
Abstract
Myotonic dystrophy type 1 (DM1) is a dominantly inherited, multisystemic disorder characterized clinically by delayed muscle relaxation and weakness. The disease is caused by a CTG repeat expansion in the 3′ untranslated region (3′ UTR) of the DMPK gene, which leads to the expression of a toxic gain-of-function mRNA. The expanded CUG repeat mRNA sequesters the MBNL1 splicing regulator in nuclear-retained foci structures, resulting in loss of protein function and disruption of alternative splicing homeostasis. In this study, we used CAG repeat antisense oligonucleotides (ASOs), composed of locked nucleic acid (LNA)- and 2′-O-methyl (2′OMe)-modified bases in a chimeric design, to alleviate CUGexpanded-mediated toxicity. Chimeric 14–18mer LNA/2′OMe oligonucleotides, exhibiting an LNA incorporation of ∼33%, significantly ameliorated the misregulated alternative splicing of Mbnl1-dependent exons in primary DM1 mouse myoblasts and tibialis anterior muscles of DM1 mice. Subcutaneous delivery of 14mer and 18mer LNA/2′OMe chimeras in DM1 mice resulted in high levels of accumulation in all tested skeletal muscles, as well as in the diaphragm and heart tissue. Despite the efficient delivery, chimeric LNA/2′OMe oligonucleotides were not able, even at a high-dosage regimen (400 mg/kg/week), to correct the misregulated splicing of Serca1 exon 22 in skeletal muscles. Nevertheless, oligonucleotide doses were well-tolerated as determined by histological and plasma biochemistry analyses. Our results provide proof of concept that inhibition of MBNL1 sequestration by systemic delivery of a steric-blocking ASO is extremely challenging, considering the large number of target sites that need to be occupied per RNA molecule. Although not suitable for DM1 therapy, chimeric LNA/2′OMe oligonucleotides could prove to be highly beneficial for other diseases, such as Duchenne muscular dystrophy, that require inhibition of a single target site per RNA molecule.
Introduction
Myotonic dystrophy type I (DM1) is a dominantly inherited, multisystemic disorder characterized clinically by muscle stiffness—a result of delayed muscle relaxation—and progressive weakness [1,2]. Pathology arises from a CTG repeat expansion in the 3′ untranslated region (3′ UTR) of the dystrophia myotonica protein kinase (DMPK) gene, which transcribes into a toxic gain-of-function mRNA [3]. The unusually long CUGexp region in the mutant transcript folds into a stable hairpin structure and sequesters the splicing regulator muscleblind-like-1 (MBNL1), forming large ribonucleoprotein (CUGexp-MBNL1) aggregates (or foci) inside the nucleus [4]. MBNL1 sequestration leads to its functional depletion in the nucleus and cytoplasm causing missplicing of its own pre-mRNA and other regulatory targets [5–9]. Abnormally expressed embryonic or nonfunctional splice isoforms help explain several multisystemic symptoms of the disease, including cardiac conduction abnormalities—a result of SCN5A (cardiac sodium channel) and TNNT2 (cardiac troponin T type 2) missplicing [8,10]—and insulin insensitivity—due to aberrant alternative splicing of IR (insulin receptor) [9].
In the last decade, therapeutic approaches for DM1 have mainly been focused on the use of chemically modified antisense oligonucleotides (ASOs) that either target the expanded repeat tract or bind the mutant DMPK mRNA outside the CUGexp region. Depending on the type of chemistry introduced and the design of the antisense construct, ASOs can mitigate RNA toxicity by two possible mechanisms: (1) competing with MBNL1 for CUGexp binding and/or displacing prebound MBNL1 proteins (steric block mechanism); (2) inducing degradation of DMPK mRNA via the RNase H machinery. Both mechanisms would result in functional restoration of MBNL1 activity, and consequently, correction of missplicing.
