
Abstract
The development process of antisense oligonucleotides (ASOs) as therapeutic agents in humans has advanced through the implementation of chemical compound modifications as well as increasingly sophisticated toxicological preclinical screening techniques. The Ionis Integrated Safety Database was utilized to determine if advances in ASO screening and clinical lead identification methods have improved the tolerability profiles of 2′-O-methoxyethyl (2′MOE)-modified ASOs as a class, relative to the first 2′MOE ASO approved for use in humans, mipomersen. Tolerability was assessed by the incidence and percentage of subcutaneous doses leading to adverse events at the injection site or flu-like reactions (FLRs), as well as by the incidence of dose discontinuations due to these events. In randomized placebo-controlled phase 1 and phase 2 trials, the incidence of each measure of tolerability was lower in the test group of 12 ASOs (713 ASO-treated subjects) compared with the reference, mipomersen (266 ASO-treated subjects); with the most marked reduction in the incidence of FLRs (0.6% vs. 9.4%). A similar reduction in the incidence of dose discontinuation due to FLRs was also observed (0.2% vs. 0.9%). When compared with mipomersen, 8 of 12 ASOs showed significant improvements in their respective mean percentage of doses leading to adverse events at the injection site, whereas 7 ASOs showed a significant improvement in mean percentage of doses leading to FLRs. These results support an overall improvement in the tolerability profile in 2′MOE ASOs that entered development after mipomersen, in parallel with advances in the drug discovery screening process as well as the gains in clinical experience during development of each ASO.
Introduction
Antisense oligonucleotides (ASOs) are single-stranded, synthetic oligonucleotides. which selectively bind to messenger ribonucleic acid (mRNA) to modify or reduce protein expression. A growing number of ASOs are now approved for use as antisense medicines [1–3]. The 2′-O-methoxyethyl (2′MOE)-modified chimeric ASOs are a chemical class of similar biochemical and biophysical properties, differing only in nucleotide sequence that have been extensively investigated in both the preclinical and clinical settings. Data collected from these investigations have been integrated and harmonized to systematically assess the safety and tolerability profiles as a class. In previous publications, we have utilized this integrated safety database to examine the overall safety profile in healthy volunteers, effects on platelet count, and effects on renal function of 2′MOE ASOs in completed human clinical trials [4–6].
Mipomersen is the first 2′MOE ASO approved by the United States Food and Drug Administration in 2013 as an orphan drug for the treatment of homozygous familial hypercholesterolemia [7,8]. The most common adverse events reported with mipomersen treatment were injection site reactions (ISRs) and constitutional symptoms characterized by influenza like illness and related symptoms, for example, chills and myalgia. In a limited number of cases, these adverse events have been the basis for discontinuation of treatment.
Local injection site and systemic flu-like reactions (FLRs) are both considered attributable to sequence and/or chemistry-dependent proinflammatory properties of oligonucleotides [9–14]. Since the development of mipomersen, advances in screening for sequence-dependent proinflammatory effects have been incorporated into the preclinical ASO lead identification process of oligonucleotide development and further refined (Fig. 1). This improved screening process includes algorithms and methods created to filter out sequences, which have a higher propensity to hybridize to nontarget RNA or contain sequences known to cause an inflammatory response, such as unmethylated CpG motifs [15]. More recently, due to inherent limitations of preclinical animal models to predict certain responses in the clinic [16], an in vitro inflammation assay has been incorporated into the process of development candidate selection [17].
