs with defined sequence (Fig. 1B), when introduced during the T4-DNA ligase ligation step.
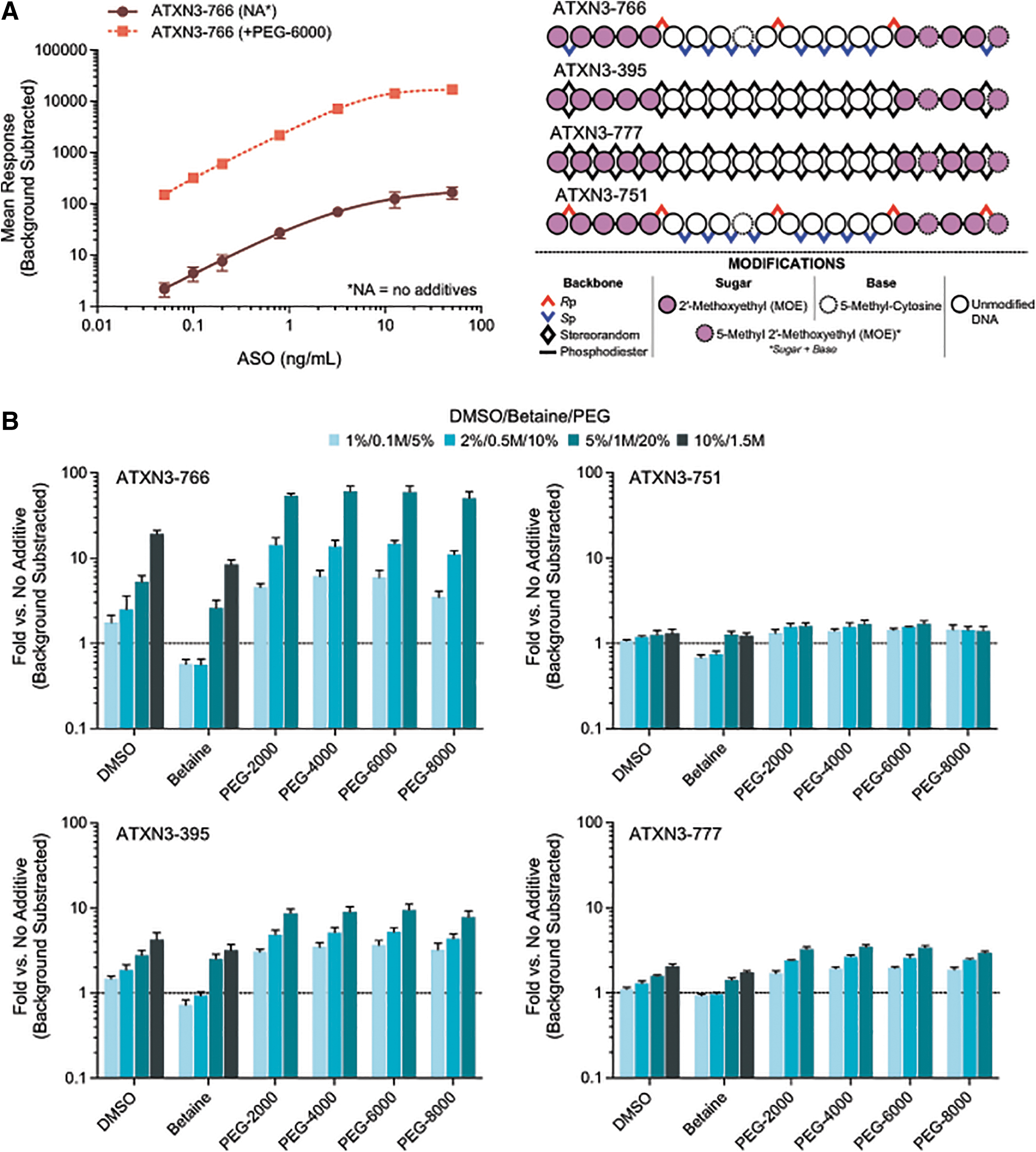
HL-ELISA assay performance enhancement dependent on DMSO, betaine, and various PEGs.
