
Abstract
Human genome wide association studies confirm the association of the rs738409 single nucleotide polymorphism (SNP) in the gene encoding protein patatin like phospholipase domain containing 3 (PNPLA3) with nonalcoholic fatty liver disease (NAFLD); the presence of the resulting mutant PNPLA3 I148M protein is a driver of nonalcoholic steatohepatitis (NASH). While Pnpla3-deficient mice do not display an adverse phenotype, the safety of knocking down endogenous wild type PNPLA3 in humans remains unknown. To expand the scope of a potential targeted NAFLD therapeutic to both homozygous and heterozygous PNPLA3 rs738409 populations, we sought to identify a minor allele-specific small interfering RNA (siRNA). Limiting our search to SNP-spanning triggers, a series of chemically modified siRNA were tested in vitro for activity and selectivity toward PNPLA3 rs738409 mRNA. Conjugation of the siRNA to a triantennary N-acetylgalactosamine (GalNAc) ligand enabled in vivo screening using adeno-associated virus to overexpress human PNPLA3I148M versus human PNPLA3I148I in mouse livers. Structure–activity relationship optimization yielded potent and minor allele-specific compounds that achieved high levels of mRNA and protein knockdown of human PNPLA3I148M but not PNPLA3I148I. Testing of the minor allele-specific siRNA in PNPLA3I148M-expressing mice fed a NASH-inducing diet prevented PNPLA3I148M-driven disease phenotypes, thus demonstrating the potential of a precision medicine approach to treating NAFLD.
Introduction
The single nucleotide polymorphism (SNP), in the gene patatin like phospholipase domain containing 3 (PNPLA3), rs738409 (I148M), is associated with hepatic triglyceride content and, over the 10 years since its identification, is now known to be the strongest genetic determinant for all stages of the nonalcoholic fatty liver disease (NAFLD) spectrum: nonalcoholic fatty liver (or steatosis), nonalcoholic steatohepatitis (NASH), cirrhosis, and hepatocellular carcinoma (HCC) [1–8].
With an estimated 50% of NAFLD patients carrying at least one allele for PNPLA3 rs738409 [7], and a global prevalence of NAFLD escalating to >30% of the population, PNPLA3I148M presents itself as a compelling therapeutic target for multiple stages of the disease. Furthermore, carriers of the rs738409 SNP also carry a synonymous SNP, rs738408, three nucleotides downstream of rs738409 [9,10]; rs738408 occurs with strong linkage disequilibrium with rs738409 (Fig. 1A). Altogether, the attributes of PNPLA3I148M present an opportunity for a minor allele-selective, precision medicine approach to therapeutically target a common genetic variant with a strong causal link to NAFLD.
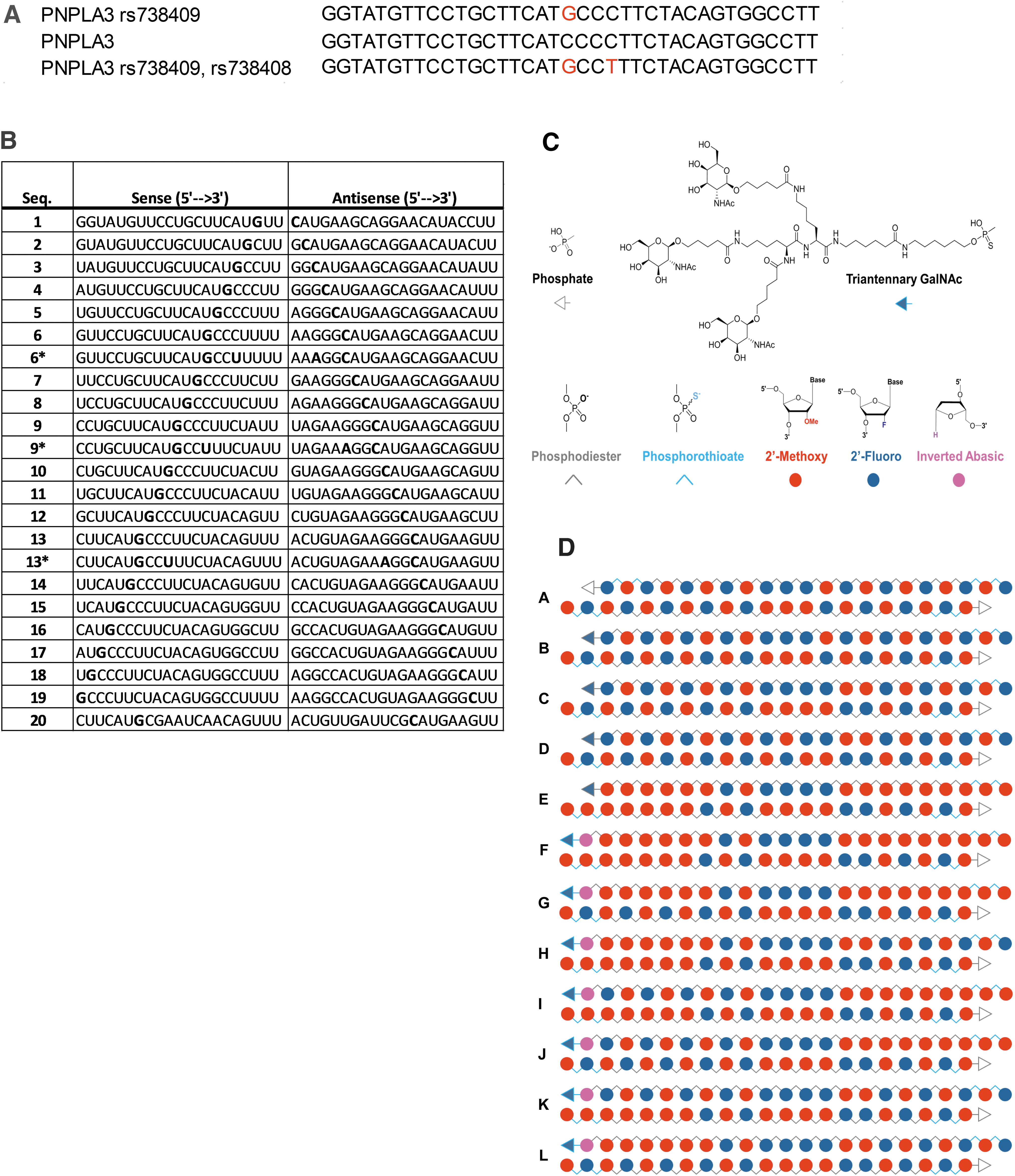
Sequences and designs of PNPLA3 siRNA.
PNPLA3 is a hydrophobic protein that associates with membranes and belongs to the patatin like phospholipase family [11–13]. Initially identified in adipocytes, PNPLA3 mRNA and PNPLA3 protein expression have been confirmed in other tissues, are enriched in the liver [12–14], and are regulated by nutritional status [14–18]. The nutritional control of PNPLA3 is the result of a feed-forward loop, driven by carbohydrates and the activation of sterol regulatory element binding protein 1c (SREBP-1c) [17]. SREBP-1c is essential for the genomic actions of insulin on carbohydrate and lipid metabolism [19]. In hepatocytes, PNPLA3 protein localizes to lipid droplets and, historically, has been presumed to possess triglyceride hydrolase activity [20–22]. However, to date, no detectable metabolic phenotype has been identified as the result of Pnpla3-deficiency or wild type human PNPLA3 overexpression in mice [23–25]. In contrast, both human PNPLA3I148M transgenic mice and Pnpla3I148M knock-in mice develop elevated hepatic triglyceride levels and NAFLD-related phenotypes [23,26]. More recently investigators reported that PNPLA3I148M protein possesses a unique ability to evade proteasomal degradation leading to its accumulation on lipid droplets [11,27]. Taken together, the reported in vivo mouse model data point to the aberrant expression of the mutant PNPLA3I148M protein, and not overexpression of the wild type protein or the loss of the wild type protein, as the driver of the disease phenotype.