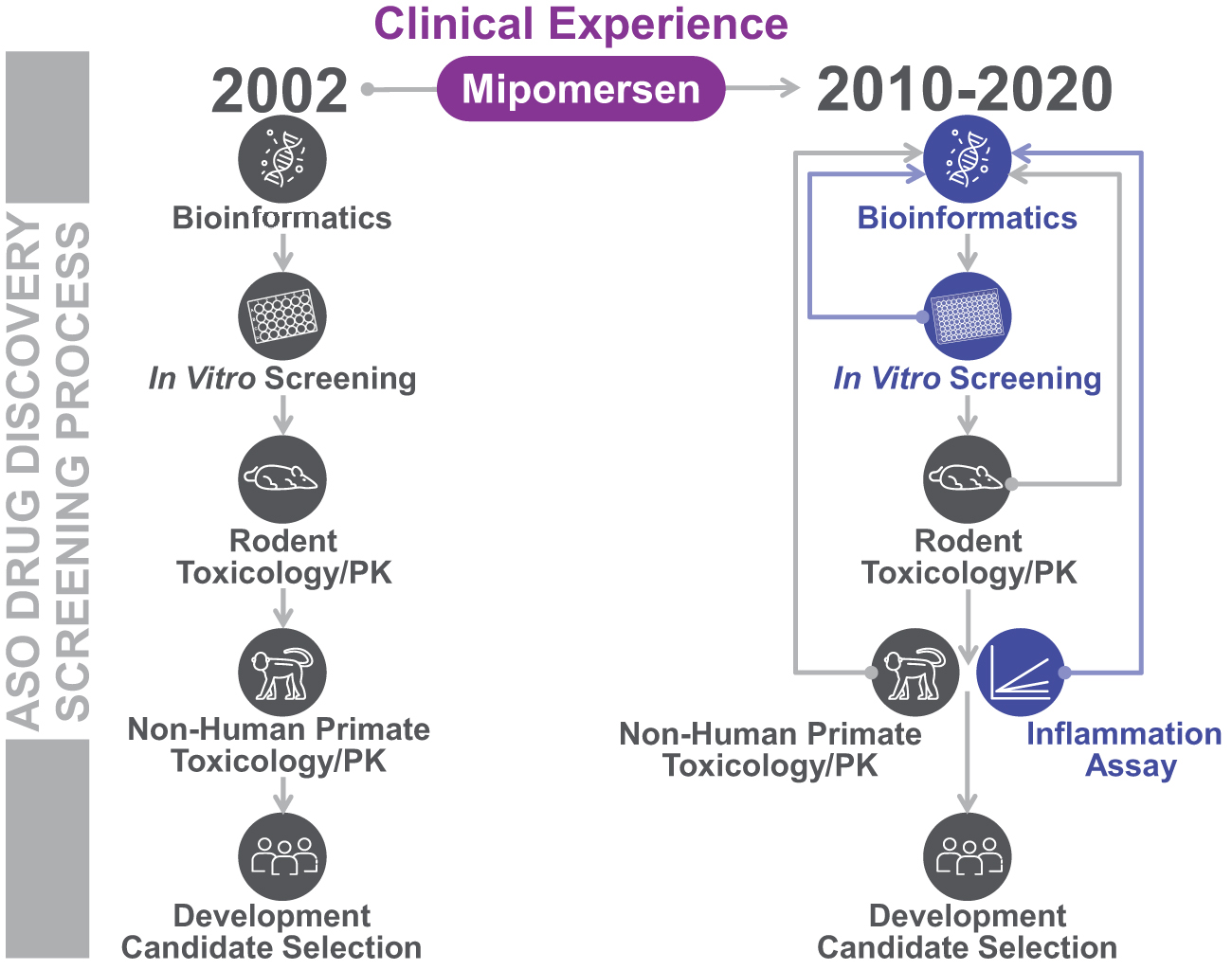
Advancement of ASO drug discovery and drug development lead identification process over time. Key advancements in steps of the antisense drug discovery and lead identification process over the last decade (2010–2020) promoted in part by the clinical experience from mipomersen trials are indicated in blue. ASO, antisense oligonucleotide.
The current investigation of integrated data explores the clinical experience of subjects who participated in early stage clinical trials of 2′MOE-modified ASOs that were identified and selected for drug development after mipomersen. The ASO drugs considered in this investigation were developed for a variety of disease indications and conditions. Thus, the principal assessment in this investigation was limited to data from phase 1 and phase 2 placebo-controlled trials, for which there was no presumption of therapeutic effect, and the tolerability profile of each compound was considered as independent of its efficacy. This limitation also minimized variability in adverse event reporting in subjects receiving self-administered doses, as well as any differences in reporting conventions that occurred across the late-stage trials.
Local adverse events at the subcutaneous (SC) injection site and systemic constitutional events, previously identified and characterized in clinical investigations of mipomersen, were used as the measures of tolerability for this integrated assessment. Of principle interest was assessment of the tolerability profile of ASO drugs that advanced through development based on the insight gained from development of mipomersen. Only subjects who were administered study drug by SC injection and assigned to multiple dose cohorts were considered. The primary analysis was a comparison of incidence and percentage of injections of these measures of tolerability in the reference group (mipomersen) and the test group (12 ASOs). Additional analyses included the incidence of events by severity, a subgroup analysis by trial phase, characterization of adverse events over time, and co-occurrence of these adverse events, respectively, and the incidence of dose discontinuation due to these events as a confirmatory measure of tolerability. It was assumed in this assessment that fewer local injection site and systemic constitutional adverse events per SC dose implied an improvement in drug tolerability.
Materials and Methods
Clinical data
Data collected from 31 completed, randomized, placebo-controlled trials were analyzed to assess the differences in clinical tolerability between mipomersen and 12 subsequent ASO development compounds (Supplementary Table S1). Among the 31 trials (Supplementary Table S2), there were 14 phase 1 trials in healthy volunteers and 17 phase 2 trials in subjects who exhibited mild symptoms of the condition, which the study drug intended to treat. Data from 7 phase 3 trials were utilized to confirm results from the assessments of the phase 1 and 2 data. Evaluated subjects were assigned to multiple-dose cohorts (vs. single-dose cohorts) and received at least one dose of study drug.
Clinical trial protocols were approved by the respective Institutional Review Boards, or independent Ethics Committees. All studies complied with the guidelines of the Declaration of Helsinki and the International Conference on Harmonization Guidelines on Good Clinical Practice. Written informed consent was obtained from all subjects before participation in the trial.