Dose-dependent increases in performance were seen for each additive, with each of the PEGs (of varying average molecule weights) yielding the highest overall enhancement under the conditions evaluated. For ATXN3-766, 50.4–60.9 × higher background subtracted signals were seen when 20% (w/v) PEG was included, with DMSO and betaine yielding maximal increases of 19.2 × and 8.4 × , at 10% (v/v) and 1.5M, respectively. While similar trends were seen for ATXN3-395, ATXN3-777, and ATXN3-751, the overall impact on relative responses were tempered.
The differential impact of these additives stem from the stereochemistry differences among the molecules surveyed. Both ATXN3-395 and ATXN3-777 represent sequence matched diastereomeric mixtures (“stereorandom”), whereas ATXN3-766 and ATXN3-751 are stereopure. The mean responses of these molecules in the absence of any additives are significantly different. The stereochemistry of ATXN3-751 yields the best background subtracted mean response; changing the terminal PS backbone linkages from the Rp configuration to Sp results in an ∼99.9% drop in mean response [from 4,992 to 135.1 relative fluorescence units (“RFU”)] at 12.6 ng/mL (Fig. 2A).
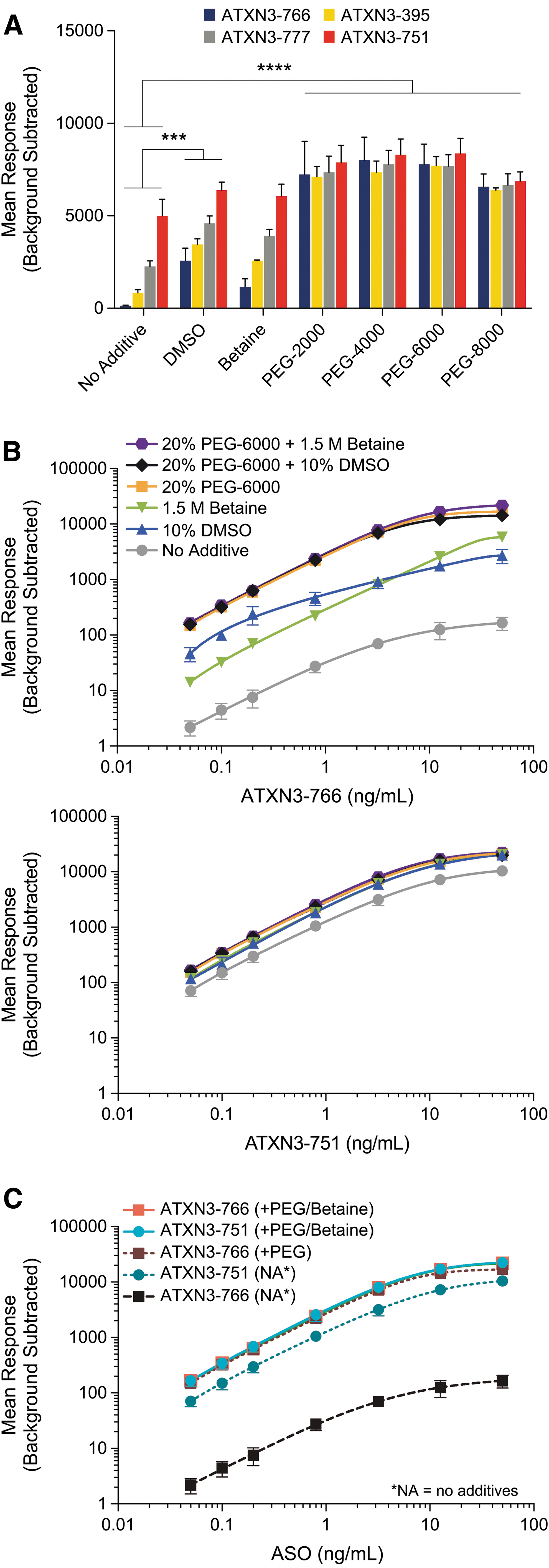
Inclusion of additives reconcile major performance differences between diastereomers, yielding an optimized quantitative assay. All data shown represent the mean background subtracted responses from independently assayed replicates.
