
Abstract
This white paper summarizes the current consensus of the Japanese Research Working Group for the ICH S6 & Related Issues (WGS6) on strategies for the nonclinical safety assessment of oligonucleotide-based therapeutics (ONTs), specifically focused on the similarities and differences to biotechnology-derived pharmaceuticals (biopharmaceuticals). ONTs, like biopharmaceuticals, have high species and target specificities. However, ONTs have characteristic off-target effects that clearly differ from those of biopharmaceuticals. The product characteristics of ONTs necessitate specific considerations when planning nonclinical studies. Some ONTs have been approved for human use and many are currently undergoing nonclinical and/or clinical development. However, as ONTs are a rapidly evolving class of drugs, there is still much to learn to achieve optimal strategies for the development of ONTs. There are no formal specific guidelines, so safety assessments of ONTs are principally conducted by referring to published white papers and conventional guidelines for biopharmaceuticals and new chemical entities, and each ONT is assessed on a case-by-case basis. The WGS6 expects that this report will be useful in considering nonclinical safety assessments and developing appropriate guidelines specific for ONTs.
Introduction
The development of oligonucleotide-based therapeutics (ONTs) is a rapidly evolving scientific field with more than 100 clinical trials already conducted worldwide [1]. The number of consultations increases each year in the Pharmaceuticals and Medical Devices Agency (PMDA) of Japan as well as in the European Medicines Agency (EMA) (Fig. 1). In January 2013, Kynamro® (mipomersen sodium) was the first systemically administered ONT approved for marketing by the US Food and Drug Administration (FDA). Macugen® (pegaptanib sodium), Spinraza® (nusinersen), and Onpattro® (patisiran) have been approved for marketing in three regions, the United States, Europe, and Japan. In addition, several more ONTs have recently been approved (Table 1).
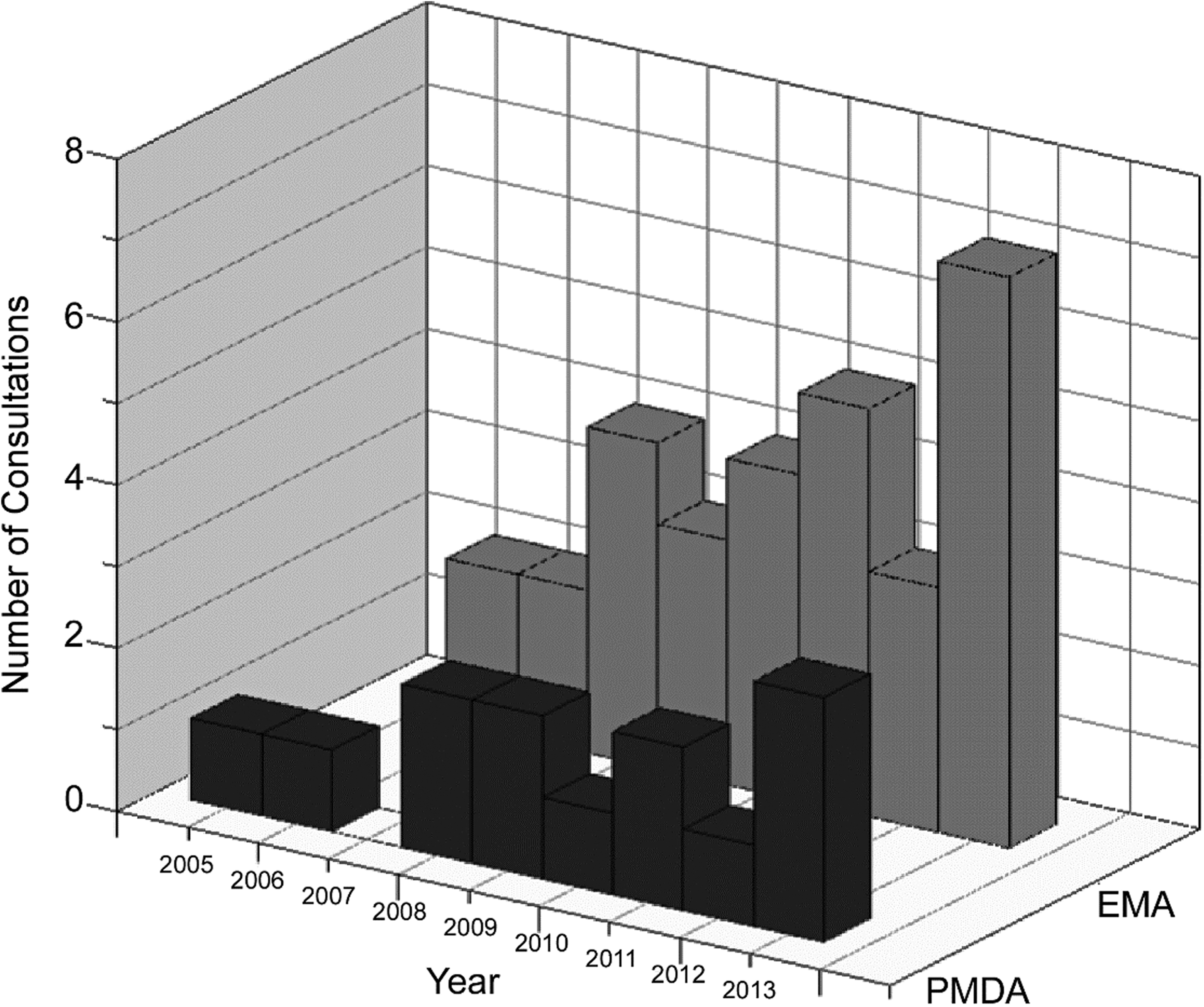
Annual change in number of consultations on oligonucleotide therapeutics. This graph shows a recent increasing trend in both Japan (PMDA) and the EU (EMA). EMA, European Medicines Agency; PMDA, Pharmaceuticals and Medical Devices Agency.
Summary of Approved Oligonucleotide Therapeutics (as of November 30, 2020)
mRNA, messenger RNA; siRNA, small interfering RNA.