Measures of tolerability
Specific groups of adverse effects have been observed throughout development of antisense therapies and categorized using standard MedDRA queries under precise event definitions. Three such definitions were utilized in the current assessment of ASO tolerability; “injection site reactions,” “local cutaneous reactions at the injection site” (LCRIS), and “flu-like reactions.”
ISR events were defined as events belonging to the System Organ Class “General disorders and administration site conditions” with the MedDRA Preferred Term starting with “Injection site” that started the day of SC injection and persisted (start to stop) for 2 days or more. Events with onset date on the day of injection and missing resolution date were also included.
LCRIS events were defined by the presence of injection site erythema, injection site swelling, injection site pruritus, or injection site pain, which started the day of SC injection and persisted (start to stop) for 2 days or more. Events with onset date on the day of injection and missing resolution date were also included. The LCRIS definition was originally developed to characterize ISRs observed from SC administration of mipomersen. In support of the current assessment, we confirmed that it remained reflective of the clinical experience of subjects exposed to ASOs developed post-mipomersen, utilizing a permutation test for pairwise co-occurrence (see Supplementary Table S3).
FLR events were defined by the MedDRA Preferred Terms of either: (1) influenza like illness starting on day of injection or the next day; or (2) pyrexia or feeling hot or body temperature increased starting on day of injection or the next day, plus at least two of the following terms: chills, myalgia, or arthralgia starting on day of injection or the next day.
ASO tolerability was summarized by the incidence and mean percentage of doses leading to each of the three defined measures of tolerability (ISR, LCRIS, and FLR) across all subjects assigned to multiple dose cohorts who received at least one dose of study drug by SC injection, and by the incidence of discontinuation of treatment due to these types of adverse events. The tolerability profile of individual test group ASOs relative to mipomersen was determined using the mean percentage of doses leading to ISR, LCRIS, and FLR events.
Additional analyses included characterization of (1) the severity of events by the incidence of worst ISR and FLR event, (2) the incidence, severity, and duration of ISR events over time by dose number, and (3) the percentage of injections leading to ISR and LCRIS events in phase 1 and phase 2 data separately to identify any differences in tolerability between healthy volunteers in phase 1 and subjects with a mild or stable state of the planned indication in phase 2.
Statistics
The mean percentage of doses leading to each of the three adverse event reactions was utilized to compare the tolerability of the test group to the mipomersen reference group. The percentage of doses leading to an adverse event reaction was defined as the number of doses leading to the given reaction type (ISR, LCRIS, or FLR) divided by the total number of doses received by the subject.
Individual ASO tolerability was categorized as improved, worsened, or inconclusive relative to mipomersen by testing the mean percentages of doses leading to each of the three reactions for each of the 12 ASOs against the corresponding set of percentages in mipomersen using a two-sided Wilcoxon rank-sum test for independent groups. Characterization of ISR events by dose number was evaluated as a time series using the Mann–Kendall Test for monotonic trends. This nonparametric test categorizes a given time series as increasing, decreasing, or having no trend. In addition to the three adverse reactions, the dimension of adverse event co-occurrence was also analyzed. A Monte Carlo permutation test was used to differentiate which adverse events co-occurred nonrandomly within a given ASO or class of ASOs. This test established a P value for a given prespecified adverse event pair (such as injection site erythema and injection site swelling) by permuting and resampling all observed co-occurring adverse events under specific conditions [18]. All statistical tests were performed at a 5% significance level. Data from placebo-control subjects were excluded from the comparative statistical analyses.
Results
Characteristics of study groups
Demographics and baseline characteristics of the mipomersen reference and test groups are shown in Table 1. The mipomersen group comprised 353 subjects (87 placebo, 266 ASO) with 64% male, and a median age of 54. The test group comprised 1010 subjects (297 placebo, 713 ASO) with 56% male, and a median age of 53. Age was relatively balanced between groups, with similar mean and median ages between the mipomersen reference and test groups while body mass index (BMI) was found to differ with significance between the two groups (Supplementary Fig. S1). The test group exhibited a higher mean and median BMI, however, both the mean and median BMI in each study group were within the BMI range of 25.0–29.9, or overweight [19].
Study Population Characteristics
Q1, lower quartile; Q3, upper quartile.
ASO, antisense oligonucleotide; BMI, body mass index; CI, confidence interval; SD, standard deviation.
The characteristics of treatment and drug exposure were compared between the mipomersen reference and test groups. There were no significant differences between the ASO-exposed subjects in the mipomersen reference and test groups in number of doses received and weekly exposure (Table 2). The two groups, however, showed significant differences in the amount per dose, treatment duration, and total exposure (Supplementary Fig. S2). Although the median amount of study drug per dose was equal between groups at 200 mg, the mean amount per dose was lower in the mipomersen group (178.4 mg vs. 202.5 mg). The mipomersen reference group also had a shorter mean duration of treatment and lower mean total exposure when compared with the test group. Although the mipomersen reference and test groups differed in these three parameters, the percentage of doses per subject leading to a given tolerability event was not skewed or influenced by increased exposure.