RNA interference (RNAi) is the process of introducing exogenous RNA into a cell leading to specific degradation of the mRNA encoding the targeted protein, with a resultant decrease in protein expression. Advances in both RNAi technology [28,29] and hepatic delivery [30–32] and mounting positive outcomes with other RNAi-based therapies [33–37] suggest RNAi as a compelling means to therapeutically treat NAFLD by directly targeting PNPLA3I148M.
A nonallele selective PNPLA3 antisense oligonucleotide (ASO) was shown to reduce inflammation and fibrosis in the livers of Pnpla3I148M knock-in mice fed a NASH-inducing diet, as well as in wild type mice fed the same diet [38], the latter of which was inconsistent with what would have been predicted from Pnpla3-knockout studies [24]. Despite N-acetylgalactosamine (GalNAc) conjugation to specifically direct delivery to hepatocytes, the PNPLA3 ASO also knocked down endogenous Pnpla3 in the adipose tissue of both wild type and knock-in mice [38], which may confound data interpretation.
Although homozygous carriers of PNPLA3 rs738409 may benefit from knockdown of the mutant protein, the effects of knocking down wild type PNPLA3 protein is unclear given that its biological function remains to be elucidated. Hence, an RNAi therapeutic designed to selectively knockdown PNPLA3 mutant protein without altering wild type PNPLA3 protein expression would be desirable for accessing the larger heterozygous population who are still at increased risk of poor outcomes while aiming to minimize additional safety concerns [39].
Silencing mutant PNPLA3 using allelic discrimination as a strategy to reduce hepatic triglyceride levels in PNPLA3I148M carriers and provide therapeutic benefit to both homozygotes and heterozygotes builds upon recent progress in the development of small interfering RNA (siRNA). The first step toward SNP-selective oligonucleotide therapeutics was to overcome the inherent metabolic liabilities of natural RNA through the development of a fully chemically modified siRNA with increased nucleolytic and thermal stability that retained biological activity [40]. This is typically accomplished by substitution of the native 2′-hydroxyl of the ribose ring with either a 2′-fluoro or 2′-methoxy group at all positions within the oligonucleotide and replacement of select phosphodiester internucleotide linkages near the termini with phosphorothioates (Fig. 1C, D) [41].
Second, the conjugation of a triantennary GalNAc moiety has been shown to be effective for delivery of siRNA to hepatic targets [32]. The GalNAc scaffold binds to the highly expressed and rapidly recycling asialoglycoprotein receptor (ASGPR) on the surface of hepatocytes, leading to internalization of the construct through receptor-mediated endocytosis [42]. A small portion of the siRNA-GalNAc conjugate escapes from the endosome where the duplex may engage with the RNA induced silencing complex (RISC) [43–45]. Dissociation of the sense or “passenger” strand leaves the antisense or “guide” strand loaded in the Argonaute 2 protein for recognition and catalytic degradation of the complementary mRNA. These chemical modification and delivery strategies allow the intact siRNA to efficiently reach its target in the liver following subcutaneous (SC) administration enabling this therapeutic modality [30].
A remaining challenge for the development of hepatic RNAi therapeutics is engineering the allele-specific siRNA needed to address the disease-causing SNPs identified by modern genomic analysis [46]. For typical gene targets, hundreds to thousands of unique ∼19 base pair (bp) trigger sequences may be evaluated for in vitro activity, but allele-specific siRNA is limited only to those that span the SNP (Fig. 1B) [47]. Among this handful of sequences, few will likely demonstrate the required potency to achieve a durable in vivo response [48]. It is even more rare that one of the active siRNAs will also contain the SNP at a position where the mismatch with the native sequence produces the desired selectivity profile [49].
Finally, the successful candidate sequence must have a suitable bioinformatics profile, for example, no other 1 bp mismatches and ideally <20 two base pair mismatches to other gene sequences. The selection of such a trigger sequence most often sacrifices any species cross-reactivity and requires the development of additional cell lines, tools, assays, and models for evaluation. Herein we report our efforts to discover a minor allele-specific siRNA to PNPLA3I148M and its therapeutic effects in ameliorating NAFLD-related phenotypes in mice overexpressing the mutant human protein.
Materials and Methods
Materials
Acetonitrile (DNA Synthesis Grade, AXO152-2505), Capping Reagent A [80:10:10 (v/v/v) tetrahydrofuran/lutidine/acetic anhydride, BIO221/4000], Capping Reagent B (16% 1-methylimidazole/tetrahydrofuran, BIO345/4000), Activator Solution [0.25 M 5-(ethylthio)-1H-tetrazole in acetonitrile, BIO152/0960], Detritylation Reagent [3% dichloroacetic acid in dichloromethane (DCM), BIO830/4000], Oxidation Reagent [0.02 M iodine in 70:20:10 (v/v/v) tetrahydrofuran/pyridine/water, BIO420/4000], and diethylamine solution (20% DEA in acetonitrile, NC0017-0505) were obtained from EMD. Thiolation Reagent ((dimethylamino-methylidene)amino)-3H-1,2,4-dithiazoline-3-thione was obtained from ChemGenes and prepared at 0.05 M in 40:60 (v/v) pyridine (Aldrich)/acetonitrile. 5′-Aminohexyl linker phosphoramidite, phosphorylating phosphoramidite, reverse abasic phosphoramidite, and 2′-methoxy and 2′-fluoro phosphoramidites of adenosine, guanosine, cytosine, and uridine from Thermo Fisher Scientific or ChemGenes were prepared at 0.10 M in acetonitrile over ∼10 mL of molecular sieves (J.T. Baker). Universal UnyLinker Support 500 or 1,000 Å controlled pore glass (CPG) was from ChemGenes. Ammonium hydroxide, concentrated, was from J.T. Baker.
Oligonucleotide synthesis
Synthesis of chemically modified siRNA sequences was performed on the MerMade 12 (Bioautomation). Reagent solutions, phosphoramidite solutions, and solvents were attached to the instrument. Solid support (10 μmol) was added to each column (4 mL SPE tube with top and bottom frit), and the columns were affixed to the instrument. The columns were washed twice with acetonitrile. The phosphoramidite and reagent solution lines were purged. The synthesis was initiated using the Poseidon software.
The synthesis was accomplished by repetition of the deprotection/coupling/oxidation/capping synthesis cycle. To the solid support was added detritylation reagent to remove the 5′-DMT protecting group. The solid support was washed with acetonitrile. To the support was added phosphoramidite and activator solution followed by incubation to couple the incoming nucleotide to the free 5′-hydroxyl group. The support was washed with acetonitrile. To the support was added oxidation or thiolation reagent to convert the phosphite triester to the phosphate triester or phosphorothioate, respectively. To the support was added capping reagents A and B to terminate any unreacted oligonucleotide chains. The support was washed with acetonitrile. The cycle was repeated as required. After the final reaction cycle, the resin was washed with diethylamine solution to remove the 2-cyanoethyl protecting groups. The support was washed with acetonitrile and dried under vacuum.
Sense strands for GalNAc conjugation were prepared with a 5′-MMT-aminohexyl linker. After automated synthesis, the column was removed from the instrument and transferred to a vacuum manifold in a hood. The 5′-MMT protecting group was removed from the solid support by successive treatments with 2 mL aliquots of 1% trifluoroacetic acid (TFA) in DCM with vacuum filtration. When the orange/yellow color was no longer observable in the eluent, the resin was washed with DCM. The resin was washed with 5 mL of 2% diisopropylethylamine in N,N-dimethylformamide (DMF). In a separate vial was prepared a solution of GalNAc3-Lys2-Ahx (67 mg, 40 μmol) in DMF (0.5 mL) with 1,1,3,3-tetramethyluronium tetrafluoroborate (TATU, 12.83 mg, 40 μmol) and N,N-diisopropylethylamine (DIEA) (10.5 uL, 360 μmol). The activated coupling solution was added to the resin, and the column was capped and incubated at room temperature overnight. The resin was washed with DMF, DCM, and dried under vacuum.