Unsurprisingly, the performance of stereorandom molecules falls in between ATXN3-766 and ATXN3-751. For ATXN3-777 and ATXN3-395, the highest affinity members of the diastereomeric population are likely responsible for most of the response.
Both DMSO and betaine partially rescue the responses of all molecules, relative to ATXN3-751 (Fig. 2A). Importantly, each of the PEGs completely rescues the response of ATXN3-766, ATXN3-777, and ATXN3-395 when compared to ATXN3-751. Furthermore, the mean response of ATXN3-751 is increased from 4,992 in the absence of any additive to between 6,875 and 8,377 RFU (37.5%–67.6%), dependent on the specific PEG used (Fig. 2A).
To better understand whether multiple mechanisms are at play across the additives surveyed, and if they are additive, optimal concentrations of each were assessed together over a standard curve from 0.0500 to 50.0 ng/mL ATXN3-766 (Fig. 2B, C). Although DMSO and betaine have differential relative impacts between the lower and upper limits of the range, their combination with 20% (w/v) PEG-6000 yields only a modest improvement near the upper limit of the curve. For ATXN3-751, there is no discernible difference (Fig. 2B). While these data do not rule out distinct molecular mechanisms for each of the additives that may be masked by inclusion of PEG, they further highlight PEG as the key contributor in optimizing HL-ELISA assays for high sensitivity and low selectivity among sequence-matched ASOs.
Nevertheless, evaluation of additives and optimization of their concentrations is recommended for each new ASO; for the ASOs discussed within this report, additives were optimized unless otherwise noted.
Specificity
While hybridization-ligation ELISA methodologies are known for their enhanced specificity compared to their nonligation-based counterparts, specificity for the parent ASO is generally only seen at the ligation site, which is not tolerant to N-X chain-length-shortened metabolites. The general lack of specificity at the 5′ end of the ASO, as shown in Supplementary Fig. S1, was hypothesized to arise from the inability for S1 nuclease to recognize capture probe-ASO duplexes with few unpaired nucleotides [10]. To increase the lability of these complexes toward nuclease activity, we characterized a set of capture probes carrying additional nucleotides at the 3′ end of the capture probe, which would be unpaired even in complex with the full-length parent ASO.
To assess the feasibility of these designs, we first evaluated capture probes of +2, +4, and +6 nucleotides against ATXN3-766 and its 5′ N-1 chain-shortened molecule WV-23449. Indeed, increasing the length of the capture probes results in enhanced specificity, whereby the relative signal of ATXN3-766 versus WV-23449 increases by up to 65% for the 29-mer +4 capture probe (Fig. 3A). Importantly, this effect is abolished when diluting out S1 nuclease, demonstrating dependence on the enzyme and supporting our original hypothesis.