Summary of Antisense Oligonucleotide Exposure by Study and Treatment Group
Q1, lower quartile; Q3, upper quartile.
Incidence and percentage of doses leading to adverse events
The incidence and mean percentage of doses leading to ISR, LCRIS, and FLR events were significantly lower in the test group, compared with mipomersen (Table 3 and Fig. 2). In the test group, a 36.1% reduction in incidence of ISR events, as well as a 39.5% reduction in mean percentage of ASO doses leading to an ISR event was observed. In the case of LCRIS events, a 37.2% reduction in incidence, as well as a 40.8% reduction in mean percentage of ASO doses leading to an LCRIS event was observed in the test group. The incidence and mean percentage of doses being lower for LCRIS since, by definition, this measurement is a subset of ISR terms. For FLR events, a 93.6% reduction in incidence, as well as a 91.5% reduction in mean percentage of ASO doses leading to an FLR event was observed. Notably, for each measure of tolerability, a higher proportion of subjects in the test group experienced no events compared with the mipomersen reference group (ISR, 54.8% vs. 29.3%; LCRIS, 62.0% vs. 39.5%; FLR, 99.4% vs. 90.6%, incidence of no events, respectively).
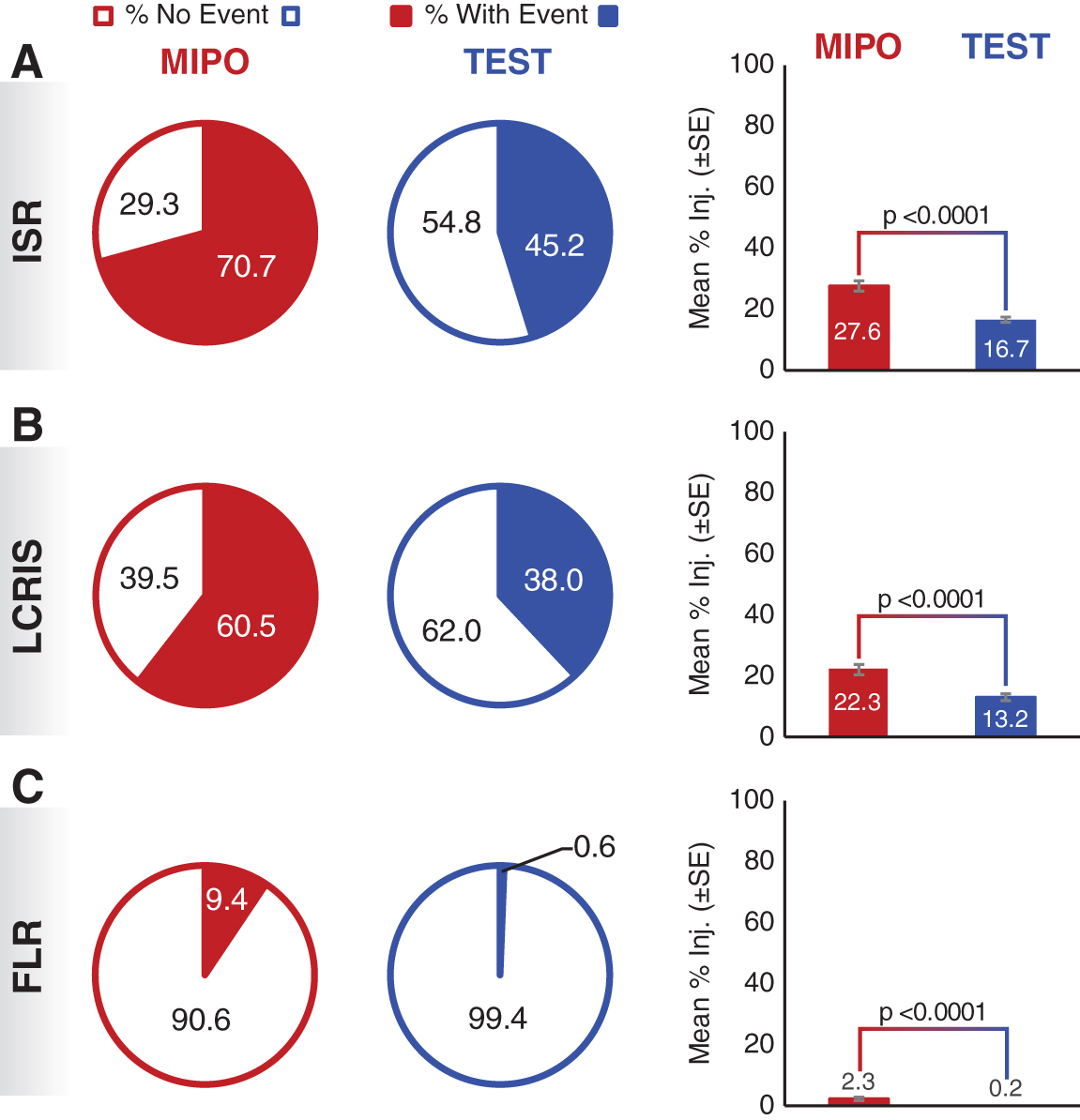
Incidence and mean percentage injections leading to an adverse event reaction.