The biggest challenge in ASO development is optimizing the right combination and precise pattern of chemical modifications to be incorporated. Chemical modifications include alterations to the phosphodiester (PO) backbone (eg, phosphorothioate, morpholino), the sugar ring [eg, 2′-O-methyl (2′OMe), 2′-O-methoxyethyl (MOE), locked nucleic acid (LNA), and 2′,4′-constrained ethyl (cEt)], and/or the nucleobases. First-generation ASOs have commonly incorporated the phosphorothioate (PS) modification, which substitutes sulfur for a nonbridging oxygen in the phosphate backbone and confers significant resistance to nuclease degradation [11]. PS linkages are cost-effective, RNase H-compatible modifications that markedly increase ASO binding to plasma proteins, thus improving drug pharmacokinetics [12,13]. Despite increasing ASO stability, PS linkages reduce the binding affinity of an oligonucleotide toward its target sequence, resulting in decreased ASO specificity and potency [11,14]. Importantly, PS-ASOs have also been shown to associate with key cellular proteins (including C23/nucleolin and paraspeckle proteins) in a sequence-independent manner [15–19], a finding that would explain the previously reported side effects of PS oligonucleotides on cells and tissues [20–24]. Indeed, a recent publication by Shen et al. provided the first direct evidence that nonspecific binding of known toxic PS-ASOs to paraspeckle proteins results in nucleolar stress, p53 activation, and apoptotic cell death [25].
To counteract the reduced specificity observed with the PS modification, second-generation ASOs have incorporated naturally occurring 2′OMe or MOE residues that increase binding affinity for an RNA target and show inherent resistance to cellular endonucleases [13]. 2′-O-alkyl-modified ASOs are still susceptible to exonuclease attack and are therefore best used in combination with a small number of PS linkages, at the 3′ and 5′ end, to block both exonuclease and endonuclease degradation. When incorporated in a gapmer design—chemically modified ASO ends flanking a central unmodified region—the 2′-O-alkyl modification is compatible with RNase H activity [26]. Nevertheless, DMPK mRNA degradation has been reported using a full 2′OMe-PS ASO, possibly via an RNase H-independent mechanism [27].
The LNA is a third-generation bicyclic sugar modification, which exhibits unprecedented thermal stability compared with other 2′-chemistries [28,29]. This feature significantly increases the binding affinity of LNA-modified ASOs, but at the same time renders them more tolerant to mismatches, leading to possible off-target interactions [30]. LNA nucleotides are resistant to nuclease degradation and can induce RNase H activity when incorporated in a gapmer design [31]. Owing to their unusually strong binding affinity, LNA monomers have been incorporated in a short (8–12 bp long), all-LNA antisense construct against CUGexp RNA with encouraging results [32]. The 10mer all-LNA ASO reduced the number of RNAexp-MBNL1 foci and specifically corrected MBNL1-sensitive splicing defects in proliferating human DM1 cells and tibialis anterior (TA) muscle of DM1 mice after a single intramuscular injection [32]. Despite encouraging data at the intramuscular level, the short all-LNA ASO has not been evaluated following systemic administration in mice. Importantly, several non-DM1-related studies carrying out short-term tolerability assays in mice have shown that LNA-modified gapmer ASOs are profoundly hepatotoxic [33–35].
The first promising ASO for DM1 therapy, targeting CUGexp RNA outside the repeat tract, was based on an MOE gapmer design with a full PS backbone [36]. Systemic delivery of this second-generation ASO in DM1 transgenic mice resulted in a significant reduction of CUGexp RNA in skeletal muscles, correction of missplicing, and elimination of myotonic discharges, an effect that was sustained for up to 1 year after the last dose [36]. Despite promising preclinical data, the MOE gapmer did not translate into human trials. Ionis Pharmaceuticals and collaborators then proceeded with the development of another RNase H-dependent ASO, modified with the cEt chemistry in a gapmer design. Similar to LNA, cEt residues show significantly increased RNA binding affinity compared with second-generation 2′-O-alkyl-modified units. Systemic administration of the cEt gapmer in human DMPK transgenic mice or cynomolgus monkeys resulted in up to 70% reduction of DMPK mRNA in skeletal muscles and ∼50% in cardiac muscle, an effect that was sustained for several months after the end of treatment [37]. This was the first ASO to progress to a phase 1–2 clinical trial for the treatment of DM1; a study that was recently concluded. Although results showed encouraging trends in biomarker and alternative splicing changes, ASO accumulation in muscle tissues did not reach sufficient concentration levels to elicit a significant therapeutic response [38].