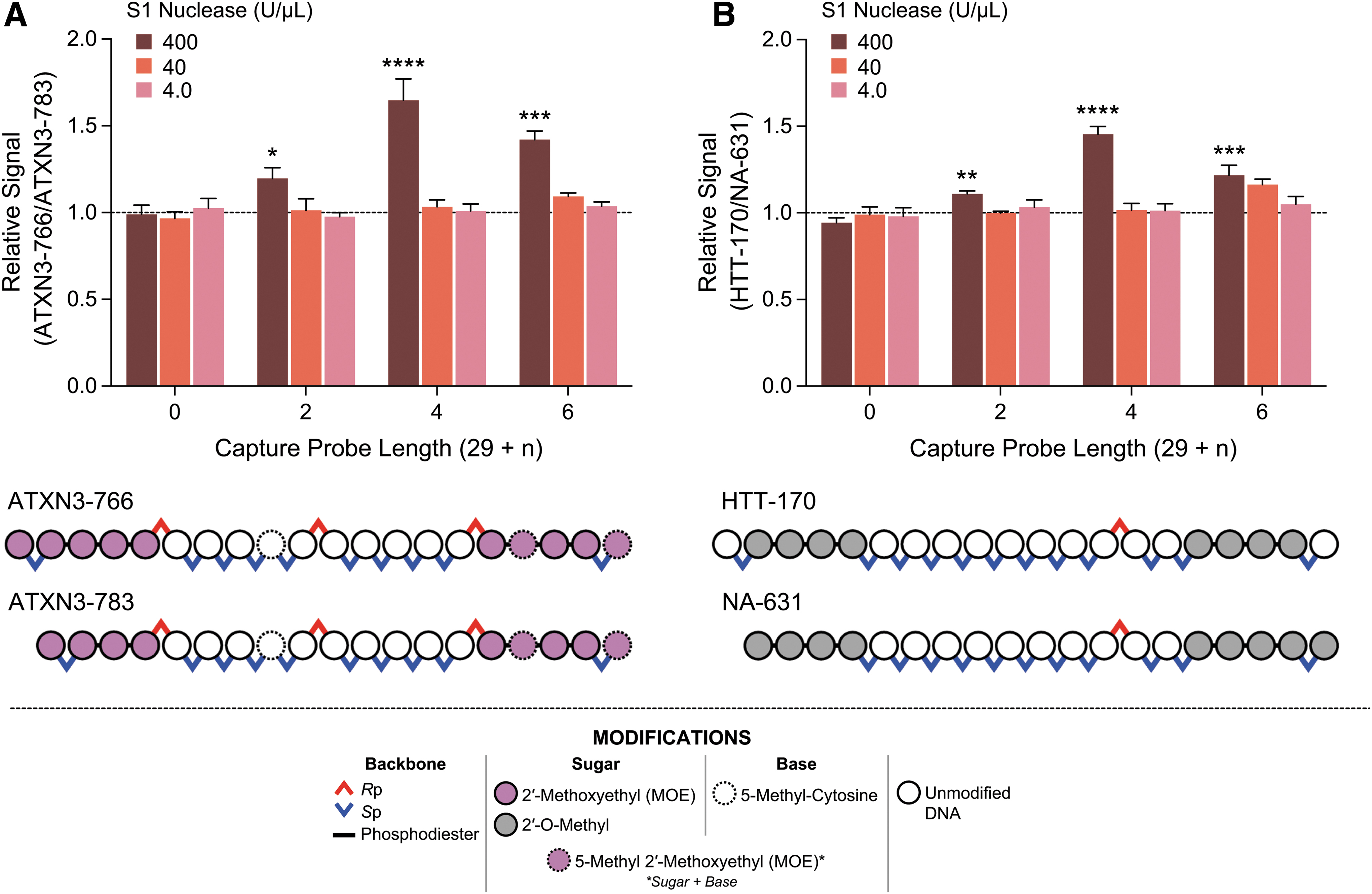
Extension of HL-ELISA capture probes with short nucleotide repeats meaningfully increases the specificity of the assay. Data shown represent the ratio of the mean background subtracted response between the parent 20-mer ASO and its respective 5′ N-1 chain-shortened 19-mer. Statistical analyses' P values shown for the given column versus its respective S1 nuclease condition for a capture probe length of 29 (“0” on the x-axis).
We further explored the generalizability of this design against another stereopure ASO, HTT-170, which differs in sequence, temperature of melting (Tm), and base modifications. Similarly, the 29-mer +4 capture probe improves specificity, whereby the relative signal of HTT-170 versus NA-631 increases by up to 45% (Fig. 3B). Again, the effect is mediated through S1 nuclease.
We further sought to better understand the ability to tune this specificity by way of introducing additional linkers into the capture probe. For the original 29-mer design, a biotinylated tri-ethylene glycol linker is used to capture the complex; in addition to S1 nuclease's ability to recognize unpaired nucleotides, it was thought that access to the 5′ end of the ASO could be sterically hindered when the complex is anchored through biotin-neutravidin interaction on the plate. A series of capture probes were assessed against the original 29-mer capture probe and the top-performing 29-mer +4 (“33-mer”) capture probe from the initial feasibility study; these probes introduce additional tri-ethylene glycol linkers, alone or in combination with extended unpaired nucleotide capture probes (Fig. 4A).