The synthesis columns were removed from the synthesizer or vacuum manifold. The solid support from each column was transferred to a 10 mL pressure vial. To the solid support was added 4 mL of concentrated ammonium hydroxide. The cap was tightly affixed to the bottle, and the mixture was heated at 55°C for 6 h. The bottle was moved to the freezer and cooled for 20 min before opening in the hood. The mixture was filtered through an 8 mL SPE tube to remove the solid support. The vial and solid support were rinsed with 1 mL of 50:50 ethanol/water.
A portion of the combined filtrate was analyzed and purified using Anion Exchange Chromatography (AEX). The pooled fractions were desalted by size exclusion chromatography and analyzed by ion pair-reversed phase high pressure liquid chromatography-mass spectometry. The pooled fractions were lyophilized to obtain a white amorphous powder.
A small amount of the sense strand and the antisense strand was weighed into individual vials. To the vials was added siRNA reconstitution buffer (Qiagen) to an approximate concentration of 2 mM based on the dry weight. The actual sample concentration was measured on the NanoDrop One (ssDNA, extinction coefficient = 33 μg/OD260). The two strands were then mixed in an equimolar ratio, and the sample was heated for 5 min in a 90°C incubator and allowed to cool slowly to room temperature. The sample was analyzed by AEX. The RNA duplex was observed to have an intermediate retention time by analytical AEX relative to the two single strands. The duplex was submitted for in vitro and in vivo testing.
Preparation of GalNAc3-Lys2-Ahx
The protected triantennary GalNAc carboxylic acid (GalNAc3-Lys2-Ahx) for amide bond coupling to the 5′ end of the sense strand is depicted in Supplementary Fig. S1.
To a 50 mL centrifuge tube was added Fmoc-Ahx-OH (1.13 g, 3.19 mmol) in DCM (30 mL) followed by DIEA (2.23 mL, 12.78 mmol). The solution was added to 2-Cl Trityl chloride resin (3.03 g, 4.79 mmol) in a 50 mL centrifuge tube and loaded onto a shaker for 2 h. The solvent was drained, and the resin was washed with 17:2:1 DCM/MeOH/DIEA (30 mL 2 × ), DCM (30 mL 4 × ) and dried. The loading was determined to be 0.76 mmol/g with ultraviolet (UV) spectrophotometric detection at 290 nm.
Three grams of the loaded 2-Cl Trityl resin was suspended in 20% 4-methylpiperidine in DMF (20 mL), and after 30 min the solvent was drained. The process was repeated one more time, and the resin was washed with DMF (30 mL 3 × ) and DCM (30 mL 3 × ).
To a solution of Fmoc-Lys(ivDde)-OH (3.45 g, 6 mmol) in DMF (20 mL) was added TATU (1.94 g, 6 mmol) followed by DIEA (1.83 mL, 10.5 mmol). The solution was then added to the above deprotected resin, and the suspension was set on a shaker overnight. The solvent was drained, and the resin was washed with DMF (30 mL 3 × ) and DCM (30 mL 3 × ).
The resin was treated with 20% 4-methylpiperidine in DMF (15 mL), and after 10 min the solvent was drained. The process was repeated one more time, and the resin was washed with DMF (15 mL 4 × ) and DCM (15 mL 4 × ).
To a solution of Fmoc-Lys(Fmoc)-OH (3.54 g, 6 mmol) in DMF (20 mL) was added TATU (1.94 g, 6 mmol) followed by DIEA (1.83 mL, 10.5 mmol). The solution was then added to the above deprotected resin, and the suspension was set on a shaker overnight. The solvent was drained, and the resin was washed with DMF (30 mL 3 × ) and DCM (30 mL 3 × ).
The resin was treated with 5% hydrazine in DMF (20 mL), and after 5 min, the solvent was drained. The process was repeated four more times, and the resin was washed with DMF (30 mL 4 × ) and DCM (30 mL 4 × ).
To a solution of 5-(((2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxy-6-(acetoxymethyl)tetrahydro-2H-pyran-2-yl)oxy)pentanoic acid (4.47 g, 10 mmol) in DMF (40 mL) was added TATU (3.22 g, 10 mmol), and the solution was stirred for 5 min. DIEA (2.96 mL, 17 mmol) was added to the solution, and the mixture was then added to the resin above. The suspension was kept at room temperature overnight, and the solvent was drained. The resin was washed with DMF (3 × 30 mL) and DCM (3 × 30 mL).
The resin was treated with 1% TFA in DCM (30 mL with 3% triisopropylsilane), and after 5 min, the solvent was drained. The process was repeated thrice, and the combined filtrate was concentrated in vacuo. The residue was triturated with diethyl ether (50 mL), and the suspension was filtered and dried to give the crude product. The crude product was purified with reverse phase chromatography and eluted with 0%–20% of MeCN in water. The fractions were combined and lyophilized to give the product as a white solid.
In vitro screening
The efficacy of each of the siRNA molecules in reducing PNPLA3 expression was assessed using a 384-well format in vitro siRNA transfection assay followed by an RNA fluorescence in situ hybridization (RNA FISH) assay to determine IC50 and maximum activity values. This assay was performed on Chinese hamster ovary (CHO) cells expressing human PNPLA3I148M (hPNPLA3I148M) or human PNPLA3I148I (hPNPLA3I148I). Cells were maintained in media containing 50% CD-CHO (Life Technologies), 50% Ex-Cell CHO 5 Medium (Sigma), 8 mM
For CHO cell assays, transfection complexes of the siRNA molecules, prepared in serial doses, and the Lipofectamine RNAiMAX transfection reagent in F12K media (MediaTech) were prepared in 384-well plates, with 0.1 μL of RNAiMAX per well. Cells were diluted to 50,000 cells/mL in antibiotic/antimycotic-free media and 30 μL added to each well, a final density of 1,500 cells/well in 40 μL media. After a 20-min incubation at room temperature, plates were transferred to a 37°C and 5% CO2 incubator. CHO human PNPLA3 I148M and I148I transfection assays were incubated for 48 h.
At harvest, the cells were fixed in an 8% formaldehyde fixative solution (Thermo Fisher Scientific) for 15 min at room temperature. The plates were then subjected to dehydration with sequential 50%, 70%, and 100% ethanol washes. Plates were then sealed and stored at −20°C.
The RNA FISH assay was performed using the Affymetrix QuantiGene® ViewRNA HC Screening Assay Kit (QVP0011), the Affymetrix View HC Signal Amplification Kit 3-plex (QVP0213), and Affymetrix gene specific probes (VX-01): PNPLA3 Human 0.44 mL ViewRNA Type 6 (650 label) VA6-20279-VC and PPIB Human 0.44 mL ViewRNA Type 1 (488 label) VA1-10148-VC.
Plates were first rehydrated with sequential 70% and 50% ethanol washes. Cells were then washed with phosphate-buffered saline (PBS) and then permeabilized and protease digested according to the kit instructions. The target Working Probe Sets were prepared according to the manufacturer's protocol, added to the wells, and incubated for 3 h at 40°C. The manufacturer's protocol was followed for the sequential hybridizations with the Working PreAmps, the Working Amps, and the Working LPs. Finally, nuclei counterstains were applied (Hoechst 33342 and CellMask Blue; Molecular Probes). Plates were incubated for 30 min at room temperature, washed with PBS, overlaid with 80 μL of PBS, and then the plates were sealed for imaging.
All plates were imaged on an Opera Phenix High Content Screening System (PerkinElmer), using the UV Channel for Hoechst 33342 and CellMask Blue, the 488 Channel for Type1 probes, and the 647 Channel for Type6 probes.
