
Abstract
Modification of specificity of T cells for the use in adoptive transfer (CAR- or TCR-redirected T cells) has revolutionized the therapy of liquid tumors and some infectious diseases. However, several obstacles are still hampering the efficacy of such potent therapy, hence concurrent modification of the function is also required to obtain successful results. Here we show the use of splice-switching antisense oligonucleotides (SSOs) as a tool to transiently modify T cell function. We demonstrate the possibility to transfect SSOs and an exogenous TCR into primary human T cells in the same electroporation reaction, without affecting viability and function of the transfected T lymphocytes. Moreover, we show that SSOs targeting T cell-specific mRNAs induce the skipping of the targeted exons, and the reduction of the protein and consequent modification of T cell function. This technical work paves the way to the use of SSOs in immune cells, not only for the knockdown of the functional isoform of the targeted proteins, but also for the protein manipulation by elimination of specific domains encoded by targeted exons.
Introduction
Adoptive T cell transfer is a versatile cell therapy modality that has the potential to address critical medical needs from chronic infections to oncology. Through the engineering of effector T cells with specific receptors, in the form of chimeric antigen receptors (CARs) or classical T cell receptors (TCRs), one can direct the lytic action of CD8+ T cells against a specific target. As examples, viral-infected host cells presenting viral antigens have been targeted by CAR/TCR-redirected T cells against human immunodeficiency virus, hepatitis B virus (HBV), hepatitis C virus, human cytomegalovirus, or opportunistic fungal infections [1–8]. Moreover, cancer cells have been targeted using CAR/TCR specific for antigens expressed on the cell of origin (eg, CD19 for B cell leukemia and lymphoma) [9,10] or targeting specific tumor antigens of self or viral origins [11–17].
To unleash the full clinical potential of adoptive T cell therapy beyond liquid tumors, two levels of cell engineering can be exploited—extrinsic and intrinsic. Extrinsic engineering, in the form of synthetic receptors, has been the hallmark of the first-generation CAR/TCR-redirected T cells. The second generation is differentiated by the modification and/or modulation of T cell endogenous factors, to induce functional features of effector T cells that are most propitious for each clinical application. As an example, for antiviral therapy the cytolytic activity and inflammatory induction of redirected T cells need to be cautiously calibrated to avert massive lysis of the infected targeted organs, and/or cytokine release syndrome [18]. On the contrary, in the context of solid tumors, first-generation adoptive CAR/TCR-redirected T cells are not equipped to bypass inhibitory factors in the tumor microenvironment or to specifically reach the target in specific organs. For this reason, efforts are ongoing to engineer TCR/CAR-redirected T cells with improved trafficking, secreting immune checkpoint inhibitors or stimulatory cytokines [19–23].
The molecular tools used in the intrinsic engineering of T cells for adoptive transfer include gene knock-in with a lenti/retroviral vector, gene knockout using TALEN or CRISPR/Cas9 technologies, and suppression of gene expression with siRNAs or GAPmers [24–27].
Here we propose and demonstrate the feasibility and flexibility of splice-switching antisense oligonucleotides (SSOs) for the intrinsic modification of T cell functions and to generate what we define armored redirected T cells (ART cells). An SSO modulates target transcript splicing by denying access of specific RNA binding proteins (RNA-BPs) to their splice-regulatory motifs through steric hindrance. Synthesized as a short single-stranded ribonucleic acid whose bases and backbones are chemically modified, the SSO is directed to bind complementarily to a pre-mRNA target sequence containing splicing silencers or enhancers motifs [28,29]. We and others have demonstrated the flexibility of SSO application in the suppression of transcript abundance by inducing nonsense-mediated decay [30], correction of aberrant and mis-splicing events [31–37], and selection of alternate splicing [38–41].
Specifically, we designed and validated novel SSOs modulating the splicing of three T cell-specific genes, namely interferon-γ (IFN-γ), perforin (PRF), and granzyme B (GZMB), for the targeted intrinsic engineering of cytotoxicity and cytokine production of primary human T cells. We demonstrate that SSOs can be efficiently transfected into primary human T cells concurrently with a synthetic TCR mRNA to create ART cells. This work paves the way for the development of a wider array of SSOs modulating T cell-relevant genes in different ways, which are key to improve adoptive T cell immunotherapy.