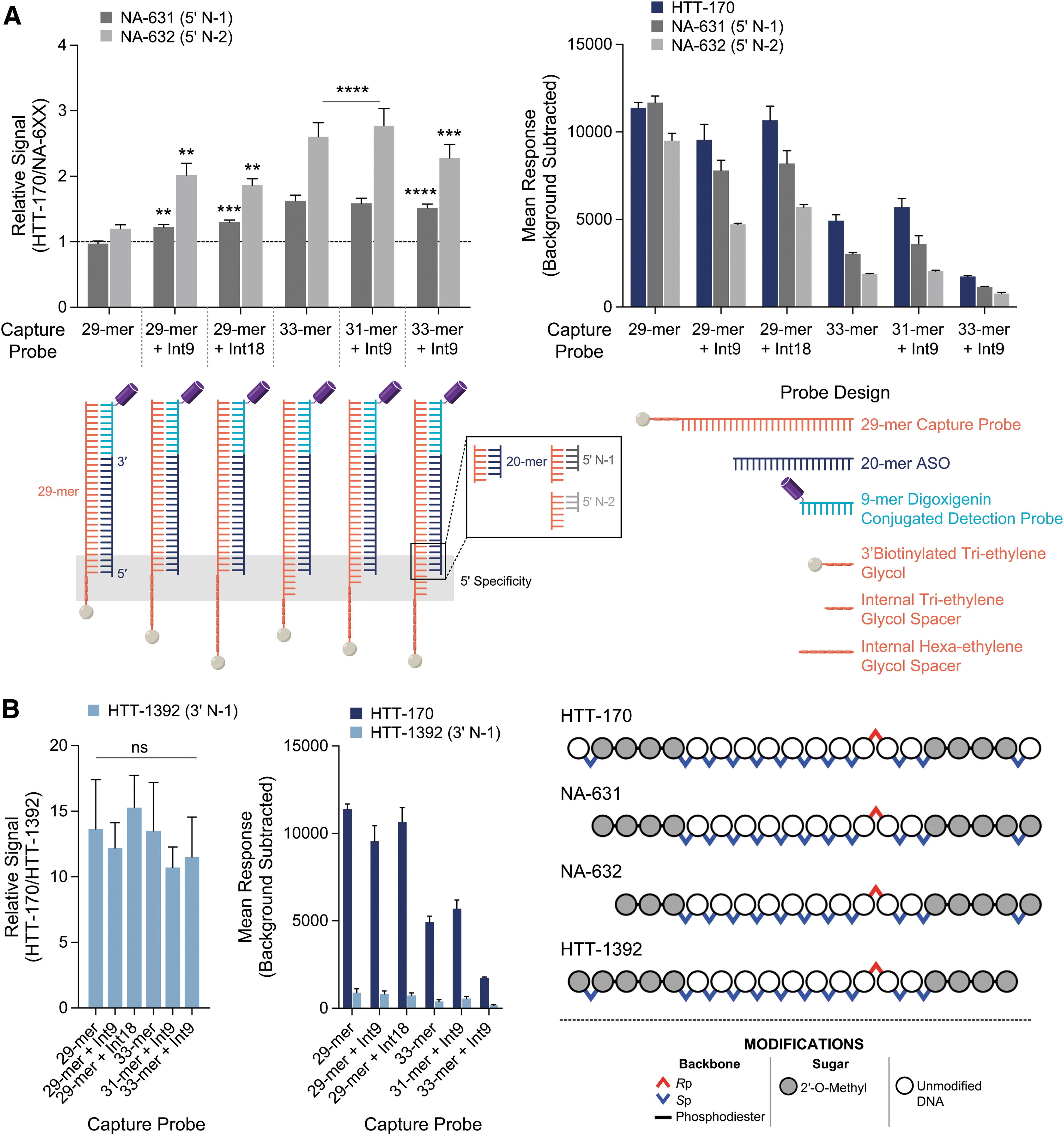
Inclusion of additional 3′ nucleotides and spacers on the capture probe engenders higher specificity with a tenable sensitivity impact. Statistical analyses' P values shown for the given column(s) versus its respective column of capture probe length of 29 (“29-mer” on the x-axis).