RNase H-dependent gapmer ASOs have been well studied in DM1 and have shown great promise for therapy. By contrast, the therapeutic potential of steric-blocking ASOs has been much less explored. Intravenous delivery of a first-generation (morpholino), steric-blocking ASO against CUGexp RNA failed to correct missplicing in skeletal muscles of DM1 transgenic mice [39], while third-generation, all-LNA ASOs did not advance beyond intramuscular evaluation [32]. In this study, we designed and tested the activity of CAG repeat steric-blocking ASOs, of variable sizes, that combine two commonly used high binding affinity modifications in a single antisense construct: the third-generation LNA chemistry, which exhibits unprecedented duplex stability, and the naturally occurring, second-generation 2′OMe chemistry, which shows high RNA binding affinity and inherent resistance to endonuclease degradation. Chimeric LNA/2′OMe oligonucleotides have previously been evaluated in microRNA inhibition studies, demonstrating superior potency compared with their LNA- or 2′OMe-modified counterparts. The combination of 2′OMe monomers with a limited number of LNA nucleotides (one at every third position) aims at minimizing potential toxicity, while retaining a high binding affinity. Furthermore, replacement of the PO backbone with a PS-modified backbone aims at enhancing ASO binding to plasma and cell surface proteins, thus increasing stability and cellular uptake in vivo [15,19].
Materials and Methods
Antisense oligonucleotides
All ASOs used in the study are listed in Table 1. Small-scale ASOs used in the in vitro experiments were synthesized in the laboratory of Professor Jesper Wengel, while ASOs delivered in vivo were purchased from RiboTask ApS. All ASOs bind directly to the expanded CUG repeats but vary in terms of their length (11–18 nucleotides long), the chemistries that they incorporate, and/or the percentage of LNA nucleotides that they hold. Three types of chemical modifications were used: LNA, 2′OMe, and PS. The LNA modification was introduced either at the cytosine or the adenine base, specified in the naming as “LNAc” or “LNAa,” respectively. All ASOs were synthesized on a fully modified PS backbone. In addition to our chimeric LNA/2′OMe ASOs, we have included two LNA/DNA mixmer ASOs, “LNAc/DNA18” and the previously published “LNA/DNA18,” to serve as functional controls.
Oligonucleotide Nomenclature, Sequence, and Chemistry
The name of each ASO depicts the sugar chemistries that it incorporates (ie, LNA, 2′OMe, and/or DNA), the position of the LNA residues (ie, “LNAc” = at every cytosine base; “LNAa” = at every adenine base), and the number of bases it consists of (11, 14, 16, or 18). Where necessary, the letters “h” and “ℓ” were added at the end of the name to differentiate same-length ASO counterparts with a high or low LNA percentage, respectively. All ASOs used in the study have a full PS backbone.
Bold and underlined = LNA, m = 2′OMe, * = PS linkages, d = DNA.
ASOs, antisense oligonucleotides; LNA, locked nucleic acid; 2′OMe, 2′-O-methyl.
Animal experiments
All animal studies were carried out at the Transgenic Mouse Facility of the Cyprus Institute of Neurology and Genetics, following evaluation by the “National Committee for the Protection of Animals Used for Scientific Purposes” and approval by the Cyprus Veterinary Services (project license approval number: CY/EXP/PR.L2/2014). Homozygous HSALR20b breeders were kindly provided by Professor Charles A. Thornton of the University of Rochester, United States, while FVB/N mice were purchased from Jackson Laboratories. Intramuscular delivery of ASOs was carried out under tribromoethanol anesthesia (Avertin). Briefly, 4 nmols of ASO was injected into the TA muscle of 2–3-month-old HSALR mice, followed by electroporation of the treated muscle using previously established parameters (100 V/cm, 20 ms pulse width, 1 Hz, 10 pulses). The contralateral TA was injected with saline only. For systemic delivery, 200 mg/kg of ASO was injected subcutaneously into the interscapular region of 2-month-old female HSALR mice, twice a week for 4 weeks. ASO pharmacokinetics was studied upon a single subcutaneous injection of the same dose (200 mg/kg). Blood sample collection was carried out from the venous orbital sinus under general anesthesia. Analyses of plasma creatine phosphokinase (CPK), aspartate aminotransferase (AST), alanine aminotransferase (ALT), blood urea nitrogen (BUN), and creatine were carried out by a private Analytical Bioscience Laboratory in Nicosia, Cyprus. All animals were sacrificed by cervical dislocation.