Materials and Methods
Institutional review board
Approval: IRB No. H-17-023E issued by National University of Singapore (NUS). All patients gave written informed consent.
Ficoll-Paque blood separation
Peripheral blood mononuclear cells (PBMCs) from healthy donors were obtained from full blood using Ficoll-Paque (GE Health Care, Chicago, IL) centrifugation. Ficoll-Paque (10 mL) was placed at the bottom of a 50 mL Falcon tube and blood was slowly layered above. After being centrifuged (600 g for 30 min at 18°C, no brakes), a layer of PBMCs would be visible and collected for further experiments.
T cell activation and expansion from PBMCs
Frozen PBMCs were thawed adding 14 mL of warm Hanks' balanced salt solution (HBSS; ThermoFisher Scientific, Waltham, MA) in a dropwise manner. After wash (427 RCF, 5 min, room temperature [RT]), PBMCs were cultured in AIM-V medium (ThermoFisher Scientific) supplemented with human AB serum (Sigma-Aldrich, St. Louis, MO) at a concentration of 1.5–2 × 106 cells/mL. T cells were activated adding 50 ng/mL of anti-CD3 (eBioscience, San Diego, CA) and 600 IU/mL of recombinant human interleukin-2 (rhIL-2; Miltenyi Biotec, Bergisch Gladbach, Germany).
Resting T cells were separated from PBMCs using the pan T cell isolation kit (Miltenyi Biotec) 1 day after thawing. Resting T cells were cultured in AIM-V supplemented with 2% AB serum and 100 U/mL rhIL-2.
HBV-specific TCR mRNA production
We derived the TCR construct from a pUC57-s183cys b2Aa vector that we had previously made, and subcloned it into the pVAX1 vector [42]. The plasmid was propagated and purified from Escherichia coli using the One Shot Top10 E. coli kit (ThermoFisher Scientific), purified using QIAGEN EndoFree Plasmid Maxi Kit (Qiagen, Hilden, Germany), and linearized using the XbaI restriction enzyme (New England Biolabs, Ipswich, MA). The linearized DNA was used to produce the TCR mRNA using the mMESSAGE mMACHINE T7 Ultra kit (ThermoFisher Scientific) following the manufacturer's instructions.
Splicing-modifying antisense oligonucleotides
SSOs were synthesized by Integrated DNA Technologies (Coralville, IA) with a phosphorothioate backbone and 2′-O-methyl ribose modifications in each position. The SSOs were resuspended in water at a final concentration of 1 mM or 500 μM and kept frozen at −20°C. The SSOs were added in the electroporation mix together with the T cells (and the TCR mRNA, eventually) in electroporation buffer at the desired concentration.
Detailed information about the utilized SSOs are given in “Modifying T lymphocytes function with Antisense Oligonucleotides (ASOs) for personalized immune therapy” (PCT/SG2018/050313; 10201705285S (IMC/Z/09724); filing date June 27, 2017; priority date June 27, 2016; licensed to IMMUNOA Pte Ltd on September 14, 2018).
Electroporation
T cells were transfected using electroporation method using the 4DNucleofector™ System (Lonza, Basel, Switzerland). T cells were washed twice with phosphate-buffered saline (PBS) and electroporated using the P3 Primary Cell 4D-Nucleofector® X kit following the manufacturer's instructions with a customized electroporation program. Electroporated T cells were then resuspended in warm AIM-V medium supplemented with 10% AB serum and 100 U/mL rhIL-2.
Usually, 5–10 × 106 cells were electroporated in each reaction. The TCR mRNA was added at 2 μg/106 T cells; SSOs were added at different concentrations ranging from 0.15 to 0.5 femtomoles/T cell.
Cell line culture
HepG2.2.15 were cultured in Dulbecco's modified Eagle's medium (DMEM; ThermoFisher Scientific) supplemented with 10% v/v heat inactivated fetal bovine serum (FBS; ThermoFisher Scientific), 2% v/v penicillin/streptomycin, 1% v/v MeM nonessential amino acids (ThermoFisher Scientific), 1 mM sodium pyruvate (ThermoFisher Scientific), and 200 μg/mL Geneticin reagent (ThermoFisher Scientific) to select for transgene expressing cells. THP-1 cells are cultured in Roswell Park Memorial Institute Medium (RPMI; ThermoFisher Scientific) supplemented with 10% v/v heat inactivated FBS and 1% v/v penicillin/streptomycin.