Incidence and Percentage of Antisense Oligonucleotide Doses Leading to a Tolerability Event
Q1, lower quartile; Q3, upper quartile.
P values were determined using the chi-square test (ISR, LCRIS) and Fisher's exact test (FLR) for comparison of the incidence in the ASO-treated mipomersen reference and test groups.
P values were determined using the Wilcoxon rank-sum test for comparison of the percentage of doses leading to an event in the ASO-treated mipomersen reference and test groups.
FLR, flu-like reaction; ISR, injection site reaction; LCRIS, local cutaneous reactions at the injection site.
The incidence of individual adverse event terms is shown in Table 4. Injection site erythema had the highest incidence of the ISR terms in both the mipomersen reference (55.3% ASO, vs. 2.3% placebo) and test group (27.5% ASO, vs. 0.3% placebo). FLRs were reported mostly as influenza-like illness with an incidence of 8.6% and 0.3% in the ASO-treated mipomersen and test groups, respectively.
Incidence of Injection Site Reaction and Flu-Like Reactions by MedDRA Preferred Term
ISR terms, incidence >10% in ASO-treated test group.
Severity of events
The incidence by severity for ISR and FLR events is shown in Fig. 3. The incidence of mild ISRs was 41% lower in the test group than in the reference group (63.9% vs. 37.7%), whereas the incidence of moderate ISRs was 51% higher in the test group than in the mipomersen reference group (4.9% vs. 7.4%). This increase in the incidence of moderate ISRs in the test group is attributable to the single ASO that was found to be significantly worse for both LCRIS and ISR events when compared with the mipomersen reference group. Exclusion of this ASO from the dataset resulted in an equivalent 4.9% incidence in the test group of subjects who experienced an ISR event that was moderate in severity. Severe ISRs and ISRs with missing severity data were only present in the mipomersen reference group with an incidence of 0.4% and 1.5%, respectively. The incidence of mild FLRs was 94% lower in the test group than in the mipomersen reference group (4.9% vs. 0.3%). The incidence of moderate FLRs was 93% lower in the test group than in the mipomersen reference group (4.1% vs. 0.3%). FLR events with missing severity data were only present in the mipomersen reference group with an incidence of 0.4%.
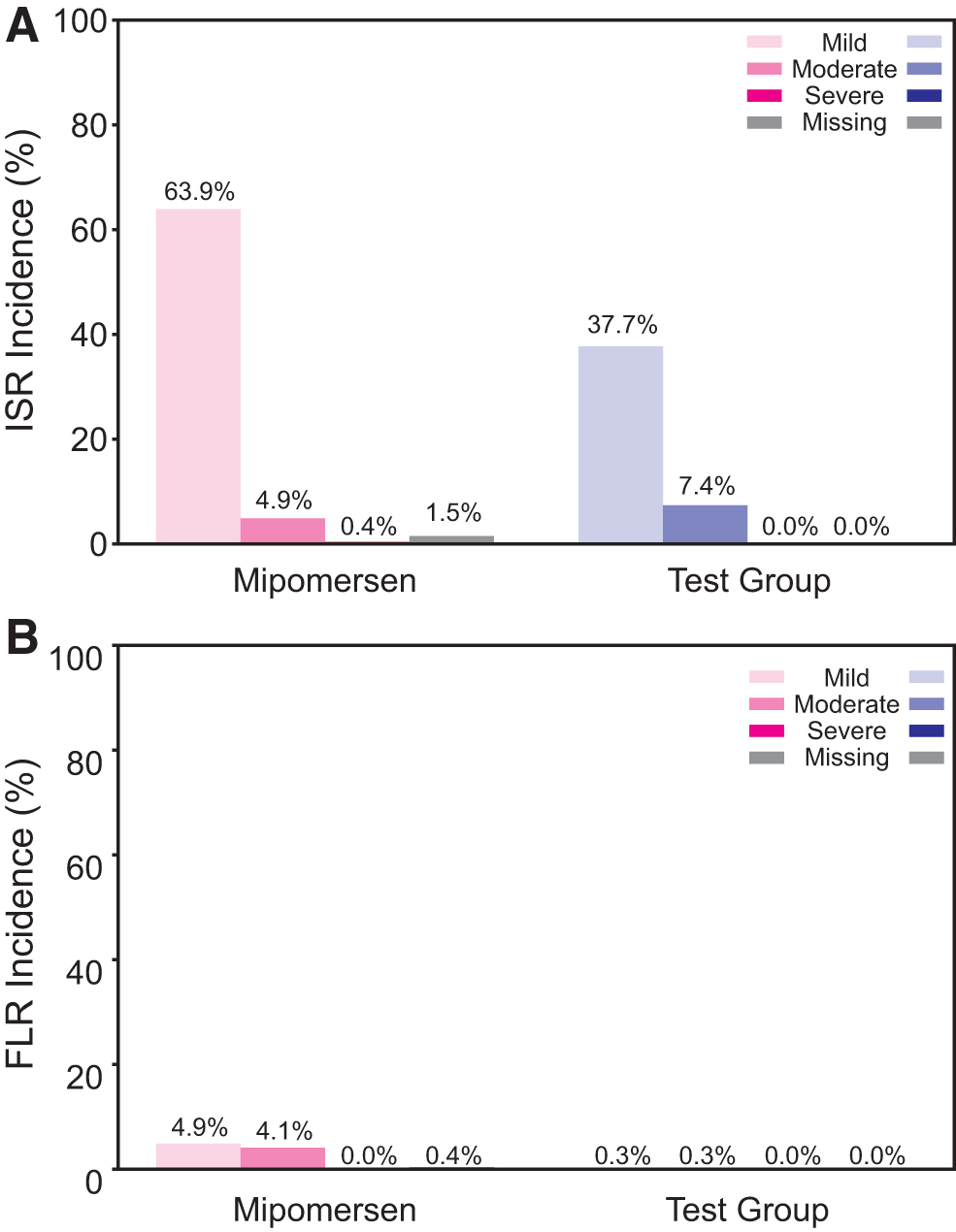
Incidence of
Duration of adverse events at the injection site