Accordingly, information regarding nonclinical safety assessments of ONTs based on actual examples is accumulating and related issues are becoming more apparent. ONTs exert medicinal effects by binding DNA or RNA sequences, and as a result, ONTs exhibit high species and target specificities, similar to biotechnology-derived pharmaceuticals (biopharmaceuticals). However, ONTs have characteristic off-target effects that are clearly different from those of biopharmaceuticals (Table 2). Thus, toxicological changes observed with ONTs are a mixture of on- and off-target effects, which make the safety assessment of ONTs more complicated than for biopharmaceuticals.
Similarities and Differences Between Oligonucleotide Therapeutics and Existing Drugs
In addition, there are various oligonucleotides that are difficult to classify (Table 3). For example, class effects, a type of off-target effects, are known to be produced by ONTs having certain chemical structures, such as containing a phosphorothioate oligonucleotide [2]. Such information is useful for the interpretation of toxicological changes of these specific ONTs; however, it cannot be generally applied to all ONTs. Furthermore, in the future, new types of ONTs will be developed as ONT technologies are rapidly being innovated. Thus, even if a specific point-to-consider can be developed on the basis of accumulated evidence from specific ONTs, it is important to define the types of ONTs.
Classes of Oligonucleotide Therapeutics
miRNA, microRNA.
There are various issues related to the safety assessment of ONTs, and to assess them, in the 1990s, there were two white papers written by FDA Pharm/Tox reviewers [3–5]. The US-based Oligo Safety Working Group (OSWG) has also been responding to these issues [6–12], and recent regional initiatives have sought to clarify the specific points for ONTs requiring consideration and to establish guidelines in individual countries [13–15] (Table 4).
Regional Initiatives Working to Clarify Specific Points to Consider for Oligonucleotide Therapeutics
OSWG, a subcommittee of the DIA (Drug Information Association, Inc.).
EFPIA, European Federation of Pharmaceutical Industries and Associations; OSWG, Oligo Safety Working Group; WGS6, Japanese Research Working Group for the ICH S6 and Related Issues.
Although the safety assessments of ONTs are principally conducted on a case-by-case basis by referring to conventional guidelines for biopharmaceuticals or new chemical entities (NCEs), only the ICH S6 guideline of the Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals (S6; part 1 of the ICH S6(R1) [16]) states that “principles outlined in this guidance may also be applicable to … oligonucleotide drugs.” This wording suggests that the basic principle of “case-by-case” in the current ICH S6(R1) guideline [S6(R1)] would be applicable to ONTs. However, ONTs are too different from biopharmaceuticals to justify a simple adoption of the S6(R1) guideline. Furthermore, other safety guidelines for NCEs either state “this guideline does not generally apply to … oligonucleotides” or provide no information regarding ONTs.
The Japanese Research Working Group for the ICH S6 and Related Issues (WGS6) was originally organized to scientifically support the Expert Working Group for ICH S6(R1). Because of the scope of S6(R1) and the lack of specific guidelines for ONTs, WGS6 expanded their focus to ONTs, in addition to biopharmaceuticals. We, the members of the WGS6, discussed the points to consider for nonclinical safety evaluations of ONTs in comparison with those for biopharmaceuticals, and published a series of papers in Japanese [13]. This white paper is a summary of those papers. We expect that this report will be helpful in considering nonclinical safety assessments for the future development of guidelines specific for ONTs.
Toxicity Classification of ONTs and Evaluation of Off-Target Effects
Most ONTs, except aptamers and decoy oligonucleotides, hybridize to target nucleotide sequences to exert their effects (known as on-target effects). Concepts provided in the S6(R1) can be applied to evaluate on-target toxicities, that is, potential adverse effects caused by an “exaggerated” degree of the intended pharmacologic activity based on the on-target effects [11] analogous to those for biopharmaceuticals. Effects occurring through unexpected actions on active sites, or mechanisms not associated with on-target effects, are generally called off-target effects. Among these off-target effects, we defined effects involving hybridization with a sequence that is the same as, or similar to, the target nucleotide sequence as “hybridization-dependent off-target effects.” Other off-target toxicities are referred to as “hybridization-independent off-target toxicities.” These latter toxicities are a result of chemical structures specific to the nucleic acid molecules or physicochemical properties not mediated by hybridization, such as changes in innate immunity mediated by toll-like receptors (TLRs), or a result of chemical modifications that were intended to improve in vivo pharmacokinetics (PK) [17] (Fig. 2).