Real-time cytotoxicity assay
The cytotoxicity assays were performed using the xCELLigence® RTCA DP (ACEA Biosciences, Inc., San Diego, CA) following the manufacturer's specifications. In brief, 105 HepG2.2.15 cells/180 μL were seeded in the specific plate (in their growth medium) and were let to adhere for 24 h. At the time of effector addition, 150 μL of medium were removed and replaced with T cells resuspended in AIM-V 2% AB serum, at a fixed effector:target (E:T) ratio. The acquisition was started at the seeding of the targets and continued for 48 h after T cell addition; the impedance measurements were acquired every 15 min. The Cell Index is a measurement of the impedance measured in each well, and the Normalized Cell Index was obtained by normalizing the Cell Index of each well to the Cell Index at a specific time point (the sweep before T cell addition); the area under the curve (AUC) was obtained using the GraphPad 7 algorithm (San Diego, CA).
Coculture experiments (PD-L1 detection)
Experiments of coculture were performed using TCR-redirected T cells specific for S183–191 peptide of HBV envelope. THP-1 cells were resuspended at 2 M/mL in medium and s183–191 peptides were added at a concentration of 1 μg/mL, at room temperature; after 1 h, the supernatant was removed, and the cells were carefully rinsed twice with warm HBSS. HBV-specific TCR-redirected T cells were cocultured with the targets in AIM-V 2% AB serum for 5 h at different E:T ratios. The supernatants of the coculture were then collected and transferred onto other THP-1 cells for 8 h. The THP-1 cells were then collected and stained for the presence of PD-L1.
Surface and intracellular staining
After culture, cells were collected and washed once (427 RCF, 3 min, 4°C) with PBS in 96-well plated (V-bottomed). Live/dead staining was performed in PBS for 10 min at RT, followed by two washes with cold PBS; MHC-I dextramer staining was performed in staining buffer (SB; PBS supplemented with 1% bovine serum albumin [BSA]; Sigma-Aldrich) and 0.1% sodium azide (Sigma-Aldrich) at RT for 15 min followed by one wash in SB; surface staining was carried out in SB for 30 min on ice, followed by two washes and 20 min of fixing and permeabilization using Cytofix/Cytoperm solution (BD, Franklin Lakes, NJ) on ice; finally, intracellular staining was performed in Permwash buffer (PBS supplemented with 1% BSA, 0.1% sodium azide, and 0.1% saponin; Sigma-Aldrich) for 30 min on ice. After intracellular staining, the sample was washed twice in Permwash buffer, and resuspended in PBS supplemented with 1% formaldehyde for flow cytometry acquisition. The samples were acquired on LSRII (BD), and analyzed using Kaluza (London, United Kingdom) or FlowJo (Ashland, OR) software.
RNA extraction and polymerase chain reaction
RNA extraction was performed using RNeasy Plus Micro kit (Qiagen) following the manufacturer's instructions. After extraction, the RNA was quantified using NanoDrop and was retro-transcribed using the cDNA iScript synthesis kit (BioRad, Hercules, CA). Polymerase chain reaction (PCR) experiments were performed to assess the effect of SSOs in skipping the target exon. Primers flanking the target exon were designed to obtain products of different sizes upon treatment with the specific SSO.
After PCR amplification, the products were run on an agarose gel (2%) and stained with SYBR Safe DNA Gel Stain (ThermoFisher Scientific) according to the manufacturer instructions. The gel was then imaged, and the images were analyzed using ImageJ software (NIH). The Percentage Spliced In was calculated after having analyzed the luminosity of the bands in the gel.
Antibody list
CD3 (Biolegend, San Diego, CA), CD274 (BD), CD8 (BD), GZMB (BD), PRF (Diaclone, Besançon, France), tumor necrosis factor-α (BD), IFN-γ (ThermoFisher Scientific), s183–191 MHC-I dextramer (Immudex, Copenhagen, Denmark), and live/dead fixable stain kit (ThermoFisher Scientific).