These data plot the relative signal of HTT-170, the full-length parent ASO, to either NA-631 or NA-632, a ratio representing the discrimination of the parent ASOs from either chain-shortened metabolite. Introduction of an additional tri-ethylene glycol (“Int9”) or hexaethylene glycol (“Int18”) spacer results in modest, but significant, relative signal of 22% or 30%, respectively, compared to the 29-mer capture probe with a relative signal of −3%, for the 5′ N-1 chain-shortened molecule NA-631. These differences become more profound with the 5′ N-2 chain-shortened molecule NA-632, where the relative signals are 102% and 86% for the tri- and hexaethylene glycol spacers, respectively, and 20% for the 29-mer (Fig. 4A).
Combination of the Int9 spacer with the 29-mer +2 nucleotide-extended capture probe (“31-mer + Int9”) was found to perform similarly to the 33-mer, yielding relative signals of 59% and 177% for NA-631 and NA-632, respectively. No further specificity enhancement was seen with combination of the Int9 spacer with the 33-mer capture probe, where relative signals for NA-631 and NA-632 were 52% and 128%, respectively. Ultimately, implementation of these capture probes does come with a hit to sensitivity, whereby the background subtracted responses decreases with enhanced specificity (Fig. 4A, right).
Evaluation of the 3′ N-1 chain-shortened molecule HTT-1392 with these capture probes yielded no material enhancement of specificity when compared to the original 29-mer capture probe; all capture probes surveyed yielded over 10-fold relative signal of HTT-170 to HTT-1392 (Fig. 4B). These results were expected, as T4 DNA ligase requires the hybridized 3′ end of the ASO for ligation to the detection probe, yielding a complex stable toward S1 nuclease.
Taken altogether, these data showcase the ability to tune these hybridization-ligation ELISA assays for 5′ specificity, using a combination of spacers and unpaired nucleotide extensions. Specificity can be balanced with sensitivity, which is an important consideration depending on availability of information for expected metabolites.
To demonstrate the ultimate impact of these designs, cross-reactivity and interference of the 5′ N-1 (NA-631) and N-2 (NA-632) chain-shortened molecules was assessed using the 31-mer + Int9 against the 29-mer capture probe. Percent difference from theoretical (%DFT) values were calculated at each of 3 standard concentration levels, interpolated against the respective standard curve using either capture probe. For NA-631, cross-reactivity is decreased significantly from a range of 10.4% to 13.7%, across all levels, to −30.8% to −32.7%. For NA-632, cross-reactivity decreased from a range of −16.9% to −27.3%, across all levels, to −57.8% to −60.6% (Fig. 5A).