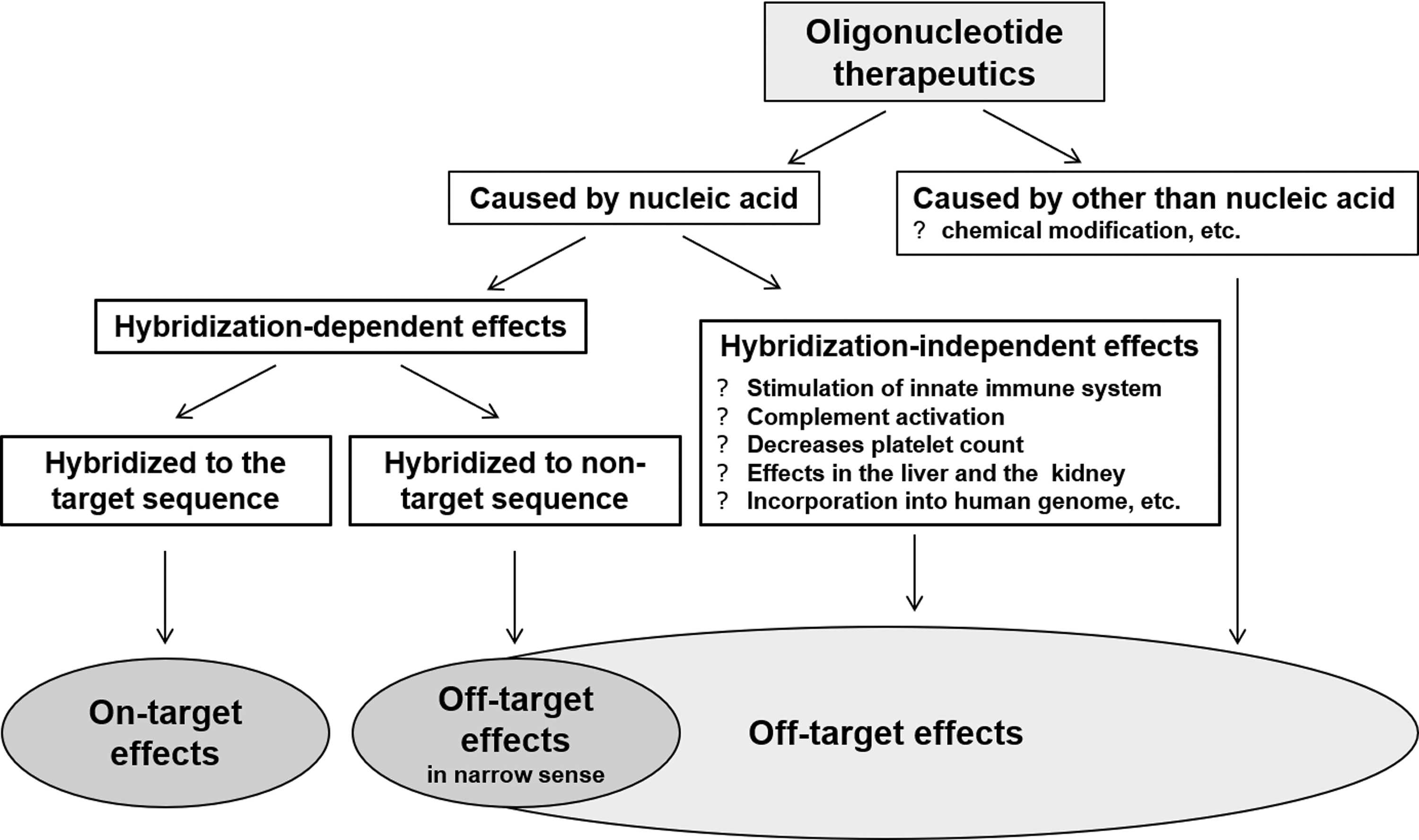
Classification of toxicities induced by oligonucleotide therapeutics.
A hybridization-dependent off-target effect is considered to occur by hybridization with a completely or almost completely matched nucleotide sequence. First, the WGS6 discussed the onset of hybridization-dependent off-target effects, focusing on the length of the nucleic acid sequence. The results from many in vitro and in vivo studies indicated that the length of sequences necessary for such effects depended on the ONT structure. The antisense oligonucleotides approved to date are between 18 and 30 mer in length, while those with high binding affinity for RNA, such as LNA gapmers, exert effects even if they have a short length of 12 to 13 mers [18]. One or two mismatches can decrease these effects [11,18–20]; however, recent reports showed that 13-mer or 16-mer antisense oligonucleotides with one or more mismatches could silence off-target genes in vitro [21,22]. An association between the effects on off-target genes and relative hybridization free energy has also been reported [23,24]. With small interfering RNA (siRNA) oligonucleotides, because the homology of “seed” sequences (positions 2–8 from the 5′ end) is important for hybridization with the target nucleotide sequence, expression of genes other than the target is considerably suppressed even with a homology of about 11 of 15 mers [25] and two to four mismatches are permitted [26].
Next, the WGS6 considered the probability of the occurrence of sequences matching the ONT target nucleotide sequence in humans. If an ONT with a specific sequence has sufficient length, it can be simply calculated that the number of potential off-target genes in the entire set of human pre-RNA/messenger RNA (mRNA) is theoretically predicted to be one or fewer. Conversely, in a report where the theoretical number of potential off-target genes was compared with the actual case-based number for the same number of nucleotides, mismatches in the sequences were calculated to generate a greater number of potential off-target genes than calculated for matched sequences [27].
Hybridization-dependent off-target toxicities may be avoided to a certain extent by using in silico and/or in vitro analyses to exclude sequences that could potentially cause off-target effects. Probably because of such efforts to exclude potential off-target sequences in the early development phase, no serious adverse event likely to have been caused by hybridization-dependent off-target effects has been reported to date for marketed ONTs. Despite such efforts, cases may occur in which a sequence essential for efficacy affects an off-target gene. To avoid such effects on off-target genes in evaluating potential toxicity risks in humans, background information, including the biological characteristics of the genes, should be rigorously and comprehensively investigated (Fig. 3).
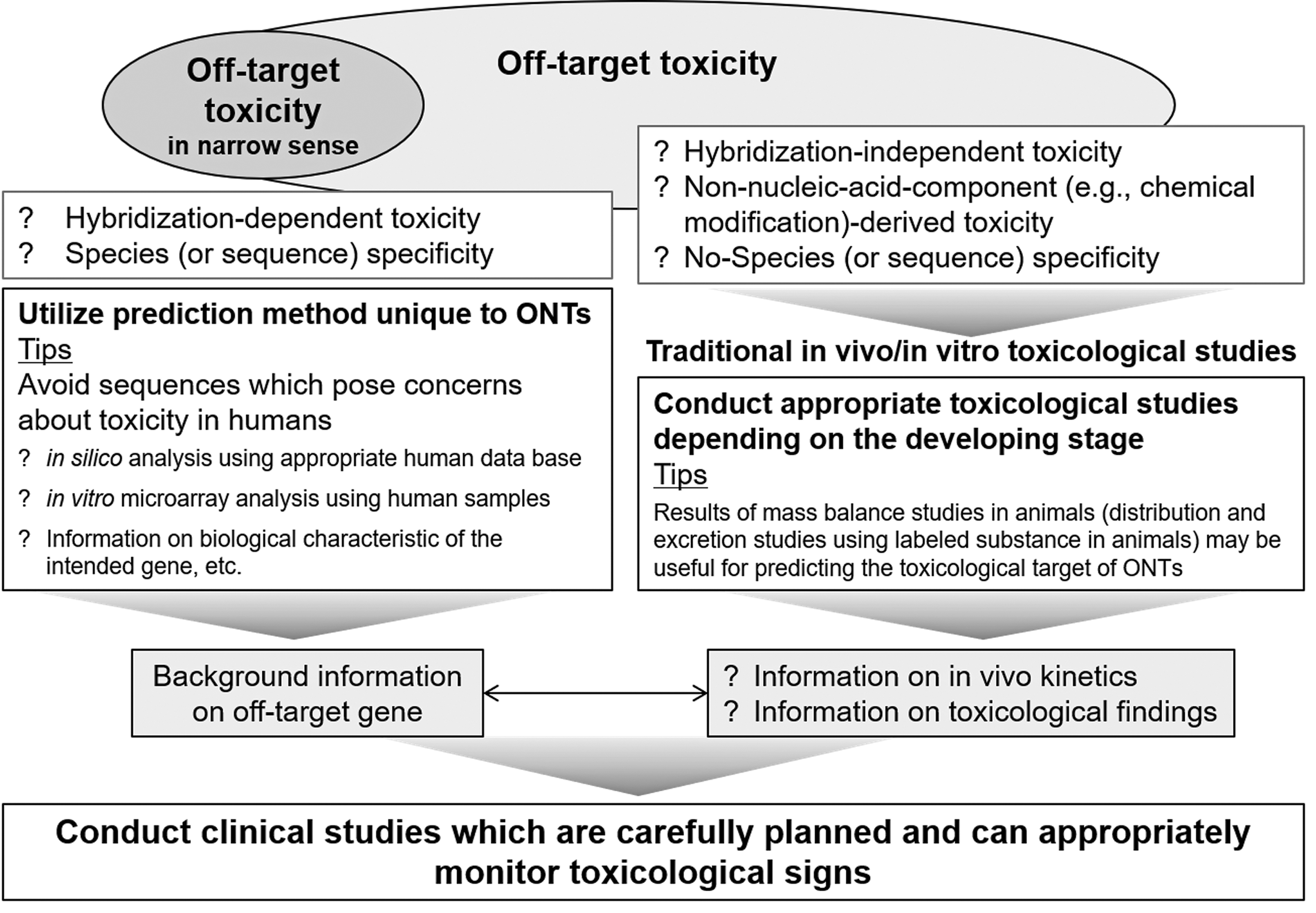
Prediction of adverse effects caused by oligonucleotide therapeutics and their metabolites.
Hybridization properties are currently predicted by quantitative measurement methods, such as quantitative real-time polymerase chain reaction, microarrays, and RNA sequencing, which allow cost-effective measurements of the global transcriptome, including all unintended RNA targets [23]. However, to address the toxicity risks in humans caused by off-target genes that cannot be excluded, rigorous and comprehensive investigation are needed as described above (Fig. 3). Although associating genotype with phenotype, and specifically predicting a toxic phenotype, still remains an intricate challenge, integrated toxicogenomics data generated at the transcriptome level, including RNA-seq and microarrays, may be useful for more comprehensive analyses in the future [28].
Evaluation of Class Effects of ONTs