Results
TCR mRNA and SSOs can be concurrently electroporated into T cells with no adverse effect
To test our approach of concurrent extrinsic and intrinsic engineering to modulate specificity and function of primary human T cells, respectively, we electroporated in the same reaction an mRNA coding for a cognate-specific TCR and SSO (Fig. 1a).
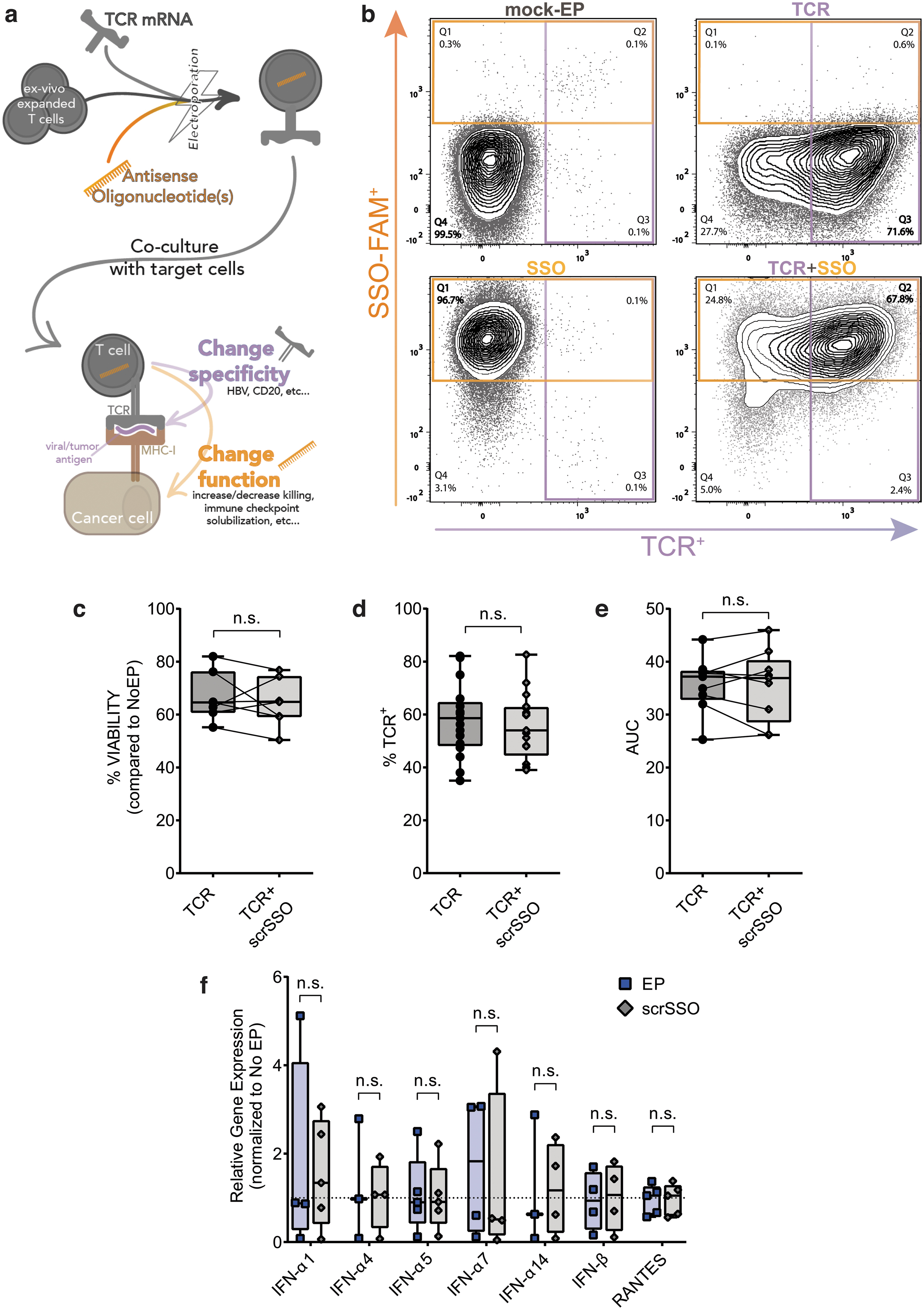
T cell specificity can be transiently modified by electroporating an mRNA encoding for an exogenous TCR. We utilized an mRNA coding for a TCR specific for HBV epitopes restricted by HLA-class I molecule A0201; we had previously demonstrated that these mRNA-electroporated T cells transiently expressed HBV-TCR up to 72 h. The HBV-TCR expressing T cells were observed to lyse HCC cells expressing HBV antigens or inhibit HBV replication both in vitro and in vivo [4,42,43].