Isolation of single muscle fibers and associated satellite cells
The protocol for isolating satellite cells from single muscle fibers was adapted from Rosenblatt and colleagues [40]. In brief, extensor digitorum longus (EDL) muscles were isolated from 6-week-old HSALR mice and incubated in 0.2% type I collagenase in Dulbecco's modified Eagle's medium (DMEM, high glucose, pyruvate, no glutamine; Thermo Fisher Scientific), supplemented with 1 × penicillin/streptomycin (P-S; Thermo Fisher Scientific) and 1 × GlutaMAX solution (Thermo Fisher Scientific), at 37°C for 1 h. Following digestion, single muscle fibers were liberated by repeatedly triturating the EDL muscle using a wide-mouth glass pipette. Viable single muscle fibers were carefully transferred to a new culture dish, in three rounds of washing, to discard hypercontracted fibers and tissue debris. Finally, viable muscle fibers were transferred to a new culture dish coated with 1 mg/mL Matrigel (Corning) in DMEM (1 × P-S/1 × GlutaMAX) and incubated in “plating medium” [DMEM supplemented with 1 × P-S, 1 × GlutaMAX, 10% horse serum (HS; Thermo Fisher Scientific), and 0.5% chick embryo extract (CEE; MP Biomedicals)] at 37°C for 3 days, to allow myogenic satellite cells to dissociate from their fibers. Three days after plating, the muscle fibers were removed from the dish, and the “plating medium” was replaced with “proliferation medium” consisting of 20% fetal bovine serum (FBS; Thermo Fisher Scientific), 10% HS, 1% CEE, and 2.5 ng/mL recombinant murine fibroblast growth factor-basic (FGF-b; PeproTech) in DMEM (1 × P-S/1 × GlutaMAX). Myogenic cells were allowed to grow in 5% CO2 and 37°C for 2–3 days, before plating for transfection.
Cell culture and ASO delivery
Primary HSALR myoblasts were seeded in 12-well plates coated with 1 mg/mL Matrigel and transfected at 60% confluency in DMEM supplemented with 10% FBS, 1 × P-S, and 1 × GlutaMAX. ASOs were diluted in Opti-MEM™ Reduced Serum Medium (Thermo Fisher Scientific), complexed with Lipofectamine 3000 reagent (Invitrogen), and delivered at a final concentration of 50 nM per well. Cells were collected 24 h post-transfection and processed for RNA extraction using the RNeasy Mini Kit (Qiagen).
DM1 (cell line 10009) human myoblasts were purchased from EuroBioBank and grown in DMEM (high glucose, pyruvate, no glutamine; Thermo Fisher Scientific) supplemented with 20% FBS, 1 × P-S solution (Thermo Fisher Scientific), 1 × GlutaMAX solution (Thermo Fisher Scientific), 10 μg/mL insulin, human recombinant, zinc solution (Thermo Fisher Scientific), 25 ng/mL recombinant human FGF-b (Thermo Fisher Scientific), and 10 ng/mL recombinant human epidermal growth factor (Thermo Fisher Scientific). Cells were incubated in 5% CO2 at 37°C. For ASO delivery, DM1 myoblasts were seeded in six-well plates so that they would be 60% confluent at the time of transfection. ASOs were diluted in Opti-MEM Reduced Serum Medium, complexed with Lipofectamine 3000 reagent, and delivered at a final concentration of 125 nM per well. Treated cells were either collected for RNA extraction using the RNeasy Mini Kit or seeded on Poly-Prep slides (Sigma) for subsequent fluorescence in situ hybridization (FISH), 48 h post-transfection.
Fluorescence in situ hybridization
RNA FISH was carried out using a (CAG)10 probe labeled with Atto 550 at the 3′-end (Eurofins MWG Operon). Transfected DM1 myoblasts were seeded on Poly-Prep slides (Sigma), fixed in 4% (w/v) paraformaldehyde in phosphate-buffered saline (PBS) for 15 min at 4°C, washed three times in PBS for 5 min at room temperature, permeabilized in 0.4% Triton X-100 for 5 min at room temperature, and washed three times in PBS for 5 min at room temperature. Cells were prehybridized in 40% deionized formamide/2 × SSC in PBS for 15 min at room temperature followed by hybridization in buffer containing 1 ng/μL probe, 40% deionized formamide, 10% dextran sulfate, 1 mg/mL salmon sperm DNA, 1 mg/mL yeast tRNA, 0.2% BSA, 2 mM vanadyl ribonucleoside complex, and 2 × SSC, for 2 h at 50°C. Posthybridization, washes were carried out in 40% deionized formamide/2 × SSC in PBS for 30 min at 53°C, followed by 2 × SSC in PBS for 30 min at 43°C. Cells were counterstained with DAPI II mounting medium and visualized on a Zeiss fluorescent microscope equipped with the AxioCam MRm Zeiss camera. The number of nuclear-retained foci was counted in at least 180 nuclei from two independent transfection experiments.