Images were analyzed using Columbus software (PerkinElmer), and the dose–response data were further analyzed using Genedata Screener (Genedata) to obtain IC50 and maximal activity values. The results of the assay for CHO transfected hPNPLA3I148M and hPNPLA3I148I cells are shown in Fig. 2 and Table 1. PNPLA3 mRNA knockdown provides a percentage of knockdown compared to the nontargeting siRNA control (hPPIB). Negative values indicate a decrease in PNPLA3 levels.
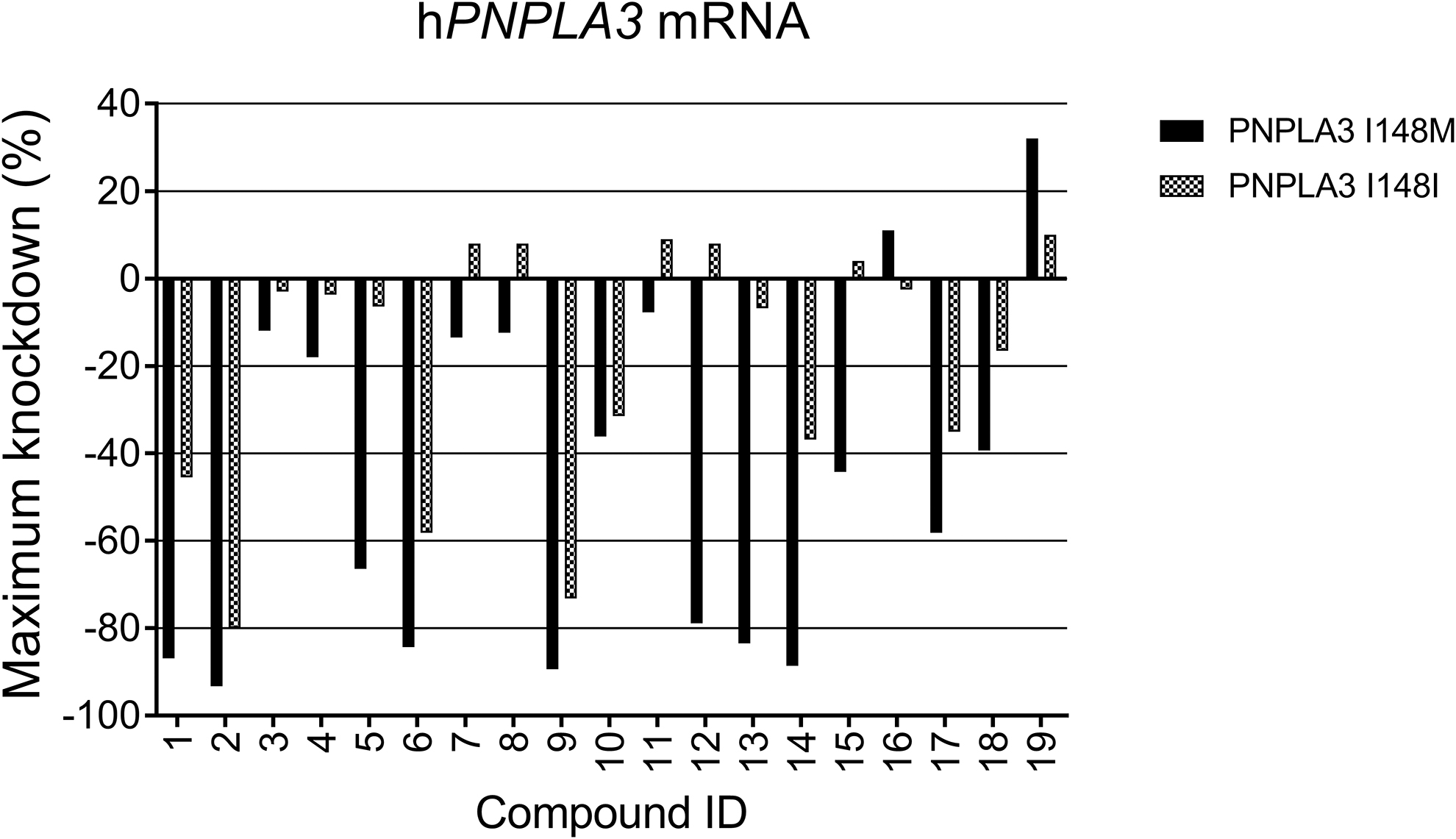
In vitro activity of PNPLA3 siRNA. In vitro screen for mRNA knockdown of hPNPLA3I148I and hPNPLA3I148M with transfected compounds
In Vitro Screening Data for mRNA Knockdown of
The bold letters in the sequence lists represent the location of rs738409. Max activity represents the target mRNA knockdown at 0.5 μM.
PNPLA3, patatin like phospholipase domain containing 3.
Mice
Ten- to 12-week-old C57BL/6N male mice (Charles River Laboratories) were used for in vivo studies. All procedures were conducted in compliance with the Guide for the Care and Use of Laboratory Animals (National Research Council, 2010) and IACUC protocol number 2019-01417. Mice were singly housed in sterilized cages with a full cage; changes were performed once weekly. Envigo 2920X Irradiated Rodent Diet (CHOW) and reverse-osmosis water from the Amgen water supply system were supplied ad libitum. The housing environment was maintained between 20°C and 22.2°C with relative humidity between 34% and 73%. Light was provided for 12 continuous hours followed by 12 h of darkness. When indicated, mice were converted to a NASH-inducing diet (American Lifestyle-Induced Obesity Syndrome model diet, “ALIOS,” Teklad; TD.190883), ad libitum.
In vivo studies
Before study initiation, mouse body weights were collected and then the mice were randomized, by weight, into the experimental groups. At study initiation, mice received a 0.2 mL intravenous (IV) injection, through the tail vein, of endotoxin-free adeno-associated virus (AAV) containing either an empty vector (EV), the full-length coding region of wild type human PNPLA3 (AAV-hPNPLA3I148I), the full-length coding region of human PNPLA3rs738409 (AAV-hPNPLA3I148M), or the full length coding region of human PNPLA3rs738409rs738408 (AAV-hPNPLA3I148M DM). AAVs were administered at a concentration of 1e12 viral particles per mouse, diluted in Dulbecco's phosphate-buffered saline (DPBS, #14190-136; Thermo Fisher Scientific), which was also used as the vehicle control, as indicated. All plasmids cloning and AAV packaging (serotype AAV7) were performed internally (Amgen, Inc.). Plasmids contained an EF1a promoter and the human PNPLA3I148I, PNPLA3I148M, or PNPLA3I148M DM full-sequence coding regions.
Two weeks following AAV administration, mice were administered a SC injection of siRNA into the left abdomen region. Mice described in Fig. 4 received either compound
At study termination, mice were weighed and then anesthetized with isoflurane. From each animal, blood was collected through intracardiac puncture using a 1 mL 25-gauge Tuberculin syringe and transferred to Microtainer SST tube (365967; Molecular Devices). Mice, still under deep anesthesia, were then euthanized by a secondary physical method and the left and right liver lobes and epididymal white adipose tissue (EpWAT) were collected, weighed, and immediately snap frozen in liquid nitrogen for RNA, protein, and content analysis. The median liver lobe was fixed with 10% neutral buffered formalin, followed by paraffin processing and embedding. Filled SST tubes stood at room temperature to clot for a minimum of 30 min and then spun, at room temperature, for 10 min at 10,000 rpm in a centrifuge. Serum was then transferred to a 96-well DeepWell™ plate, sealed, frozen, and stored at −80°C.
RNA/quantitative polymerase chain reaction
RNA was extracted from snap frozen liver and EpWAT. Both were homogenized in TRIzol™ Reagent (15596026; Thermo Fisher Scientific), followed by chloroform extraction. Lysate samples were centrifuged for 15 min at 12,000 g at 4°C, and then, the aqueous phase containing the RNA was transferred to a new tube for RNA isolation using QIAcube System (9001292; Qiagen) and RNeasy Mini Kit reagents (74106; Qiagen), both according to manufacturer's instructions. Isolated RNA samples were analyzed using a QIAxpert system. RNA samples were subsequently treated with RQ1 RNase-Free DNase (M6101; Promega) and prepared for real-time quantitative polymerase chain reaction (qPCR) using the TaqMan™ RNA-to-Ct™ 1-Step Kit (4392656; Thermo Fisher Scientific). Real-time qPCR was performed using a QuantStudio Real-Time PCR system (7 Flex; Thermo Fisher Scientific).