To assess the efficiency of SSO delivery in primary human T cells, an FAM-tagged SSO with a scrambled nontargeting sequence (scrSSO) was co-electroporated with the HBV-TCR mRNA [11]. An average of 70% cotransfection efficiency was achieved (Fig. 1b).
Of note, the concurrent transfection of HBV-TCR mRNA (TCR for short) and scrSSO does not impinge on the expected biophysical and biological properties of each other. The kinetics of HBV-TCR transfection (Supplementary Fig. S1a) and scrSSO transfection (Supplementary S1b) were not affected. Of importance, viability (Fig. 1c), TCR protein expression as assessed by MHC multimer staining in flow cytometry (Fig. 1d), and T cell antiviral activity as measured in a 2D killing assay (Fig. 1e) were not affected. The concomitant mRNA TCR and scrSSO electroporation was efficient not just on activated proliferating T cells but also in resting human primary T cells (Supplementary Fig. S1e, f). Moreover, scrSSO does not induce an elevation of TLR-related proinflammatory genes usually upregulated in the presence of naked nucleic acids (Fig. 1f) [44] up to 72 h after electroporation (Supplementary Fig. S1c, d).
Generation of ART cells with reduced IFN-γ secretion capacity
IFN-γ is a proinflammatory cytokine secreted mainly by Th1-type T cells. Besides playing roles in antiviral function [45], IFN-γ can promote activation-induced cell death of T cells [46] and is the main inducer of both PD-L1 and PD-L2 expression [47], and thus participates in inducing an immunosuppressive environment [48,49].
Exon 2 of IFNG codes for part of the “interferon-γ domain” (the specific cytokine domain), and we hypothesized that its exclusion will result in the expression of a shortened IFN-γ protein with attenuated cytokine function. We designed and synthesized an SSO to induce specific IFN-γ exon 2 skipping (Fig. 2a) and transfected through electroporation on activated primary human T cells, at three different concentrations. Figure 2b shows the temporal exon skipping efficiencies after transfection: exon 2 was skipped as early as 6 h after electroporation, and the level of exon skipping, as well as the duration of the effect, are dose dependent (as the skipping is reduced faster with lower doses of SSOs) (Supplementary Fig. S2c). At all the tested IFN-γ SSO concentrations, cell viability and TCR expression in primary human activated T cells were not affected (Supplementary S2a, b) while inducing skipping of exon 2 (Fig. 2c) 24 h after transfection (0.25 femtomoles/T cell). Similarly, viability and TCR expression are not affected by the SSO transfection in resting TCR-redirected T cells (Supplementary Fig. S2d, e) in the presence of IFNG exon 2 exclusion (Supplementary Fig. S2f).