Class effects are defined as the effects of drugs of a specific category and are dependent on the physicochemical properties of the drugs. Class effects of ONTs are off-target effects common to nucleic acid molecules and are independent of hybridization (Fig. 2). Known class effects include changes caused by binding to plasma proteins [prolonged activated partial thromboplastin time (aPTT) and complement activation]; immunostimulatory and inflammatory responses, including effects on innate immunity mediated by TLRs; and effects on organs, such as the kidney and liver, to which the drugs are distributed at high concentrations. These effects are known to have the following properties: (1) they each occur at a particular concentration; (2) they are observed in cases where the products contain a particular nucleic acid structure or sequence, such as a phosphorothioate oligonucleotide (eg, prolonged aPTT [29] and complement activation [30]), siRNA (eg, immunostimulation mediated by TLR3 [31]), or CpG-rich sequences (eg, complement activation [32] and immunostimulation mediated by TLR9 [33]); and (3) they can be decreased by chemical modifications, such as 2′-O-methoxyethyl (eg, immunostimulation and complement activation).
Interestingly, a detailed analysis of class effects caused by a particular category of ONTs has suggested that class effects are not always caused by all the ONTs in the category and that some toxicological changes induced by an ONT are not only a result of the class effect but also because of exaggerated pharmacology [34]. Thus, toxicological changes should be carefully evaluated compound-by-compound for ONTs, although the information available on the class effects for a specific category of ONTs will be helpful to understand these changes.
Class effects can be detected in conventional toxicity studies. Several reviews describing species sensitivity to class effects have been published [2]. In a comparison of the toxicity of ONTs in individual animal species, rodents were found to be more susceptible than non-human primates (NHPs), with the exception of complement activation. However, the clinical toxicity of ONTs is not always predictable from nonclinical data. Continued investigation of class effects is warranted to further our understanding of these primary manifestations of ONT toxicity.
Use of Surrogates
Many biopharmaceuticals show species differences in the responsiveness to pharmacological effects and, thus, NHPs are often the only relevant species for toxicity studies. In such cases, the original S6 document encouraged the use of a homologous protein or surrogate that is pharmacologically active in an animal species commonly used in toxicity studies. However, evidence acquired over the 10 years since the issuance of the S6 guidelines has indicated that it is difficult to interpret toxicity results obtained with surrogates. For example, toxicological changes induced by a surrogate in rodents are sometimes different from those of the corresponding clinical candidate in NHPs. This difference is probably because of differences in the potency and/or duration of the pharmacological effects between the clinical candidate and surrogate, or differences in the postreceptor signal transduction pathways between NHPs and rodents. The causes of such differences vary depending on the situation, making it difficult to conclude which study results are appropriate to consider. In addition, the safety margins of the clinical candidate cannot be calculated from the results of toxicity studies using a surrogate. Thus, because of such difficulties, the S6(R1) suggests that the use of surrogates is of limited value.
ONTs are highly target specific and, therefore, often species specific. When no appropriate pharmacologically responsive animal species for the clinical candidate can be identified, on-target toxicity may be evaluated using an animal species-specific surrogate oligonucleotide [11]. Surrogate oligonucleotides can be made more quickly and at lower cost than surrogate biopharmaceuticals, which makes it possible to more easily select a surrogate that is similar to the clinical candidate in terms of structure, PK, and pharmacodynamics (PD). The comparability of PD between a clinical candidate and an animal surrogate can be determined by in vitro assessment of receptor binding (affinity and selectivity) and functional (potency) assays. However, it is important to note that any toxicological finding with a surrogate will represent a mixture of on-target effects, surrogate molecule-specific hybridization-dependent off-target effects, surrogate molecule-specific hybridization-independent off-target effects, and hybridization-independent class effects. Therefore, such findings must be interpreted carefully. Furthermore, off-target effects of a clinical candidate should not be evaluated based on findings from a surrogate.