Results are presented as the raw Ct value and relative fold change in mRNA expression. Relative fold change mRNA values were determined by normalizing the gene of interest Ct value to that of a housekeeping gene, mouse hydroxymethylbilane synthase (mHmbs) for liver samples and the housekeeping gene mouse TATA-box binding protein (mTbp) for EpWAT samples, and then calculating the ddCt value. All samples were run in duplicate. qPCR primer and probe assays: human PNPLA3 (nonallele-selective assay; Hs.00228747_m1), mHmbs qPCR (IDT, Mm.Pt. 58.43648767), mouse Pnpla3 (Mm00504420_m1), mouse Hprt (Mm03024075_m1), mTbp (Mm 01277042_M1), mouse Timp1 (Mm01341361 m1), mouse Timp2 (Mm00441825_m1), mouse Tnfα (Mm00443258_m1), mouse Tgfβ (Mm01178820_m1), mouse Col1a1 (Mm00801666_g1), mouse Col3a1 (Mm01254471_g1), mouse Cd68 (Mm03047343_m1), and mouse Samm50 (Mm00618120_m1); Hmbs purchased from IDT, all other assays purchased from Invitrogen.
Protein
For protein extraction, ∼50 mg of frozen liver tissue from each animal sample was independently homogenized, in 1 mL glass homogenizer tubes (6479001; Electron Microscopy Sciences), in cold Pierce IP buffer (87787; Thermo Fisher Scientific), including 1 × Halt™ Protease and Phosphatase Inhibitor Cocktail (78441; Thermo Fisher Scientific) to make a 50 mg tissue per milliliter lysis buffer solution. Samples were spun at 14,000 rcf for 15 min at 4°C and transferred to clean tubes. Protein concentrations were determined using a Direct Detect™ infrared spectrometer (Millipore). Protein analysis was performed on a Simple Western “Wes™” instrument (ProteinSimple) using a 12 to 230 kD Wes™ separation module (SM-W004; ProteinSimple) and a 12 to 230 kD biotinylated ladder (PS-ST01EZ-8; ProteinSimple).
For Fig. 4E: Previous sample optimization determined the capillary loading concentrations for detection: PNPLA3 and HPRT samples were both loaded at 0.25 mg/mL, and β-actin samples were loaded at 0.1 mg/mL. To obtain these concentrations, samples were diluted in 0.1 × sample buffer (SM-W004; Protein Simple). 1 × Master Mix and DTT (PS-FL01-8; Protein Simple) were added to lysates and boiled at 95°C for 5 min as per vendor's instructions. Recombinant proteins were included as positive controls for antibody detection: recombinant human PNPLA3-TAG C-Myc/DDK 52.7 kD (TP309577; OriGene) and recombinant human HPRT His tag25KD (10314-HP-050; R&D Systems). Antibodies and optimized dilution are as follows: rabbit anti-PNPLA3, 1:50 (ab81874; Abcam); rabbit anti-HPRT, 1:30 (ab10479; Abcam); mouse α-actin, 1:25 (A5316; Sigma).
For Fig. 5B: Optimized capillary loading concentrations for detection: PNPLA3 samples: 5-fold diluted stock lysate and β-actin sample: 10-fold diluted stock lysate. Samples were diluted as described for Fig. 4. The recombinant PNPLA3-tag N-GST protein 79 kD (ab132787; Abcam) was included as a positive control for antibody detection. Antibodies and optimized dilution are as follows: rabbit anti-PNPLA3, 1:50 (SAB1401851; Sigma); mouse β-actin, 1:25 (A5316; Sigma).
All Wes™ processing and data analysis were performed using Compass for SW, Version 5.0.0 software.
Triglyceride content
To determine hepatic triglyceride content, ∼50 mg of frozen liver tissue from each animal was weighed, on ice, and then homogenized independently in 2 mL Eppendorf™ Snap-Cap Microcentrifuge Safe-Lock™ Tubes (05-402-8; Thermo Fisher Scientific) with a 5 mm stainless steel bead (69989; Qiagen) and 1 mL of isopropanol alcohol (I9526; Sigma). Samples were lysed using a TissueLyser II (85300; Qiagen) for 2 min and then incubated on ice for ∼1 h, vortexing two to three times during the incubation. After cold incubation, samples were centrifuged at 10,000 rpm × 15 min at 4°C, and then, the supernatant was transferred to a new tube.
Triglyceride concentration was determined using the Infinity™ Triglyceride reagent (TR22421; Thermo Fisher Scientific) and Triglyceride standard (T7531-STD; Pointe Scientific) according to the manufacturer's instructions. Absorbance was determined using a SpectraMax® M Series Multi-Mode Microplate Reader (Molecular Devices) and SoftMax Pro6 Software, V1.0 (Molecular Devices).
Serum analysis
Serum tissue inhibitor of metalloproteinases 1 (TIMP1) was determined using a Mouse TIMP1 Quantikine ELISA Kit (MYM100; R&D Systems) according to manufacturer's instructions. Absorbance was determined using a SpectraMax M Series Multi-Mode Microplate Reader (Molecular Devices) and SoftMax Pro6 Software, V1.0 (Molecular Devices). Frozen serum was sent to IDEXX BioAnalytics (West Sacramento, CA) for determination of the following clinical chemistry end points: alanine aminotransferase (ALT), aspartate aminotransferase (AST), albumin, and triglyceride. All data reported came directly from the delivered report.
Histopathology
From each animal, the median liver lobe was collected, fixed with 10% neutral buffered formalin for no more than 24 h, and then transferred to 70% ethanol. The fixed median lobes were embedded in paraffin, sectioned, and then stained with hematoxylin and eosin to evaluate steatosis and inflammation/degeneration. Microscopic evaluation was conducted by a board-certified veterinary pathologist. Tissues were evaluated by light microscopy and graded on a 4-point scale based on lesion severity.
Steatosis scoring system: Grade 0 (Absent), Grade 1 (minimal; in ≤5% hepatocytes), Grade 2 (mild; in 6%–33% hepatocytes), Grade 3 (moderate; in 34%–66% hepatocytes), and Grade 4 (severe; in ≥67% hepatocytes). Inflammation/degeneration scoring system: Grade 0 (Absent), Grade 1 (minimal; ≤1 inflammatory focus per 200 × field), Grade 2 (mild; 1–2 inflammatory foci per 200 × field), Grade 3 (moderate; 3 inflammatory foci per 200 × field), and Grade 4 (Severe, ≥4 inflammatory foci per 200 × field).
Results
Identification of minor allele specific active sequences
A series of 19 rs738409 SNP-spanning human PNPLA3 siRNA were prepared and tested in vitro through transfection for mRNA knockdown in a RNA FISH assay in two different CHO cell lines, one overexpressing the wild type form of human PNPLA3 (hPNPLA3I148I) and the other overexpressing the mutant form of human PNPLA3 containing the single nucleotide change, rs738409 SNP (hPNPLA3I148M). The compounds for in vitro assays were prepared with a standard chemical modification pattern (
Seven sequences, including
Only
Optimization for in vivo potency and durability
Select PNPLA3I148M siRNA sequences showing activity and/or selectivity in the in vitro screening were resynthesized with a triantennary GalNAc moiety conjugated to the 5′ end of the sense strand to facilitate the hepatic delivery required for in vivo testing with SC dosing. For in vivo screening, mice were administered, through IV tail vein injection, AAV containing a vector for hPNPLA3I148M. Following a 2-week transduction and viral shedding period, mice received a 5 mg/kg SC dose of GalNAc-siRNA conjugate. One-week post-siRNA dose, the livers were harvested, and the level of hPNPLA3 mRNA was measured by a nonselective qPCR assay (Fig. 3A).