The duration of ISR and LCRIS events was analyzed under the assumption that longer durations are less tolerable. The median durations for the most common type of ISR and LCRIS event, injection site erythema, were 8 and 5 days for mipomersen and the test group, respectively. The difference in duration between the two ASO groups was significant, with a 37.5% reduction in the test group relative to the mipomersen reference group. Statistical differences in median duration for the remaining ISR and LCRIS adverse event types were found to be insignificant.
Characterization of events over time
The incidence, severity, and duration of ISRs in ASO-treated subjects who completed treatment were evaluated over time by dose number. The incidence of ISRs decreased with increasing dose number in both the mipomersen group and the test group (Supplementary Fig. S3). At the individual sequence level, including mipomersen, the ISR incidence exhibited a decreasing trend in 6 of 13 compounds, and no trend in 7 of 13 compounds with increasing dose number. The incidence of moderate and severe ISR events also exhibited a decreasing trend in 3 of 13 compounds, an increasing trend in 1 compound, and no trend in 9 of 13 compounds with increasing dose number (Supplementary Fig. S4). Testing of the sequential duration of ISR symptoms yielded no evidence of increasing trends of median event duration from one ISR event to the next in both the mipomersen reference and test groups (Supplementary Fig. S5).
Subgroup analysis of phase 1 and phase 2 data
The mean percentages of doses leading to each type of adverse event was also assessed by phase 1 and phase 2 trial data separately. As observed in the pooled analysis, the test group subjects showed an improved mean percentage of doses leading to ISR and LCRIS events with statistical significance compared with mipomersen in both phase 1 and phase 2. In the case of FLR events, which are less common than ISR or LCRIS events, inconclusive evidence was found when comparing phase 1 test group data to mipomersen reference data. No FLR events were experienced by test group subjects from phase 2 trials, which can be said to have significantly improved when compared with phase 2 mipomersen data.
We also investigated the percentage of injections leading to an LCRIS event in the subset of subjects who experienced an event (Fig. 4). Nonparametric testing found a significant difference between the mipomersen and test groups in phase 2 (P < 0.001). This difference may reflect improvements in injection method and technique stemming from gains in clinical experience during development of ASO drugs and other biologicals.