A satellite group receiving a single dose of a surrogate to evaluate on-target toxicities may be useful for hazard identification when no appropriate pharmacologically responsive animal species for the clinical candidate can be identified. A similar study design has been proposed by S6(R1) and in a white paper by Kornbrust et al. [11]. Furthermore, a rodent developmental and reproductive toxicity (DART) study with a surrogate may provide information that is as useful as an NHP DART study with the clinical candidate. This is because an NHP DART study can be used for hazard identification, but not for risk assessment because of the limited number of NHPs that can be used.
Safety Assessments of the Metabolites of the Chemical Moieties of ONTs
The degradation of ONTs to short oligonucleotides and individual nucleotides by nucleases is analogous to biopharmaceuticals being degraded to amino acids by peptidases. The degradation products of an ONT that comprises only natural nucleic acids can be physiologically reutilized through reuptake into DNA, as for endogenous nucleic acids. Therefore, no safety assessment of such metabolites is needed. However, most ONTs under development comprise nucleic acids that have been chemically modified to improve the PK, delivery to target tissues, and/or stability to nuclease degradation. Examples of such modifications include chemical modifications made to the nucleoside or phosphodiester linkages, and nucleic acids that are conjugated to other molecules (eg, peptides, sugars, polyethylene glycol, or monoclonal antibodies) [35]. When the chemically modified ONT is very stable and is eliminated from the body with no or little degradation, there is no need to consider the safety associated with the metabolites.
If metabolites of an ONT containing a chemical moiety are produced in the body, one of the potential safety concerns is the incorporation of chemically modified mononucleotides into DNA. However, the chemically modified mononucleotides that are commonly used in ONTs are known to be poor substrates for endogenous kinases and/or polymerases. Therefore, the DNA incorporation of such chemically modified mononucleotides is less efficient than for natural nucleotides, and the incorporation of these mononucleotides into newly synthesized DNA under physiological conditions is deemed of negligible probability [7,36–39]. Nucleosides are generally metabolically activated by conversion to nucleotides by phosphorylation and might also induce cytotoxicity through the inhibition of various nucleic acid metabolizing enzymes [40].
Databases on the structure–toxicity relationships of existing modified nucleic acids may be useful to assess whether the metabolites potentially formed from an ONT would cause cytotoxicity or be incorporated into human DNA or RNA. Any concern regarding the potential reincorporation of novel monomer metabolites into the host DNA based on evaluation results, including the cell viability, could be addressed by the following: examination of the DNA derived from organs and tissues to detect high accumulation following repeat administration of the compound over a particular period in vivo; genotoxicity testing of those organs and tissues; and in vitro transformation assays using the modified nucleic acid. However, not all of the above studies need to be conducted, and the safety assessment of metabolites with a novel chemical moiety should be performed based on a weight-of-evidence approach.
For ONTs conjugated to other molecules, the safety of the added molecule and its metabolites should be assessed [14,41–43]. If the unconjugated molecule is not novel, sufficient information on the safety may be available. If the unconjugated molecule is novel (ie, there is little or no safety information available), the safety of the unconjugated molecule may need to be assessed in a separate short-term study or in an arm of the short-term study with the conjugated ONT, similar to the approach described in S6(R1). The metabolism of a conjugated ONT should be considered in in vitro human and animal models and animal species. If a conjugated ONT is not metabolized in the body at all, or if it is metabolized to the same metabolites in animals as in humans, further safety studies are not warranted. The pharmacologically responsive species to be used for the study of unconjugated molecules should be selected on a case-by-case basis.
Evaluation of the characteristics of a compound with anti-nuclease activity in incubation studies using nuclease solutions and similarly, identification of its metabolites in in vitro studies using human or animal specimens may provide information on the potential metabolites in vivo. It should be noted that whole organ homogenates may be more suitable than tissue fractions for investigating the in vitro drug metabolism of ONT drugs, as there are both soluble and membrane-bound nucleases, which may have different substrate specificity [44]. The target organs for toxicity in humans can be predicted by identifying the excretion pathways, and the organs or tissues that accumulate the compounds and their metabolites, using labeled compounds in appropriate animals. The target organs are often the liver and kidneys, as described in Evaluation of Class Effects of ONTs section.
Finally, a shorter sequence decreases the hybridization potential of the ONT. Therefore, metabolites that have shorter sequences than the parent compound are predicted to be much less likely to cause hybridization-dependent toxicities.
Nonclinical Safety Studies of ONTs
Study design
Factors for the design of studies to evaluate ONT toxicity include the animal species and dosing parameters (route, period, interval, and dosage). These factors should be determined based on the compound's predicted pharmacokinetic profile in humans. The pharmacokinetic profiles (plasma maximum drug concentration (Cmax), area under the curve, and clearance) of phosphorothioate antisense oligonucleotides have been reported to show similarities among animal species, including humans [45–47]. When there is an accumulation of similarity data between species, then conventional methods for determining dosing parameters equivalent to those to be used in the clinic would also be appropriate for the toxicity evaluation of such ONTs, in principle by referring to S6(R1). However, for ONTs with pharmacokinetic profiles that have been predicted to be greatly different in humans than in animals, the toxicity evaluation must be made more carefully. For example, when the blood concentration of a compound very rapidly decreases in animals, but not in humans, maintaining the exposure levels, for example by frequent dosing, should be considered for appropriate toxicity evaluations in those animals. Another important point to note is that plasma PK may be less informative for ONTs than for biologics because most ONTs (with exceptions, such as aptamers) are effective intracellularly. Therefore, the evaluation of tissue PK using appropriate methods might need to be considered.
Selection of the highest dose
Biopharmaceuticals have high specificities for their target molecules and most toxicological changes are caused by exaggerated pharmacology (on-target toxicity). Therefore, a dose much higher than the intended clinical dose is not normally required for nonclinical toxicity evaluations. The S6(R1) states that after defining the dose–response and exposure relationships to the pharmacological effects, the following should be determined: (1) the dose causing the maximum intended pharmacological effect in the nonclinical species and (2) the dose enabling ∼10-fold greater exposure than the maximum exposure expected to be achieved in clinical use. The higher of these two doses should then be selected as the highest dose group in nonclinical toxicity studies unless using a lower dose is justified [eg, it is the maximum feasible dose (MFD)]. This rationale for dose selection is focused on on-target toxicities and, following the rationale of S6(R1), would not be sufficient for ONTs, considering the need to also detect off-target toxicities caused by chemical modifications.