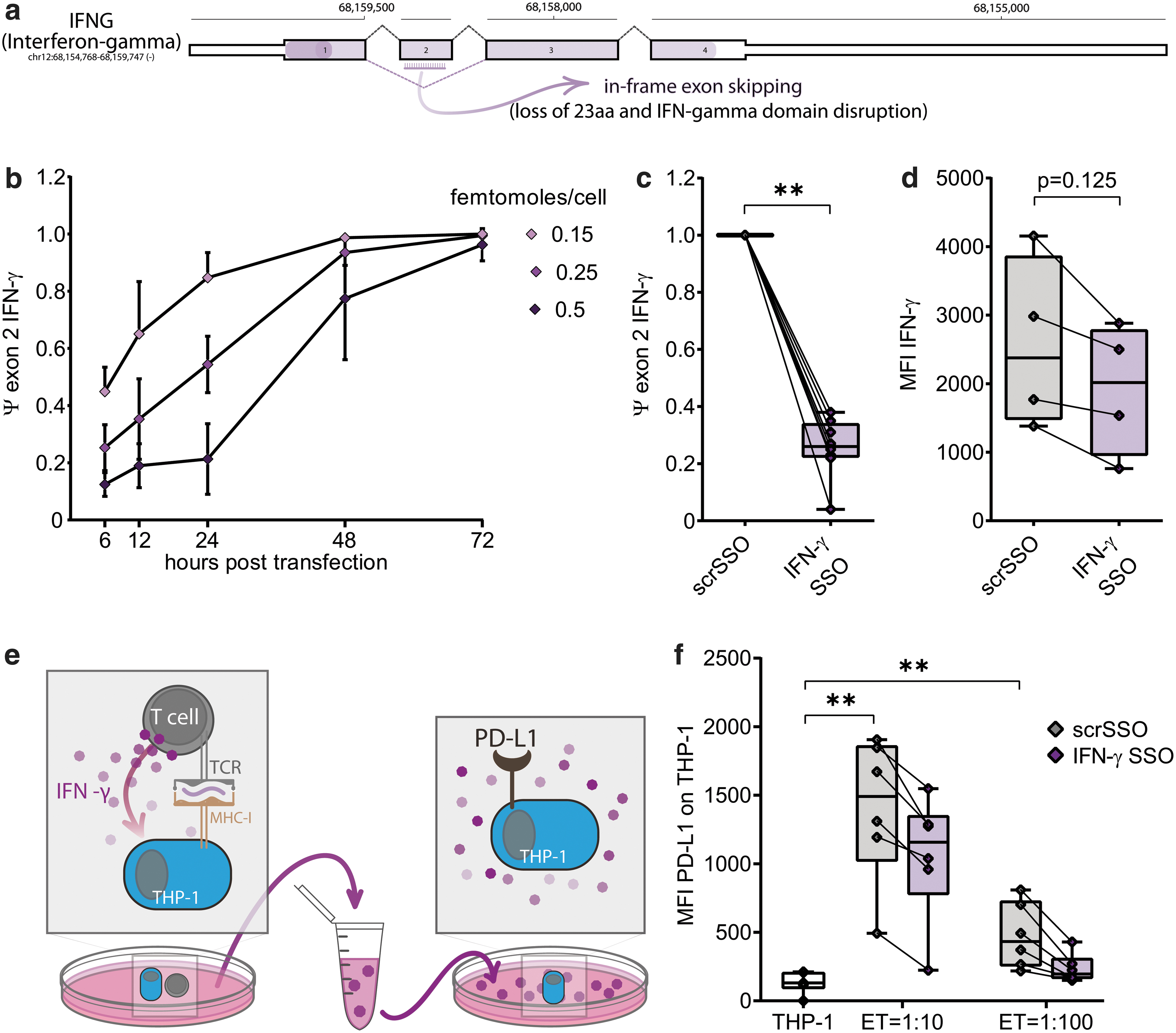
Next, we tested using flow cytometry whether the alteration induced by IFN-γ SSO would lead to a reduction of IFN-γ 24 h after transfection with 0.25 femtomoles/cell (Fig. 2d). We then assessed the ability of the T cells supernatant to induce PD-L1 expression in a monocytic cell line (THP-1) [50]. IFN-γ ART cells resulted in monocytes producing up to 40% less PD-L1 protein when cultured with supernatants derived from activated IFN-γ ART cells (Fig. 2e, f), demonstrating a functional difference compared with the scrSSO controls.