To carry out FISH on muscle tissue sections, quadriceps muscles were snap-frozen in liquid nitrogen-cooled isopentane and stored at −80°C. Frozen tissues were cryosectioned in the coronal plane, at 5-μm thickness, and collected at 100-μm intervals. Tissue cryosections were fixed in cold 4% paraformaldehyde in PBS for 10 min at room temperature, washed three times in PBS for 5 min, permeabilized in prechilled 1:1 acetone/methanol mixture for 10 min at −20°C, and washed three times in PBS for 5 min. Tissue sections were prehybridized in 50% deionized formamide/4 × SSC in PBS for 10 min at room temperature followed by hybridization in a buffer containing 1 ng/μL probe, 50% deionized formamide, 4 × SSC, 10% dextran sulfate, 0.1 mg/mL salmon sperm DNA, 1 mg/mL yeast tRNA, 0.1% BSA, and 2 mM vanadyl ribonucleoside complex, for 2 h at 40°C. Tissue sections were then washed twice in 1 × SSC in PBS for 20 min at 55°C, and once in 0.5 × SSC in PBS for 15 min at 55°C. Nuclear counterstaining was carried out in Hoechst solution (1:10,000) for 5 min at room temperature, followed by two washes in PBS for 5 min. Tissue sections were visualized under a TCSL confocal microscope (Leica, Germany).
Semiquantitative alternative splicing assays
Total RNA was extracted from untransfected and ASO-transfected myoblasts (human DM1 and primary HSALR) using the RNeasy Mini Kit (Qiagen). First-strand cDNA was synthesized using 500–1,000 ng total RNA, 0.25 mM dNTPs, 0.75–3 μM random hexamer primers (MWG-Biotech), 200 U M-MuLV reverse transcriptase (NEB), and 1 × M-MuLV transcription buffer (NEB), at 37°C for 1 h. Alternative splicing of hMBNL1 and mMbnl1 exon 7 was assessed by polymerase chain reaction (PCR) amplification of cDNA, using upstream (5′-GCTGCCCAATACCAGGTCAAC-3′) and downstream (5′-TGGTGGGAGAAATGCTGTATGC-3′) primers that bind to exon 6 and 8, respectively. These primers generate either a 216-bp fragment, lacking exon 7, or a 270-bp fragment, which includes exon 7. After 27–28 cycles of amplification, PCR products were electrophoretically resolved on 1.8% agarose gels and visualized by ethidium bromide staining. Band intensities were measured using ImageJ software and the percentage of exon 7 inclusion was calculated in Microsoft Excel. PCR experiments involved three independent transfection replicates.
To assess alternative splicing changes following in vivo ASO delivery, skeletal muscle tissues were isolated and homogenized in TRIzol reagent (Ambion). Total RNA was then extracted according to the manufacturer's instructions, treated with DNAse 1 (NEB) and further purified using acid phenol:chloroform (1:1) extraction. First-strand cDNA was synthesized using 1,000 ng total RNA, 0.5 mM dNTPs, 6 μM random primer mix (NEB), 200 U M-MuLV reverse transcriptase (NEB), and 1 × M-MuLV transcription buffer (NEB), at 42°C for 1 h. Alternative splicing of mSerca1 exon 22 and mMbnl1 exon 7 was assessed by PCR amplification of cDNA using upstream (for mSerca1: 5′-GCTCAT GGTCCTCAAGATCTCAC-3′, for mMbnl1: see paragraph above) and downstream (for mSerca1: 5′-GGGTCAGTGCCTCAGCTTTG-3′, for mMbnl1: see paragraph above) primers, for 21 and 24 cycles, respectively. PCR products were electrophoretically resolved on 5% polyacrylamide gels, stained with SYBRSafe (Thermo Fisher Scientific), and analyzed using ImageJ.