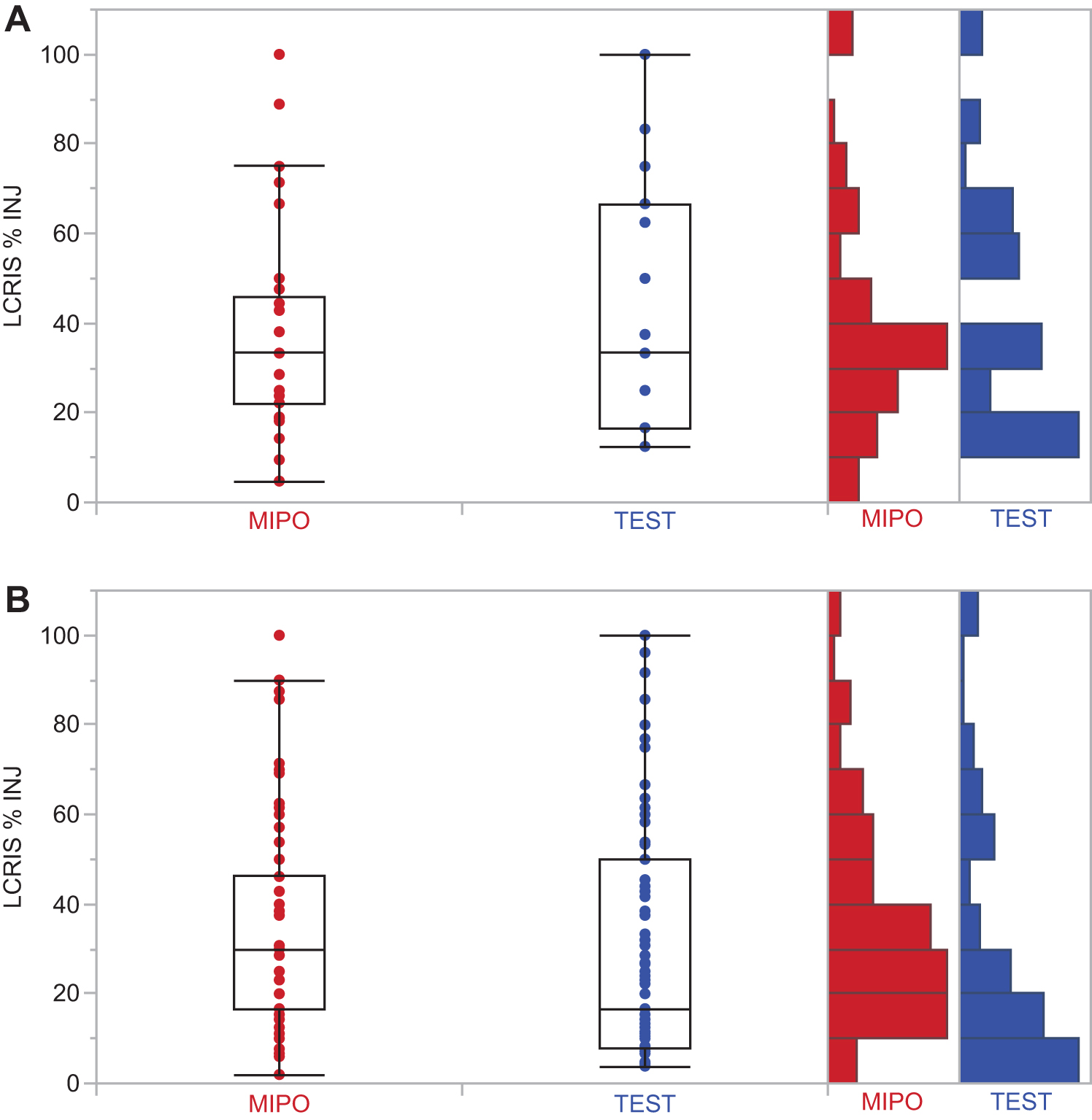
Significant reduction in percentage injections leading to LCRIS in phase 2 trials of test group. Quantile plots of mipomersen and test group subjects who experienced at least one LCRIS event in
Discontinuation of treatment due to adverse events
The incidence of dose discontinuation due to adverse events is an industry standard with an unambiguous endpoint for measuring the tolerability of an investigational drug in the clinic. Initial assessment of the incidence of discontinuation of dosing due to constitutional symptoms found insufficient data for comparison of the reference to the test group. To address this limitation, we included phase 3 data to increase the sample size (Supplementary Tables S4 and S5). Including data from phase 3 trials, 16 of 527 (3.0%) mipomersen-treated subjects discontinued dosing due to adverse events at the injection site, and 23 of 933 (2.5%) subjects in the test group discontinued treatment due to these events (Table 5). Five subjects (0.9%) from mipomersen trials discontinued dosing due to FLR events, compared with two subjects (0.2%) in the test group.
Incidence of Subject Dose Discontinuations Due to Tolerability Events
Results shown include data from three phase 3 trials.
Reporting terms with incidence 0.5% or greater in any treatment group. For dose discontinuations by all reported reason, see Supplementary Table S6.
Tolerability profile of individual test group ASOs in relation to mipomersen
The 12 ASO drugs within the test group were individually categorized for each tolerability event type as having an improved, worsened, or inconclusive change to its tolerability profile when compared with mipomersen. This categorization was based on a comparison of their mean percentage of doses leading to the given event type and whether this set of percentages differed with statistical significance from that of the mipomersen reference group. Those drugs which had a lower mean percentage of SC doses leading to the given event and differed from mipomersen with significance were said to have an improved tolerability profile (Table 6).
Comparison of Individual Antisense Oligonucleotides in Test Group to Mipomersen by the Mean Percentage of Doses Leading to an Adverse Event Reaction
Eleven of 12 (92%) ASOs in the test group showed a reduction in the mean percentage of doses leading to ISR events, and 10 of 12 (83.3%) showed a reduction for LCRIS events compared with the reference, mipomersen. Eight ASOs were categorized as having an improved tolerability profile as measured by ISRs and LCRIS compared with mipomersen. A direct correlation in rank order of ASOs was found between phase 1 and phase 2 trial data in the mean percentage of doses for both ISR and LCRIS events (r = 0.697, P < 0.05). These results indicate a similar tolerability profile for an ASO from phase 1 in healthy volunteers to phase 2 in subjects with early or controlled clinical signs of the planned indication.