Generation of ART cells with reduced cytotoxic activity: PRF and GZMB
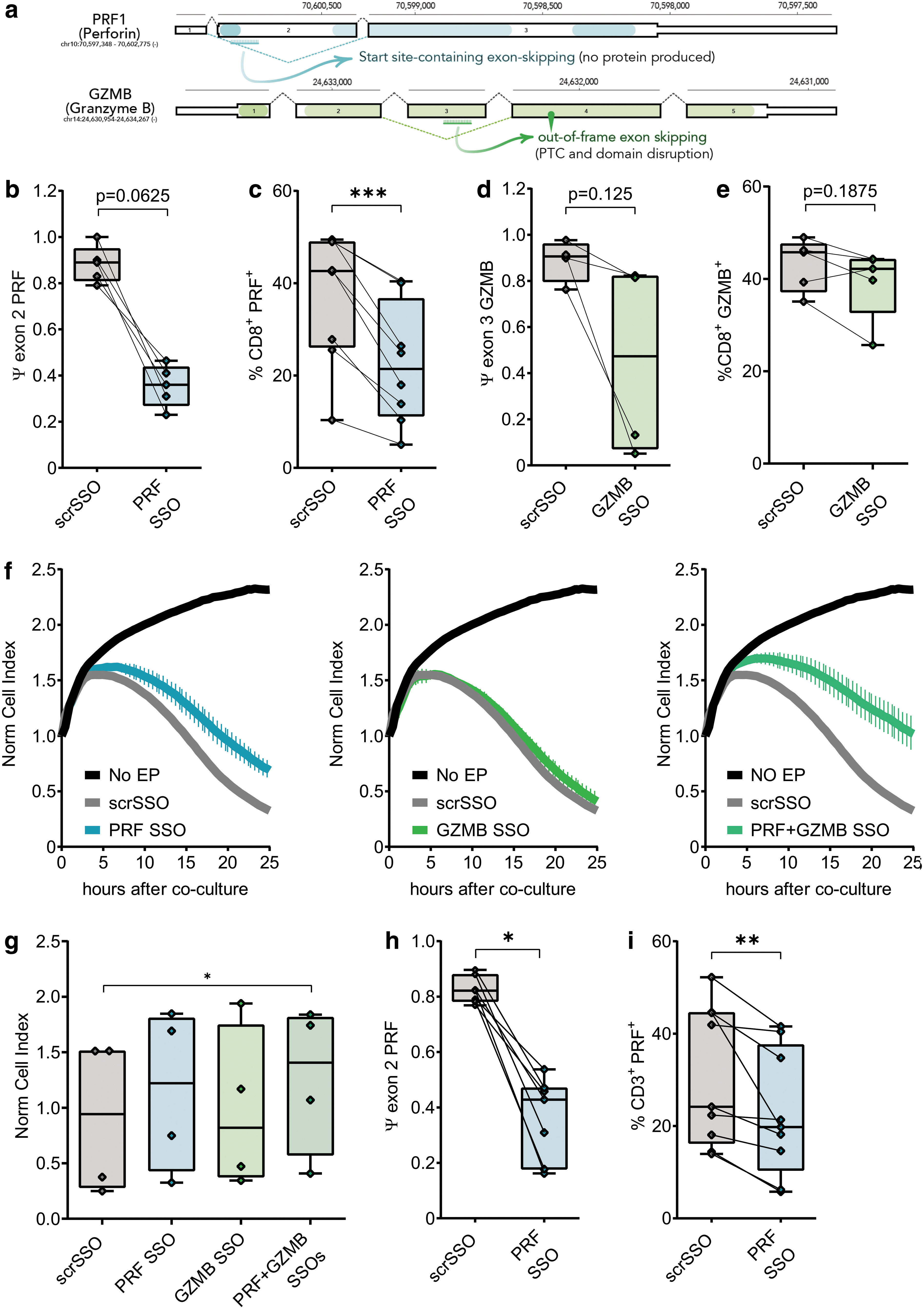
To further test the ability of SSOs to modulate essential T cell functions, we designed SSOs to suppress the expressions of PRF1 and GZMB. For the former, the PRF SSO induces the skipping of exon 2b where the translation start codon resides, whereas the GZMB SSO induces exon 3 skipping that generates a frameshifted transcript (Fig. 3a); in both cases, no protein product is expected from the respective resultant transcripts. We quantified the exon skipping efficiency of each SSO 24 h after electroporation as PSI, shown in Fig. 3b and d. Intracellular cytokine staining (ICS) of PRF and GZMB ART cells shows a reduction of the respective proteins (Fig. 3c, e and Supplementary Fig. S3e). Again, both PRF and GZMB SSOs do not affect T cell viability (Supplementary Fig. S3a, b) and TCR expression (Supplementary Fig. S3c, d) compared with the controls, but inhibit their cytolytic ability (Fig. 3f, g) in T cells cotransfected with both the SSOs. Of importance, modulation of T cell function with SSOs was not demonstrated only on T cells of healthy individuals but on T cells of patients with inherent pathology (ie, chronic hepatitis B infection) (Fig. 3h, i, and Supplementary Fig. S3f).