Quantification of ASOs in mouse plasma and tissues using liquid chromatography tandem mass spectrometry
Quantification of LNAc/2′OMe18h and LNAc/2′OMe14 ASOs in the skeletal muscle (TA, quadriceps, and gastrocnemius), diaphragm, heart, liver, and kidney tissues was carried out by liquid chromatography with tandem mass spectrometry (LC-MS/MS), as previously described in Goyenvalle et al. In brief, ∼50 mg of tissue was isolated and homogenized in 1 mL of proteinase K buffer (100 mM Tris-HCl, pH 8.5, 200 mM NaCl, 5 mM EDTA, and 0.2% SDS) containing 2 mg/mL of proteinase K (Roche Applied Science). To enhance enzyme activity, the samples were incubated at 55°C for 2 (liver) or 4 h (kidney, heart, diaphragm, and skeletal muscle) and then centrifuged at 16,000g for 15 min. The supernatant was collected and prepared for LC-MS/MS analysis by solid-phase extraction (SPE) on Oasis HLB cartridges (10 mg; Waters, United Kingdom). First, 100 μL of the supernatant sample was spiked with 100 μL of internal standard (IS) solution (0.025 mg of 16mer LNAc/2′OMe16 ASO in 100 mM triethylammonium bicarbonate buffer) and incubated at room temperature for 30 min. The SPE cartridges were preconditioned with 1 mL of methanol followed by 2 × 500 μL of buffer A [100 mM 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP; Honeywell Fluka) and 8.6 mM triethylamine (TEA; Sigma) in ultrapure water]. Subsequently, 200 μL of buffer A was added to the ASO-IS mixture and the sample was loaded on the SPE cartridge. The cartridges were washed twice with 250 μL of buffer A and once with 300 μL of 100 mM triethylammonium bicarbonate. ASO elution was carried out using 2 × 200 μL of 40% buffer A in methanol. The samples were dried in a rotational vacuum concentrator (RVC 2–25 CDplus, CHRIST) coupled to a freeze dryer (Alpha 1–4 LSCbasic, CHRIST) and solubilized in 50 μL mobile phase A (15 mM TEA/400 mM HFIP) just before LC-MS/MS analysis.
Sample analysis was performed on a Xevo TQD MS instrument (Waters), coupled to an Acquity UPLC I-class system (Waters), and operated in the negative ion multiple reaction monitoring (MRM) mode (capillary voltage −2.9 kV, source temperature 400°C). Chromatographic separations were carried out on an Acquity BEH OST C18 1.7 μm, 2.1 × 100 mm analytical column (Waters) with gradient elution using mobile phase A and mobile phase B (50% mobile phase A, 50% methanol). Gradient elution was performed at a flow rate of 200 μL/min, starting with 42% B, followed by a linear increase to 66% within 4 min and then to 80% within the next 1 min. The column temperature was kept at 60°C throughout the analyses.
The concentrations of the experimental ASOs (LNAc/2′OMe18h and LNAc/2′OMe14) and IS ASO (LNAc/2′OMe16) were determined according to the peak area ratios AAnalyte/AIS for the following MRM transitions: LNAc/2′OMe18h [M-9H]9−, m/z 711.5 > 346.1; LNAc/2′OMe16 [M-9H]9−, m/z 629.9 > 346.0; LNAc/2′OMe14 [M-8H]8− m/z 618.5 > 346.2 (additional transitions for each precursor ion were used as confirmation ions, see Supplementary Table S1). Calibration was based on a twofold serial dilution of all three ASOs (LNAc/2′OMe18h, LNAc/2′OMe14, and LNAc/2′OMe16) in mobile phase A, over a concentration range of 3.9–1,000 μg/mL.
Drug pharmacokinetics was studied upon a single subcutaneous injection of 200 mg/kg of the respective ASO, followed by blood collection at 0, 30, 60, 90, and 120 min postinjection. A fixed volume of plasma (15 μL) was isolated from each blood sample and spiked with 0.05 mg of IS ASO (LNAc/2′OMe16). Plasma samples were then treated with 200 μL of 5% ammonium hydroxide solution, mixed together by vortexing for 10 min, to denature plasma proteins and displace ASO molecules from their binding sites. An equal volume (200 μL) of cold PCA buffer (phenol:chloroform:isoamyl alcohol, 25:24:1; Sigma) was then added to the sample, mixed by vortexing for 10 min, and then centrifuged at maximum speed for 10 min. The aqueous phase was carefully collected and mixed with an equal volume of 100 mM TEA bicarbonate solution for 30 min, to allow ion-pair formation between the ASOs and triethylammonium ions. The SPE method and LC-MS/MS analysis that followed are the same as described for the biodistribution study.
Histology
ASO toxicity was evaluated in the liver and kidney tissues of HSALR mice treated with 200 mg/kg of the respective ASO, twice a week for 4 weeks. Liver and kidney tissue samples were isolated 2 days after the last dose, fixed overnight in 4% PFA, and embedded in paraffin. The samples were sectioned, stained with hematoxylin and eosin, and examined blindly by a single veterinary pathologist.