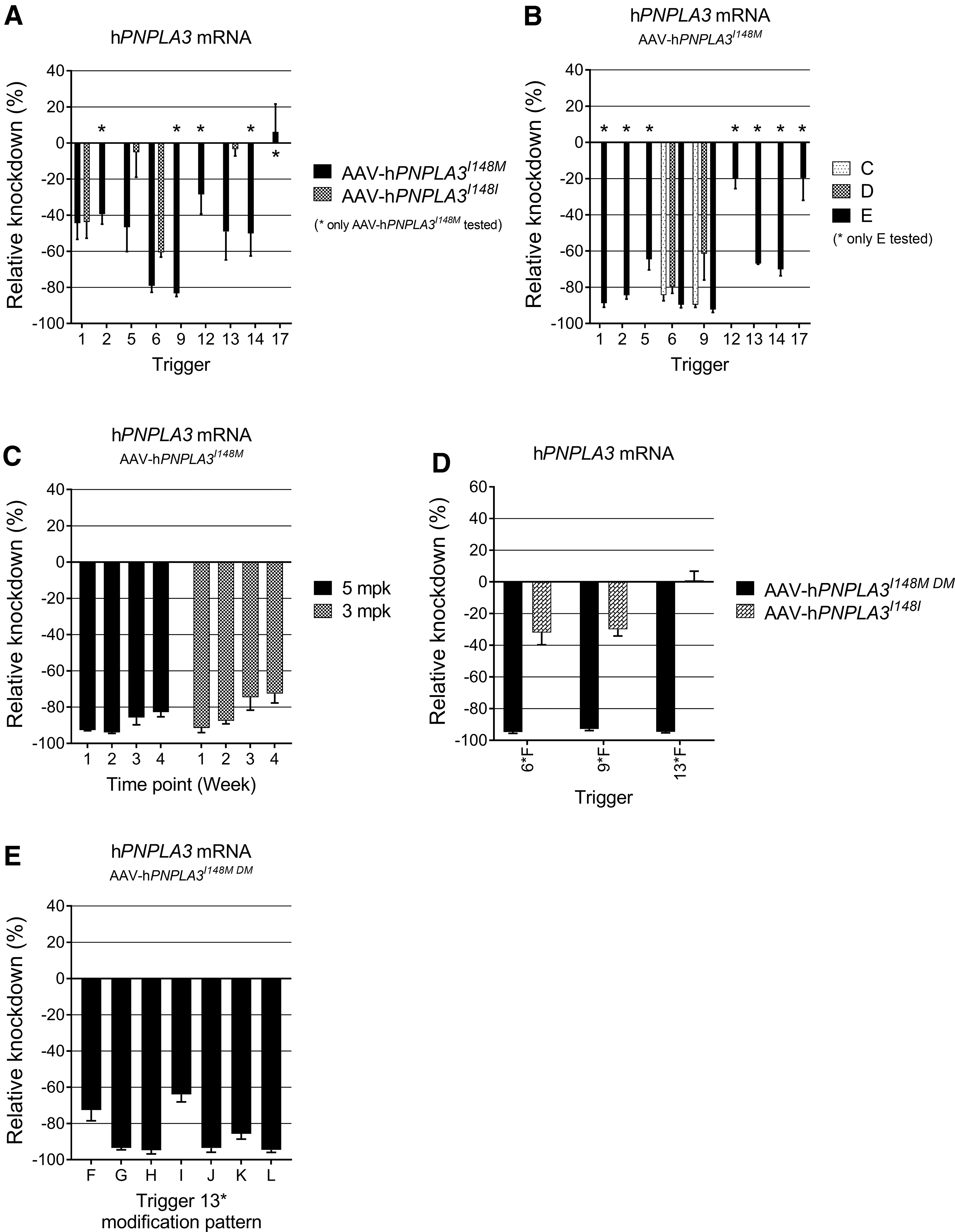
In vivo optimization of a minor allele-specific siRNA-GalNAc conjugate.
Initial results showed that nearly all compounds selected based on in vitro potency achieved ≥40% mRNA knockdown at this early time point. One exception was the potentially selective sequence
The structure–activity relationship of the chemical modification pattern was explored to optimize potency and duration of action of the initial compounds. Using triggers
It has been reported that siRNA with an increased proportion of 2′-methoxy nucleotides is less susceptible to nucleolytic degradation and may be more long lived in vivo [54]. Accordingly, substituting several 2′-methoxy nucleotides into the 5′ and 3′ terminal segments of the sense strand and the 3′ end of the antisense strand improved the initial potency of compounds
A nearby synonymous SNP, rs738408, is in complete linkage disequilibrium [10,55] with rs738409, resulting in a “double minor allele” that may make identification of a selective siRNA more likely due to the close proximity of two distinct base pair mismatches between the wild type and mutant alleles (Fig. 1A). We sought to determine whether siRNAs designed to target both SNPs would retain or even improve the selectivity for the mutant allele without affecting on-target gene silencing activity. An additional change was made to generate modification pattern
Thus, the AAV DNA plasmid for hPNPLA3I148M was reengineered to express the double minor (rs738409, rs738408) version of PNPLA3, henceforth referred to as AAV-hPNPLA3I148M DM (DM for “double minor”). The lead sequences
A final round of chemical modification pattern optimization was performed with the intention of identifying an analog of
Validation of minor allele specificity in vivo
To validate double minor allele specificity in vivo, mice were administered AAV to overexpress hPNPLA3I148I or hPNPLA3 I148M DM. Control animals were generated by injecting either vehicle alone (veh) or AAV containing an empty vector (EV). Two weeks after administration of AAV, mice were administered a single SC dose of compounds
Comparison of qPCR mRNA Ct values for mouse Pnpla3 (open bars) and hPNPLA3 (black bars) suggests robust overexpression of hPNPLA3, compared to endogenous mPnpla3 expression, in both AAV-hPNPLA3I148I/veh and AAV-hPNPLA3I148M DM/veh mice (Fig. 4A). While
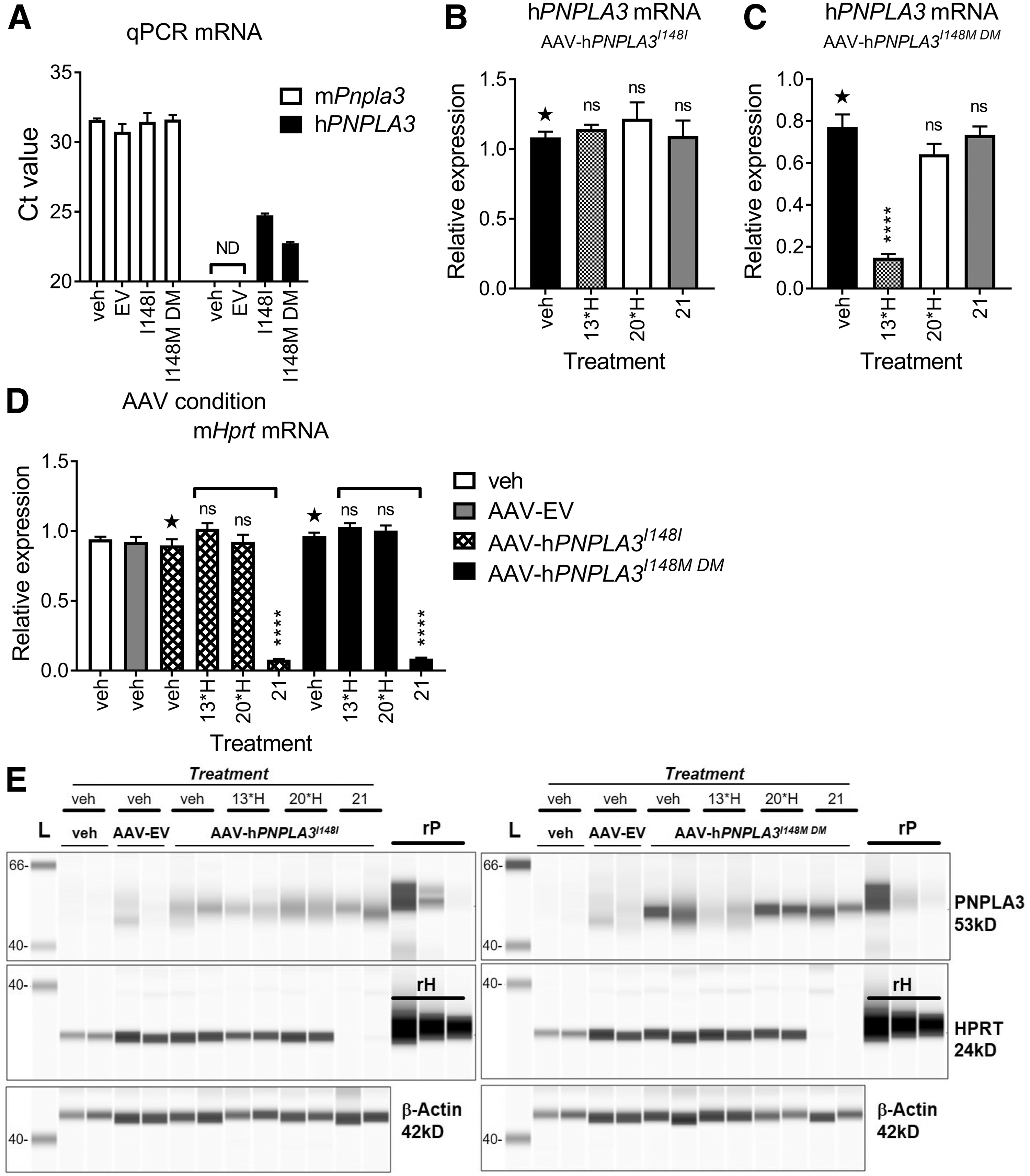
Validation of compound
Two representative liver lysate samples from all AAV groups were processed for protein analysis by WES™. Staining with an anti-hPNPLA3 antibody confirmed compound
A minor allele-specific siRNA ameliorates PNPLA3I148M DM -driven NASH phenotypes
To demonstrate a functional effect of hPNPLA3I148M DM modulation on NASH phenotypes, mice were administered AAV-hPNPLA3I148M DM, AAV-EV, or vehicle control, through tail vein injection, and placed on a NASH-inducing diet (American Lifestyle-Induced Obesity Syndrome model diet, “ALIOS”) [58] for 8 weeks to boost hepatic triglyceride levels, liver inflammation, and markers of early fibrosis. Two weeks after AAV administration and diet initiation, mice were administered a SC dose of compound
Comparison of qPCR mRNA Ct values for hPNPLA3 from AAV-hPNPLA3I148M DM liver tissue and EpWAT indicated an estimated Ct value of ∼22 in liver and 28 in EpWAT (Fig. 5A). Administration of siRNA