In addition, the ICH M3(R2) guidance [M3(R2)] [48] states that the use of doses in toxicity studies that will not help predict clinical safety should be avoided from the perspective of animal welfare (replacement, reduction, and refinement: 3Rs). Thus, the recommended highest doses are those achieving saturated exposure, the MFD, and the maximum tolerated dose, as well as doses providing a 50-fold margin over the expected clinical exposure. This recommendation also states that an upper limit of 1,000 mg/kg/day is to be used if no other information is available. For the time being, the M3(R2) should be applied to chemically modified ONTs as it is to small-molecule drugs. Novel approaches for determining the highest doses for ONT toxicity evaluations may be identified once more information on the relationships between doses and toxicity becomes available. For example, if the on-target toxicity was sufficiently examined and the class effects of a series of chemical modifications or specific sequence structures were well understood, then the dose producing a class effect might be a useful choice as an upper limit.
Selection of animal species
Two animal species, rodent and non-rodent, should, in principle, be used in ONT toxicity studies. M3(R2) should be the basis for assessing the off-target toxicities of ONTs because of the similarities between ONTs and small-molecule NCEs. For example, ONTs often cause hybridization-independent off-target toxicities and sensitivity to these effects may vary with species.
S6(R1) should be referred to for assessing on-target toxicities of ONTs. When two or more species are available that are appropriate for testing biopharmaceuticals, toxicity studies should, in principle, be conducted in both, one rodent and one non-rodent. However, if there are no differences in the nature or intensity of the toxicity in the two species in short-term studies, then long-term toxicity studies using only one of the two species would be acceptable [16]. The reason that only studies in one species are necessary is because, based on findings to date, most toxicological changes caused by biopharmaceuticals have been demonstrated to be caused by exaggerated pharmacology, mediated by known mechanisms of action, that is, on-target effects. Moreover, the degradation products of biopharmaceuticals are naturally occurring amino acids. Conversely, these characteristics do not apply to ONTs. Therefore, even if similar toxicological profiles were shown in two species in short-term toxicity studies, two animal species should also be used for long-term toxicity studies of ONTs. If a clinical trial sponsor has sufficient information regarding the chronic toxicities of a particular category of ONTs, provided there is scientific justification, the sponsor can propose a chronic toxicity study using only one species.
Animal species responsive to the pharmacological effects or biological activities of ONTs are considered relevant for evaluating on-target toxicities. When only one relevant species is identified, on-target toxicity should be evaluated only in that species. Additional toxicity studies using a surrogate in another species should not be performed as discussed in the Use of Surrogates section and a previous report [49]. Conversely, two species, rodent and non-rodent, should be used to evaluate off-target toxicities without regard to the responsiveness to the pharmacological effects and biological activities of the ONTs.
A pharmacologically responsive animal species may not be able to be identified for some ONTs targeting human-specific sequences. For such ONTs, the risk of on-target toxicities could be assessed by in silico and/or in vitro microarray analyses, as well as using background information on the biological function encoded by the target sequence. The sponsor can propose whether to conduct an additional in vivo toxicity study with a surrogate before clinical studies or proceed to the clinical stage without such surrogate data. This decision may be made on a case-by-case basis depending on the available information. Furthermore, it is recommended that the sponsor discusses with the regulatory agency approaches for addressing possible on-target toxicities and communicating risk, considering the clinical indication, patient population, and any information regarding potential on-target toxicities.
Information on class effects [50], if available, may be useful for selecting an animal species for toxicity studies and interpreting the results. Species differences in ONT metabolism would be one factor affecting the species selection. Because the nuclease resistance and the similarity of the metabolites of ONTs can be determined in vitro or in vivo [51], selecting an animal species with a metabolic profile similar to that of humans would be appropriate if species-specific metabolite differences are observed.
In addition, an animal species showing a pharmacokinetic profile similar to that in humans should be selected if such information is available for an ONT in the same class. For example, an antisense oligonucleotide with a phosphorothioate modification showed pharmacokinetic profiles in nonclinical animal studies, except in mice, which correlated well with the PK in humans. This information was useful for extrapolating the toxicity findings in nonclinical animal studies to predict the onset of adverse reactions in humans [17].
Toxicity evaluation (specific considerations)
Safety pharmacology
The purpose of the core battery of safety pharmacology tests is to investigate the effects of a compound on the major physiological functions (central nervous, cardiovascular, and respiratory systems). This evaluation must be conducted before initiation of clinical studies for all agents, including for ONTs. M3(R2) states that consideration should be given to including such evaluations, in addition to general toxicity studies, to the extent this is feasible based on the 3Rs principles. Similar statements are also provided in S6(R1) [16,48]. For drugs, including ONTs, conducting independent studies is not always needed in accordance with the concept of these two guidelines based on compliance with the 3Rs principles.
The investigation timing in core battery studies should be considered. For example, for ONTs targeting mRNA, peak concentrations in blood or target organs may not be consistent with peak pharmacological effects because of the time lag between hybridization with the target sequence and suppression of the formation of proteins encoded by the target sequence [52]. Therefore, the timing for safety pharmacology evaluations should be selected with an understanding of the characteristics of each compound.
There are several potential approaches for evaluating the effects on ion channels, including the human ether-a-go-go-related gene (hERG) channel, in vitro. For biopharmaceuticals, in vitro studies on ion channels are not needed because it is unlikely that high molecular weight biopharmaceuticals will cross cell membranes and inhibit hERG channels [16]. Some oligonucleotide products, including mipomersen sodium, were tested in hERG studies and, so far, all have shown negative results. As has been stated in previous reports, “this is not surprising, considering the chemical nature of the oligonucleotides, which are polyanionic molecules of approximately 7,200 Mw” [53–55]. The OSWG [12] also stated that “in vitro human ether-a-go-go-related gene (hERG) testing does not provide any specific value and is not warranted” [8]. WGS6 also considered that the significance of conducting the hERG assay will be low. There is not much information available regarding the effect of smaller molecules resulting from the degradation and metabolism of oligonucleotides with chemical modifications. However, the effects of such degradation products and metabolites formed in the body, together with the effects of the clinical candidate, should be revealed in appropriately designed in vivo toxicity studies.