Statistical analysis
Data are expressed as mean ± standard error, unless otherwise specified. Statistical significance was calculated using two-tailed unpaired t-test, followed by Bonferroni correction.
Results
Activity of chimeric LNA/2′OMe ASOs in vitro
To determine whether ASO delivery can improve splicing misregulation in primary HSALR myoblasts, we examined Mbnl1 alternative splicing changes in transfected and untransfected cells by two-step reverse transcription-PCR (RT-PCR). MBNL1 is a ubiquitously expressed splicing factor that autoregulates the alternative splicing of its own pre-mRNA. In healthy cells, increased nuclear MBNL1 activity enhances exon 7 skipping, resulting in MBNL1 isoforms that lack the nuclear localization signal (Fig. 1A) [7]. Reduced nuclear MBNL1 activity may in turn enhance exon 7 inclusion leading to MBNL1 nuclear localization. This feedback autoregulation is crucial for maintaining MBNL1 function in the nucleus [7]. In DM1 myoblasts, functional depletion of nuclear MBNL1 leads to enhanced exon 7 inclusion resulting in abnormal nuclear MBNL1 accumulation and enhanced CUGexp-MBNL1 interaction [7]. Correcting MBNL1 alternative missplicing will restore its function in the nucleus leading to correct splicing of other regulatory targets.
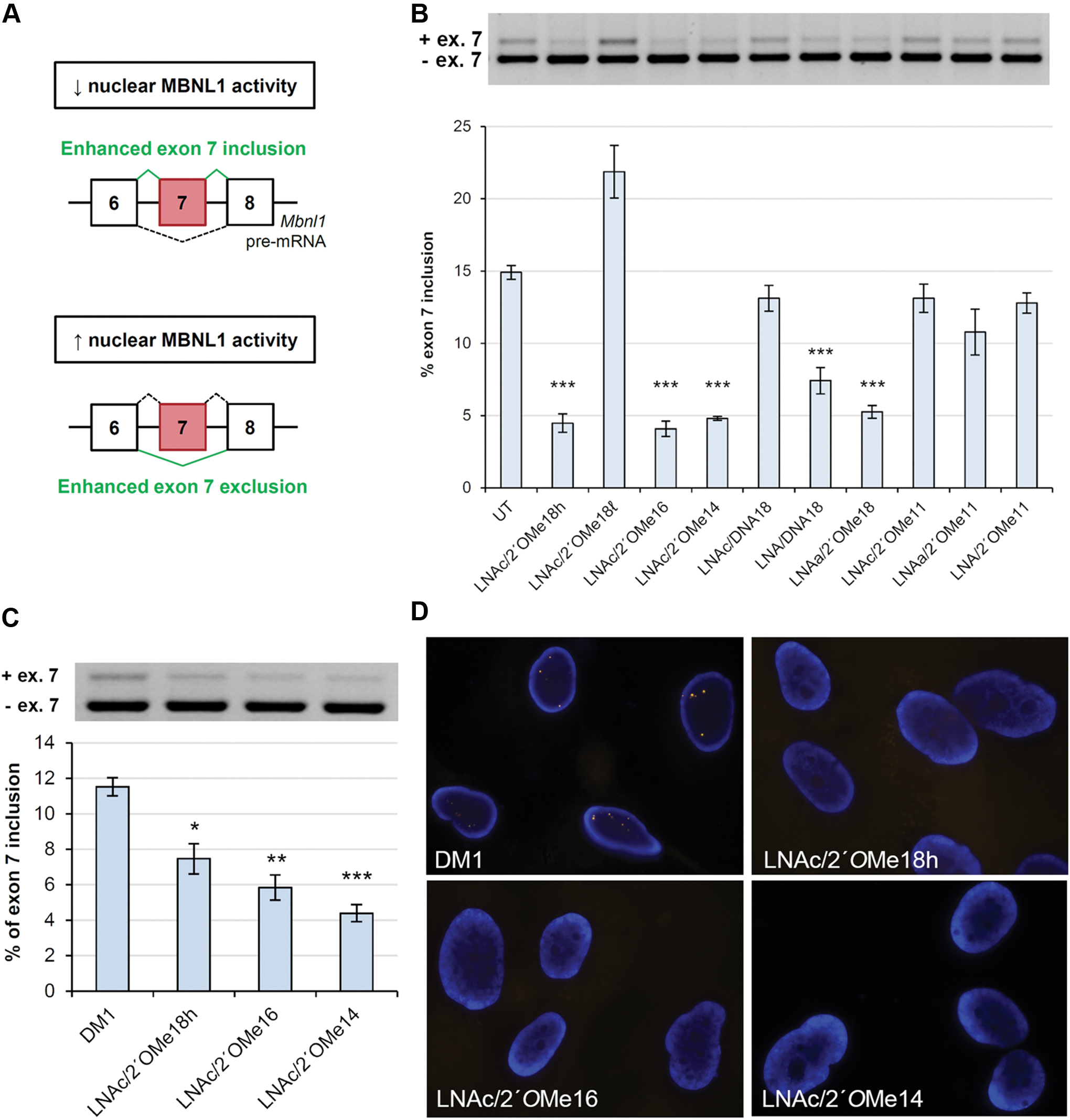
Chimeric LNA/2′OMe ASOs ameliorate Mbnl1 alternative missplicing in vitro.