Eleven of 12 (92%) ASOs in the test group had a lower mean percentage of doses leading to FLR events compared with mipomersen. Only 2 of 12 had FLR events. Neither of these two ASOs were found to differ significantly from mipomersen in percentage of doses leading to FLRs. Of the remaining 10 ASOs for which no FLR events were observed, 7 were categorized as having an improved tolerability profile with respect to FLR events.
Discussion
The advancement of antisense drug discovery and screening methods to identify and select ASOs for clinical development has resulted in significant improvements in the clinical tolerability profile of 2′MOE ASOs as measured by the incidence and mean percentage of doses resulting in local adverse events at the SC injection site and postinjection constitutional symptoms. Notably, the test group exhibited an approximate twofold increase in the number of subjects who did not experience an ISR or LCRIS event. The test group also showed an overall reduction in the duration of events as determined by injection site erythema, the most common injection site event in both the reference and test groups. A reduced incidence and significant reduction in mean percentage of doses leading to an FLR was also found in this assessment of tolerability. Although the occurrence of FLRs were relatively low, these results were supported by a reduction in the incidence of dose discontinuations due to the related adverse events.
A body of evidence indicates that the proinflammatory nature of ASOs is partially dependent on sequence [12,13,20]. Looking at each sequence in our test group, we found a lower mean percentage of SC doses leading to both local and systemic proinflammatory responses in the vast majority of ASOs (11 of 12) relative to mipomersen. Further to this result, more than half of ASOs in the test group (7 to 8 of 12) were individually categorized as an improved profile across the three measures of tolerability. Collectively, these results indicate a certain degree of progress in eliminating or mitigating sequence-dependent proinflammatory effects.
The LCRIS definition was created to distinguish the ISRs specific to this chemical class of ASOs from nonspecific responses to tissue injury at the SC injection site, based on experience with mipomersen. For this purpose, persistence of the characteristic symptoms for at least 2 days was considered a critical component for separation of the ASO-specific effects. The fact that the same LCRIS term pairs were found to significantly co-occur in both the mipomersen and test groups (ie, injection site erythema was found to co-occur with injection site pruritus in both groups) supported the effectiveness of the LCRIS definition in characterizing injection site-related adverse events in this class of compounds. In the current assessment, we also found that the same eight ASOs exhibited improved tolerability with respect to both LCRIS and ISR events, indirectly indicating that the four LCRIS-associated preferred terms are sufficient in characterizing the ISRs experienced by subjects in the test group. This validation of the LCRIS definition supports the current tolerability assessment as well as provides credence for comparisons with previous reports on individual 2′MOE ASO clinical trial results.
Implementation of additional and improved preclinical screening steps appears to yield better-tolerated 2′MOE ASO drugs in the clinic. This preclinical screening paradigm is applicable across ASO-target genes, irrespective of the RNA target or therapeutic indication [21], and may provide utility across all classes of oligonucleotide therapies. ISRs, for example, are not limited to the ASO design in the current assessment, and occur at higher incidences in other compounds such as drisapersen [22], a subcutaneously administered phosphorothioate 2′-O-methyl-modified oligonucleotide.
Other factors that may have contributed to an improved tolerability profile, and particularly in relation to administration of 2′MOE-modified ASOs by SC injection, are likely found in injection method and technique [8,23–25]. Such methods include rotating locations of SC injection sites, splitting amount of study drug injected per dose by administration in two injections instead of one, reducing the needle size and/or length, and implementing other mitigation steps such as icing.
Although the statistical tests used for evaluation of tolerability controlled for dissimilar sample sizes across clinical trials, this assessment was limited by sample size and for FLR a low occurrence of events. Due to the low incidence of FLR events, ASOs in the test group, which caused no FLR events, could not be categorized as improved or worsened with statistical significance. Differences in drug exposure, such as the difference in the amount per dose received between ASO-exposed subjects in the mipomersen reference and test groups, may have also limited the comparability of these groups. Past studies have identified these events as dose dependent [26], indicating that the test group, which received a higher mean amount per dose, may have had a proportionately higher likelihood to experience an event. Finally, improvements in screening and lead identification over time, as well as efforts to mitigate adverse events associated with tolerability to study drug, likely imparted a degree of variability in the clinical response observed for these ASO drugs.
In conclusion, the improvements made to the tolerability profile of this set of 2′MOE ASOs indicate the effectiveness of improvements made in Ionis' drug discovery and preclinical screening platform, which continues to evolve, as well as insights gained through clinical development of this class of ASOs. The potency and safety of many of the 2′MOE-modified ASOs included in this analysis have been further improved by conjugation of the triantennary N-acetylgalactosamine (GalNAc3) moiety for preferential and productive uptake by hepatocytes [27–33]. This enhancement in drug delivery increases ASO potency for RNA targets expressed by hepatocytes to support lower and less-frequent dosing, which upon integrated assessments may indicate a marked improvement in the tolerability profile for this emerging class of ASO drugs.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.