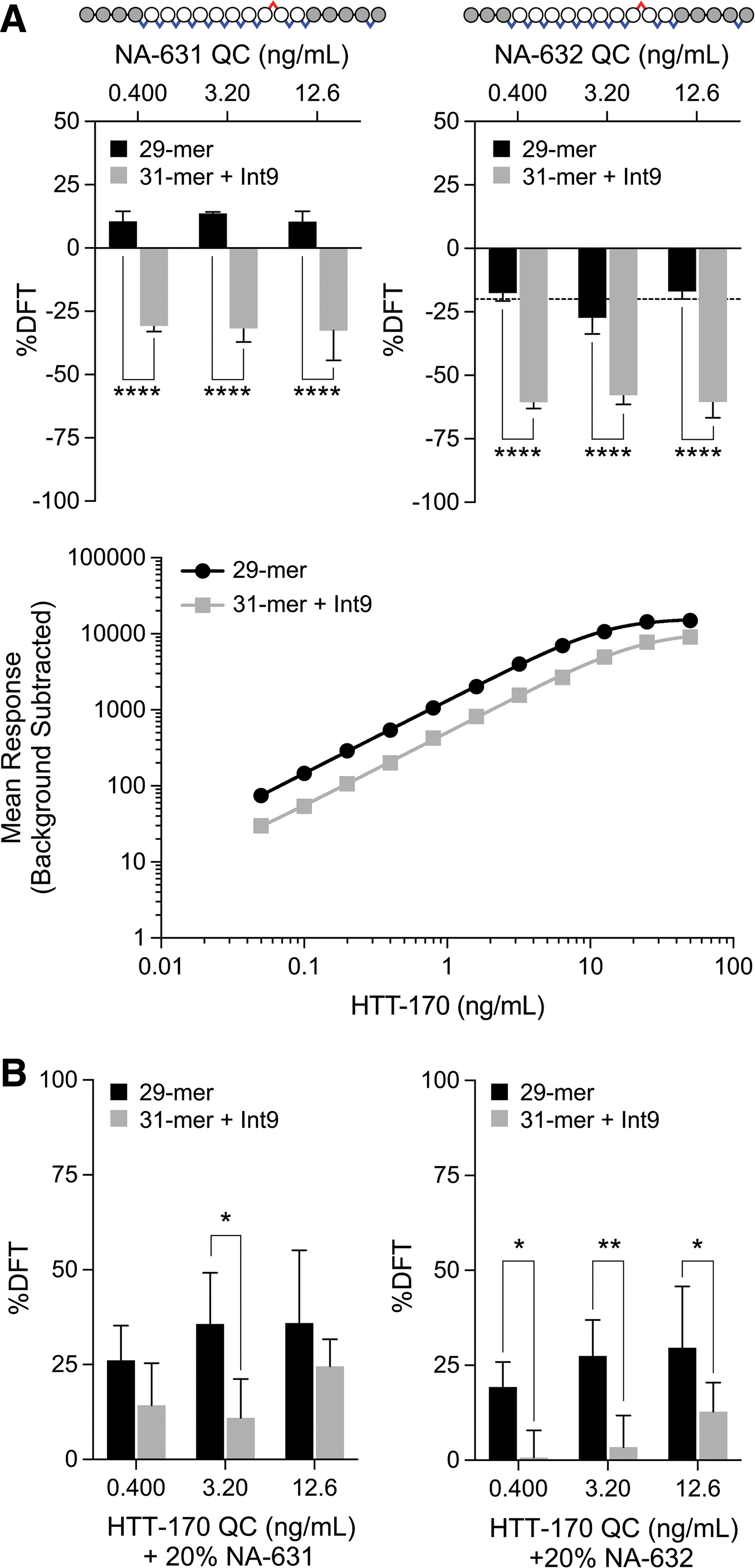
Evaluation of cross-reactivity and interference of 5′ chain-shortened ASOs further highlight meaningful improvements engendered by extensions to the capture probe.
Interference is one of the key parameters used for specificity evaluation in assay qualification and validation. Assessment of interference here was performed by inclusion of 20% (relative to HTT-170) of the indicated chain-shortened metabolite with the QC level noted. For NA-631, interference is decreased from a range of 26.1% to 35.9%, across all levels, to 11.0% to 24.6%, although significantly only for the mid QC level, which decreases from 35.7% to 11.0% (Fig. 5B). For NA-632, interference is significantly decreased from a range of 19.3% to 29.7%, across all levels, to 0.7% to 12.8%. With a typical target of 20%–25% %DFT or less to demonstrate sufficient specificity in qualification and validation contexts, these data provide strong evidence for the practical benefit provided by these extended capture probe designs.