Discussion
To engineer ART cells with definite function and specificity, we decided to utilize fully chemically modified splicing-modulating SSOs over siRNA, GAPmer, shRNA, and CRISPR/Cas9 approaches, for the following reasons: (i) superior selectivity and stability, (ii) multimodality, (iii) limited immunogenicity, and (iv) clinical compatibility owing to their transient nature and their success in clinic [36 37,51,52]. Thanks to their versatility, SSOs could be explored in other cell types also used in adoptive transfer (such as NK cells or dendritic cells) to further ameliorate cell-based therapeutic approaches. The manipulation ex vivo of cells used for adoptive cell transfer is relatively straightforward. In fact, high numbers of PBMCs can be easily obtained through phlebotomy and they can be expanded in vitro for several days before reinfusion into the patient. During the expansion phase, concurrent modifications can be implemented for intrinsic engineering of these cells: from redirection, to the boosting of their function using cytokines or other drugs [53,54].
The advantage of our cotransfection protocol is evident when considering its practicality in a clinical setting, where substantial and subsequent manipulations of the samples could lead to higher risk of contamination and/or loss of the sample itself. The choice of transfection (over transduction) offers an advantage with regard to safety. Transient redirection is safer for the patient, as the specificity receptor is lost within few days, with lower risk of collateral effects [8]. After the exogenous specificity receptor is lost, T cells still maintain their natural receptor; therefore, any stable functional modification induced would alter indefinitely their function against the natural targets. Another advantage of an SSO lies in its noncatalytic action, which does not require a functional RNAi machinery or RNase H activity, unlike siRNA and GAPmer, respectively [40]. Given the patient variability, potential deregulation of the two endogenous cellular factors in exhausted primary T cells and cytotoxicity effects from dose-induced saturation of these factors [55,56], anticipating the efficacy and the toxicity of both siRNA and GAPmer is not straightforward.
Our data showed that SSO-mediated intrinsic engineering of primary human T cells from healthy donors and chronic hepatitis B patients does not impinge on the ART cell viability and TCR expression, as well as T cell antiviral or cytotoxic function mediated by the extrinsic engineering. On the contrary, we were able to modify immune-modulatory and cytotoxic functions of ART cells targeting IFN-γ, PRF, and GZMB production singly and in combination, and in conjunction with extrinsic engineering.
In summary, this work is a technical demonstration that targeted intrinsic engineering of immune cells for adoptive immunotherapy is possible with splice-switching oligonucleotides. This work paves the way for more applications in the field, as well as, a clinical translation of the technology.
Footnotes
Acknowledgments
The authors thank the entire E.G., K.B.W., and A.B. laboratories for critical discussion.
Author Disclosure Statement
E.G., K.B.W., and A.B. are inventors on “Modifying T lymphocytes function with Antisense Oligonucleotides (ASOs) for personalized immune therapy” (PCT/SG2018/050313; 10201705285S [IMC/Z/09724]; filing date June 27, 2017; priority date June 27, 2016; licensed to IMMUNOA Pte Ltd on September 14, 2018). E.G., K.B.W., and T.T. are cofounders and scientific advisors of IMMUNOA Pte Ltd. T.T. is in the Board of Directors of IMMUNOA Pte Ltd; K.B.W. is Chairman in the Board of Directors of IMMUNOA Pte Ltd; A.B. is cofounder of Lion TCR Pte Ltd, in the Board of Directors Lion TCR Pte Ltd and in the Board of Directors of IMMUNOA Pte Ltd; E.G. has served on advisory board for Lion TCR Pte Ltd.
The rest of the authors declare no competing financial interests.
Funding Information
This work was supported by the Institute of Molecular and Cell Biology, Agency for Science Technology and Research, Singapore. E.G. acknowledges support from NMRC/OFIRG/0032/2017, NRF-CRP17-2017-06 and MSSM seed fund.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.