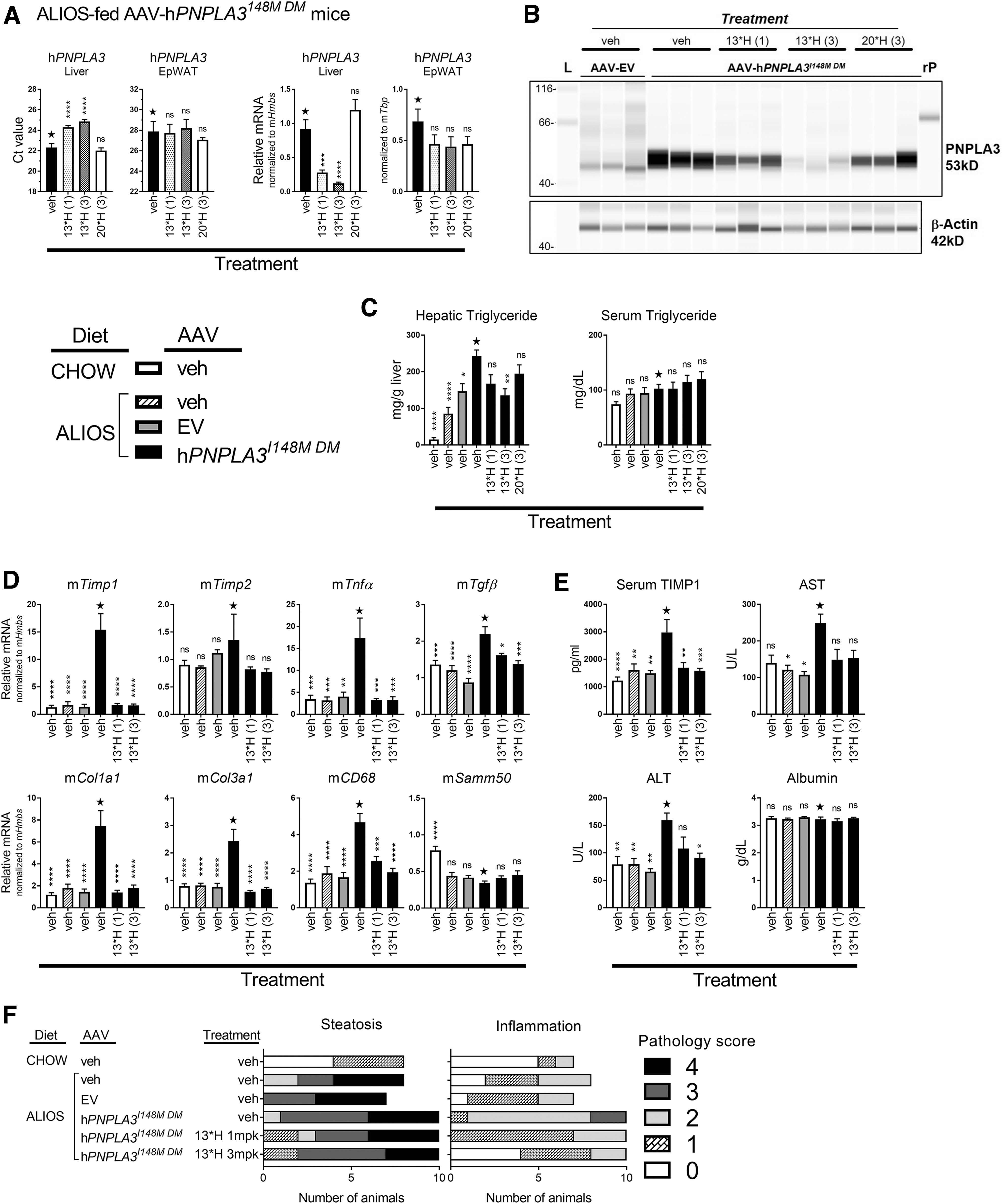
Confirmation of in vivo efficacy by a minor allele-specific siRNA. The following groups were included in this study: CHOW-fed veh/veh mice, n = 9; ALIOS fed-veh/veh mice, n = 8; ALIOS-fed AAV-EV/veh mice, n = 7; and ALIOS-fed AAV-hPNPLA3I148M DM mice administered either veh (n = 10), compound
To confirm transcriptional regulation of hPNPLA3 by siRNA
The addition of fructose and fat in the ALIOS diet induces elevated hepatic triglyceride content within weeks of feeding. This is observed by increased triglyceride content (mg triglyceride/g liver tissue) in all mice fed the ALIOS diet compared to CHOW-fed mice (Fig. 5C). Overexpression of hPNPLA3I148M DM further increased triglyceride levels over diet, while serum TG levels were not significantly altered by AAV or siRNA treatment. Administration of
There are many well described gene and serum markers that correlate with NAFLD progression and severity. Although 8 weeks is relatively short and does not induce robust late-stage markers of disease, overexpression of hPNPLA3I148M DM in liver tissue induced robust increases in the mRNA expression level of several markers of NASH-related inflammation, including Timp1, Timp2, Tnfa, Tgfb, the profibrotic markers, Col1a1 and Col3a1, and the macrophage marker, CD68 (Fig. 5D). Treatment with
The chromosomal location for SAMM50, chr22:44,351,261-44,406,41, is immediately downstream of PNPLA3, chr22:44,319,619-44,360,368, and SAMM50 variants have been identified with NAFLD [59,60]. Although the ALIOS diet reduced Samm50 mRNA expression, neither AAV-hPNPLA3I148M DM nor siRNA treatment effected expression. Thus, despite association of SAMM50 with NAFLD, these data suggest that SAMM50 expression is not linked to hPNPLA3I148M
DM
expression. The data, however, demonstrate the impact hPNPLA3I148M DM expression has on NASH-related phenotypes and that markers for these phenotypes are modulated by siRNA
Serum TIMP1 is one of the many diagnostic markers for human NASH and has been shown to correlate with stages of fibrosis [61–64]. In this model, overexpression of hPNPLA3I148M DM induced an increase in secreted TIMP1 compared to chow-fed and NASH-fed control mice (Fig. 5E). Like that observed with the genetic markers, treatment with
Serum from the mice was also evaluated for liver transaminases. Numerous published reports identify ALT, and in some cases AST, as an associated phenotypic trait of PNPLA3I148M [65–68]. Eight weeks on the ALIOS diet alone was insufficient to induce elevated secretion of either ALT or AST; however, overexpression of hPNPLA3I148M DM triggered secretion of both transaminases compared to the diet control groups (veh and EV). Treatment with
Histopathology assessment of biopsied tissue is the hallmark diagnostic for NASH; therefore, anatomical pathology was performed on tissues collected at study termination to evaluate differences in steatosis and inflammation across diet, AAV, and siRNA conditions. Formalin-fixed, hematoxylin and eosin-stained liver tissue from all groups of mice were examined by a board-certified pathologist and assigned a pathology score for lesion incidence and severity, as described in the Materials and Methods section.