Single-dose toxicity studies
M3(R2) states that when acute toxicity information is available from any study (eg, dose-escalation or short-duration dose-ranging studies), separate single-dose studies are not recommended. This concept is also presented in S6(R1), which indicates that the information obtained from pharmacology studies can be used in place of single-dose studies. The concepts in these guidance documents are also applicable to the acute toxicity evaluations of ONTs.
Repeat-dose toxicity studies
It should be noted that test compounds, including ONTs, are not distributed equally throughout the body, even when systemically administered. The recently developed oligonucleotide products with potent anti-nuclease activities may accumulate in specific tissues and induce toxicities more frequently than other ONTs [35]. ONTs are believed to accumulate in the kidney and liver. For example, the kidney is a target organ of mipomersen sodium toxicity in NHPs [53–55]. The accumulation levels of test compounds in target organs after long-term administration, and the progression of toxicity along with the accumulation, are considered important for toxicity evaluations.
For a locally administered ONT, it is necessary to evaluate toxicity, not only at the injection site but also in organs where the ONT is expected to accumulate through systemic circulation.
Genotoxicity studies
ONTs comprising exclusively natural nucleic acids are considered to have a very low risk of being genotoxic because the degradation products are the same as endogenous nucleic acids. Thus, these ONTs may not need to be tested in the battery of genotoxicity assays described in S2(R1) [56]. However, many ONTs contain chemically modified moieties, such as chemically modified nucleic acids, chemical compounds for delivery, and/or linkers to connect the delivery moieties with the ONTs. The genotoxicity risks of not only these chemically modified moieties but also the metabolites and degradation products should be considered. Therefore, ONTs with chemical modifications may need to be tested for genotoxicity.
There is controversy concerning whether oligonucleotides, including ONTs, may be genotoxic. Some studies have suggested there is a low possibility of detecting genotoxicity caused by ONTs [14,17]. Recently, many chemically modified oligonucleotides have been designed to have enzyme resistance and are less likely to be substrates for polymerases. Thus, the incorporation of these modified oligonucleotides into newly synthesized DNA is less efficient than with natural nucleotides, and incorporation under physiological conditions has been deemed of negligible probability. However, it has been shown that some oligonucleotides that were degraded and incorporated into DNA may cause genotoxicity [57]. Thus, it is necessary to continue to consider the relationship between the structure of modified nucleic acids and genotoxicity. The methods used to evaluate the genotoxicity risks of ONTs should be carefully selected as the controversial results might be partly attributed to differences in the experimental conditions. For example, an appropriate metabolism system, other than an S9 mixture, can be used to assess the potential genotoxicity of metabolites. Using in vivo tests may be useful in solving this metabolic activation problem. Furthermore, genotoxicity assays should be conducted using a drug product (ie, a formulation comprising the ONT and a delivery system) rather than a drug substance (ie, the ONT itself) if the permeability of the naked ONT into the cell membrane is limited.
DART studies
The DART of oligonucleotides should be evaluated in accordance with the ICH S5(R3) guideline [S5(R3)] and S6(R1) (Fig. 3), and the data package will depend on the intended patient population and pharmacological effects of the specific oligonucleotide. For example, for women of child-bearing potential, three separate DART studies [fertility, embryo-fetal developmental (EFD), and prenatal and postnatal developmental (PPND) studies] are generally conducted as described in the ICH M3(R2) guideline [M3(R2)]. However, for anticancer pharmaceuticals, fertility and early EFD or PPND studies are not warranted as described in the ICH S9 guideline (S9) [58]. When the weight of evidence (eg, mechanism of action, phenotype of genetically modified animals, information on human genetic diseases, and class effects) indicates hazardous effects on fertility or pregnancy outcome and there is adequate information to communicate the risk to reproduction, additional nonclinical studies might not be warranted as described in S6(R1).
In DART studies of oligonucleotides, the animal species for testing should be justified based on both the advantages and disadvantages as described in S5(R3). Namely, pharmacokinetic and metabolite profiles, reproductive physiology of the animals, historical background data, and sensitivity to teratogens (eg, thalidomide) should be considered in selecting the species for DART studies. Based on these considerations, rodents are usually used for DART studies, and rabbits are additionally used for EFD studies to detect off-target toxicities. To address the on-target DART of oligonucleotides, pharmacologically relevant species should be selected from among those commonly used in DART studies. When no pharmacologically relevant species exists or an NHP is the only pharmacologically responsive species, DART studies using a surrogate can be considered, as discussed in the Use of Surrogates section.
Dose selection in DART studies should be based on all available information (eg, pharmacology, repeat-dose toxicity, and pharmacokinetic profile). When evaluating DART, the highest dose should be determined based on various information (eg, minimal toxicity, exposure saturation, exposure margin, and MFD) as described in S5(R3).
Information on placental transfer might be useful for evaluating the effects of drugs on embryos and fetuses. Several studies have reported that the placental transfer of phosphorothioate-modified nucleic acids was very limited [59,60]. However, quantifiable oligonucleotide levels and pharmacology-related changes have been detected in the fetus in some studies [61,62].
Carcinogenicity studies
Evaluation of carcinogenicity caused by off-target toxicities attributed to chemical modifications is considered necessary for ONTs, although standard carcinogenicity studies in rodents are generally inappropriate for biopharmaceuticals. The ICH S1A guidance addressed whether carcinogenicity studies are necessary [63]. If the period of clinical use is for 6 months or more and carcinogenicity studies are recommended, these can be performed as conventional rodent studies. As evaluation of carcinogenicity requires two species of rodents, the evaluation of ONTs with chemical modifications also would conform to the current requirement [64]. However, novel approaches as alternative methods may be acceptable in the future if evaluation strategies are developed based on newly acquired data on carcinogenicity (toxicity) induced by specific chemical modifications.