Discussion
Assessment of the pharmacokinetics for ONs from preclinical to clinical development requires high sensitivity assays capable of quantifying picomolar concentrations, while also retaining high specificity for the analyte. The ability to discern the parent ONs from other chain-shortened metabolites without requiring complex extraction and purification steps, needed for LC-MS/MS analysis [26,27], or compromising specificity for sensitivity, shown with qPCR based assays[16], is an important step forward in HL-ELISA LBA methodology for quantifying ASOs. In this report, we showcase the development of a toolbox of performance-enhancing additives and probe design schemes, universally applied over a diverse range of both stereopure and stereorandom ASOs.
The usage of PEGs at optimized concentrations is an especially important advancement as it normalizes responses of stereochemically diverse ASOs with defined sequence. This is critical, enabling pharmacokinetic (PK) assays capable of quantifying multiple ASOs in a single assay, saving both assay developers and analysts considerable time throughout evolution of the assay from discovery to development.
Furthermore, the significant increase in sensitivity these methods enable is specifically relevant for development of analytical assays for stereopure ASOs. The doses administered may often be lower than their counterparts with diastereomeric mixtures, necessitating methods with higher sensitivity [1]. Especially for intrathecally administered ASOs, evaluation of systemic exposure is expected to depend on the ability of the assay to accurately quantify low analyte levels and still discriminate against chain-shortened metabolites, balancing sensitivity with specificity.
The methods presented here have been successfully transferred and validated at external partners, for multiple ASOs of varying sequence and chemistry, with “LLOQ” of 15 pM to upper limits of quantification (“ULOQ”) of 12,000 pM in human cerebrospinal fluid (data not shown). To date, no other known method has been reported capable of reaching these levels of sensitivity without compromising specificity; recent reported methods using Meso Scale Discovery (“MSD”) electrochemiluminescence are shown to yield similar sensitivities but the specificity for these assays is yet unknown and attempts to utilize the platform with chemically modified ASOs similar to those evaluated here have not yielded the sensitivity reported [28].
A previously reported dual ligation hybridization method that yielded excellent 5′ and 3′ specificity demonstrated a LLOQ of 120 pM (HL-ELISA) and moderately better sensitivity of 75 pM, when coupled to qPCR [17]. A more recent hybridization LC-MS/MS method was able to achieve LLOQs of ∼70–80 pM (0.5 ng/mL), discriminating 3′ N-1 metabolites in rat plasma and brain tissue [15]. Both methods require coupling of multiple platforms and numerous steps to achieve reasonable sensitivity, increasing the complexity of analysis, which may confound assay development of chemically diverse ONs.
Considerable improvements in sensitivity have been realized by RT-qPCR methods as evidenced by a recent method applied to the quantification of siRNA molecules, where sensitivity reaches 1.3 fg/mL in human serum [16]. However, specificity for the parent molecule is only achieved on the 3′ end; no difference was reported between the full-length and 5′ N-1 truncated molecule [16].
In conclusion, the modifications to the commonly used hybridization-ligation ELISA method presented here serve as simple, yet important, solutions in addressing long-standing challenges in the bioanalytical development of assays of increasingly diverse ONs. Through the examples provided in this report, these modifications were instrumental in rescuing the performance of sequence-matched, but stereochemically distinct, ASOs, and thereby yielded a path forward for further development of PK assays that could equally meet the minimal requirements of sensitivity required for practical application toward preclinical animal studies, without prohibitive compromises of specificity.
We anticipate that the successes demonstrated here may have further application toward the ONs field at large, where diverse internal and terminal chemical modifications, increased oligo lengths, and backbone chemistries may otherwise impact assay development.
Footnotes
Acknowledgments
The authors thank Eric Smith for assistance in preparing figures and Amy Donner for review of the article. The authors thank the following colleagues for supplying the ONs used in this study: Frank Favaloro, Richard Looby, Jeff Rossi, Amber Lindsey, Jack Marson, Rowshon Alam, Jason Williams, Chloe David, Timea Kolozsvary, Vincent Aduda, Bridget Newman, and Stephany Standley.
Author Disclosure Statement
All authors were employees of Wave Life Sciences USA, Inc. at the time of this study.
Funding Information
This work was funded by Wave Life Sciences.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.