An overall increase in score was observed for both steatosis and inflammation with the conversion from a CHOW diet to the ALIOS diet (Fig. 5F). Overexpression of hPNPLA3I148M DM led to a further mean increase in severity across animals for both steatosis and inflammation. Although the shift in scores with
Discussion
While the potential of siRNA therapy for personalized medicine was ideated over 15 years ago, progress in the treatment of diseases caused by a SNP has been elusive. Several allele-specific siRNAs have been reported that target autosomal dominant mutations [50–52], SNPs linked with aberrantly expanded trinucleotide repeats (SCA1, SCA3, and SCA7 in Huntington's Disease), and duplicated disease genes (LMNB1 in ADLD) [69]. However, the clinical translation of allele-specific siRNA proof-of-concept experiments for central nervous system (CNS) disorders of interest has been limited by the long-standing challenges of oligonucleotide stability, delivery, and safety.
While there is progress toward addressing these issues for gene variants expressed in the CNS, chemical developments have fully enabled delivery of RNAi for suitable targets in the liver [70]. siRNA backbones with 2′-fluoro and 2′-methoxy ribose modification patterns and terminal phosphorothioate internucleotide linkages may be used to confer metabolic stability and protection from endogenous nucleases encountered during SC delivery, transit through the bloodstream, and endosomal uptake. Attachment of a multivalent GalNAc ligand achieves efficient hepatocyte targeting through ASGPR binding and internalization. These advances recently culminated in the FDA approval of the first GalNAc-siRNA conjugate [71].
We sought to extend this technology to engineer a minor allele-specific siRNA for the potent and durable in vivo mRNA knockdown of a hepatic gene variant, PNPLA3 rs738409. This SNP results in production of the PNPLA3I148M mutant protein known to drive the NAFLD phenotype and is an important therapeutic target for a growing patient population of homozygous and heterozygous carriers. Most bioinformatic filters for clinical siRNA programs yield hundreds of 19 bp triggers from the gene-of-interest that meet prespecified selectivity criteria related to species cross-reactivity, avoidance of SNPs with a minor allele frequency >1%, a minimal number of mismatches with other liver-expressed genes, and a seed region that does not match known microRNAs, and may be screened to identify a set of potent lead sequences.
Although development of a human-only trigger may be accomplished with a surrogate or a humanized animal model, selection of an allele-specific siRNA represents a true precision medicine approach that circumvents the uncertainty of silencing a wild type allele in heterozygous carriers of the mutation. Specific targeting of the PNPLA3I148M DM limited the pool of triggers to a small subset of sequences spanning both the rs738409 and rs738408 SNPs. The C > G transversion of rs738409 results in only a mildly disruptive C∙C mismatch at the SNP site between the antisense strand and the wild type mRNA, leading to identification of an even smaller number of minor allele-specific sequences.
Although fully chemically modified compounds were tested in vitro, translation of activity from a cell-based assay through transfection to a free-uptake in vivo context was not assured and was dependent on development of a multivalent ligand for ASGPR. Our GalNAc construct and conjugation approach were inspired by solid-phase peptide synthesis. The branched scaffold was prepared on solid support by coupling consecutive, orthogonally protected lysine residues, followed by attachment of an alkyl-linked GalNAc to each of the three amines. The C-terminal carboxylic acid of the cleaved triantennary structure could then be coupled to the amino-C6 linker on the 5′ terminus of the sense strand oligonucleotide through amide bond formation while anchored to the CPG support. This strategy afforded GalNAc-siRNA conjugates in good yield and purity (after AEX purification of the single strands) with delivery properties consistent with literature standards (data not shown).
While some compounds fell out of the flow scheme for a lack of efficacy in our overexpression hPNPLA3I148M mouse model, in vivo potency and selectivity to the minor allele were confirmed for lead trigger sequence
Double minor allele selectivity and activity in reducing both mRNA and protein levels were confirmed in vivo. Testing lead compound
Over the last several years, meta-analyses of PNPLA3I148M carriers versus noncarriers repeatedly conclude that carriers of the variant allele are at increased risk for all stages of NAFLD, including NASH, cirrhosis, and HCC [6,7,65,72–75], among the pediatric and adolescent population [2,76], and among lean individuals with NAFLD [77]. NASH is emerging as the leading cause of liver transplant in the developed world. Currently, there are no approved therapeutic options for NASH, and treatment is largely limited to ineffective lifestyle modifications and management of comorbidities. Hence, there is a highly unmet medical need for safe and effective therapies specifically designed for the treatment of this serious, potentially life-threatening condition.
Our progress toward the identification of a potent and durable minor-allele specific siRNA for the genetically validated NAFLD target, PNPLA3I148M, exemplifies an attractive precision-medicine approach to treat a disease driven by aberrant expression of a mutant protein. Selectively knocking down the mutant allele, while maintaining the wild type allele, whose function remains uncertain, may provide meaningful benefit with less risk or harm to not only homozygous carriers but also the vast PNPLA3I148M heterozygous NAFLD patient population around the world.
Data and Material Availability
All data are available in the main text.
Footnotes
Acknowledgments
The authors thank Amgen Research for supporting this work, Melissa Thomas for overseeing development of the CHO cell lines, Christopher Hale for RNA FISH imaging and analysis support, Hans Meisen and Huiren Zhao for AAV support, Kimberly Ly for in vivo study support, Eric Zollars, Lawrence Kong, and Jason Legg for comprehensive article review, and Simon Jackson, Les Miranda, and Saptarsi Haldar for meaningful discussion and program support.
Author Disclosure Statement
M.O. is currently employed by Janssen Biopharma.
Funding Information
This work was supported by Amgen; no additional funding sources to report.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.