It is difficult to eliminate all the carcinogenicity concerns of a test compound regardless of whether existing carcinogenicity data are available if the target is human specific and the compound's on-target effects might involve carcinogenicity (eg, effects related to immunosuppression) or if toxicity findings indicating carcinogenicity were observed in genotoxicity, repeat-dose toxicity, or other studies. In such situations, carcinogenicity studies should not be conducted because negative results obtained from a conventional study would not eliminate the concern. Also, if such concerns are obvious before conducting the study, then a carcinogenicity study should not be performed because of the 3Rs principles. In such cases, by considering the clinical risks and benefits, not performing a carcinogenicity study is regarded as an acceptable option (Fig. 4). Appropriate risk communication should then be provided through package inserts and other means of conveying such information as “no carcinogenicity studies have been conducted.”
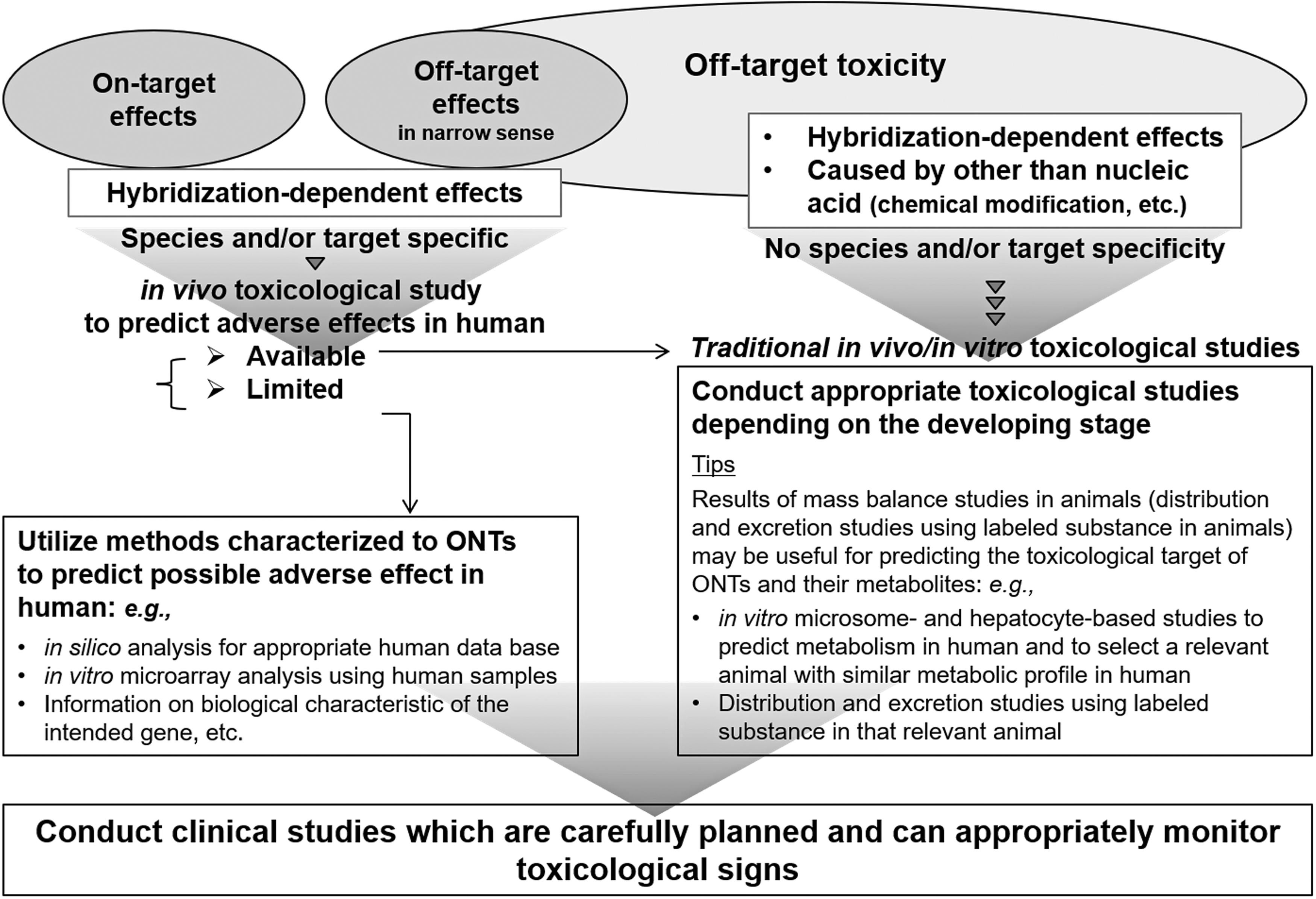
Points to consider for evaluating the carcinogenicity of therapeutic oligonucleotides.
Conclusions
Recent progress in the development of ONTs has been remarkable, but their nonclinical safety assessments are currently conducted on a case-by-case basis by referring to ICH guidelines related to nonclinical assessments, including S6(R1) for biopharmaceuticals and M3(R2). To evaluate new drug candidates, including, but not limited to ONTs, efforts to determine what is necessary for appropriate evaluation are indispensable in parallel with consideration of the limitations of traditional approaches. As described in the Introduction section, various investigations regarding the nonclinical safety assessment of ONTs are ongoing in parallel in Japan and other countries. The WGS6 expects that our efforts, along with the suggestions of the OSWG, will lead to viable guidelines in the future [6–11].
Footnotes
Acknowledgments
We thank Dr. Art Levin from Avidity Nanomedicines and Dr. Scott Henry from Ionis Pharmaceuticals for valuable comments on the article. We also thank Susan R. Doctrow, PhD, and Victoria Muir, PhD, from Edanz Group for editing drafts of this article.
Author Disclosure Statement
K.K. is an employee of MSD; K.K. and T.N. are consultants for AnGes, Inc.; S.O. is a co-founder of Luxna Biotech Co., Ltd.; K.W. is employee of Chugai Pharmaceutical Co., Ltd.; and M.S. is an employee of Kyowa Kirin Co., Ltd. All other authors have no competing financial interests.
Funding Information
This work was supported by the Japan Agency for Medical Research and Development (AMED, Promotion Project “Pharmaceutical Regulations Harmonization and Evaluation Research Project”).