Our 18mer, 16mer, and 14mer chimeric LNA/2′OMe ASOs (LNAc/2′OMe18h, LNAa/2′OMe18h, LNAc/2′OMe16, and LNAc/2′OMe14), exhibiting a relatively high LNA incorporation (33%–38%), significantly ameliorated Mbnl1 exon 7 missplicing in primary HSALR myoblasts (Fig. 1B). Control LNA/DNA18 mixmer, which contains the largest number of LNA nucleotides among all tested ASOs, was less potent than our 14–18mer LNA/2′OMe chimeras, but still significantly reduced the levels of exon 7-included Mbnl1 transcript in primary HSALR myoblasts. By contrast, control LNAc/DNA18 mixmer, with an LNA incorporation of 33.3%, and LNAc/2′OMe18ℓ, containing half the number of LNA residues than all other tested ASOs, did not improve the misregulated splicing of Mbnl1 exon 7. These results suggest that (1) the combination of LNA and 2′OMe monomers in a single antisense construct greatly enhances ASO potency compared with LNA-containing ASOs; and (2) an LNA incorporation of ∼33%, evenly distributed at every third position, shows the most potent results. Importantly, our 11mer LNA/2′OMe chimeras exhibiting a high LNA incorporation (36.3%) did not improve Mbnl1 missplicing, suggesting that the combination of LNA and 2′OMe chemistries in very short ASO designs does not enable sufficient target binding to achieve an effect.
To further evaluate ASO activity, LNAc/2′OMe18h, LNAc/2′OMe16, and LNAc/2′OMe14 chimeras were complexed with the Lipofectamine reagent and delivered in proliferating human DM1 myoblasts. All three ASOs were able to completely eliminate nuclear-retained CUGexp-MBNL1 foci and significantly improve MBNL1 exon 7 missplicing in human DM1 myoblasts, after 48 h of treatment (Fig. 1C, D). Unexpectedly, we observed that ASO activity increased with reduced ASO size. A possible explanation would be that a larger number of 14mer molecules can be accommodated per nucleic acid/cationic lipid complex than a 16mer or 18mer ASO. This trend was not seen following transfection of ASOs in primary HSALR myoblasts, probably due to differences in the efficiency of lipofection between the various cell lines.
Activity of chimeric LNA/2′OMe ASOs after intramuscular delivery
Following in vitro screening, we progressed to intramuscular delivery with the three most potent ASOs—LNAc/2′OMe18h, LNAc/2′OMe16, and LNAc/2′OMe14. All three oligomers have the same percentage of LNA-modified nucleotides but vary in size. The ASOs were delivered locally into the TA muscle of HSALR mice and electroporated to facilitate cellular uptake. Tissues were isolated 2 weeks post-treatment and alternative splicing regulation was assessed by RT-PCR analysis. The 18mer, 16mer, and 14mer LNA/2′OMe chimeras were able to reverse the misregulated alternative splicing of Mbnl1 exon 7 and Serca1 exon 22 by ∼50%, 40%, and 28%, respectively (Fig. 2). As expected, LNAc/2′OMe18h showed the highest potency in vivo, a result that contrasts to what we observed in cultured human DM1 myoblasts. A possible explanation would be the difference in the method—chemical versus physical—of ASO delivery. While cationic lipid-mediated transfection (chemical method) in cultured cells may be more sensitive to ASO size, thus favoring shorter nucleic acid molecules (see ‘Activity of chimeric LNA/2'OMe ASOs in vitro’ in the ‘Results’ section), ASO delivery by in vivo electroporation (physical method) may be less affected by molecular size.
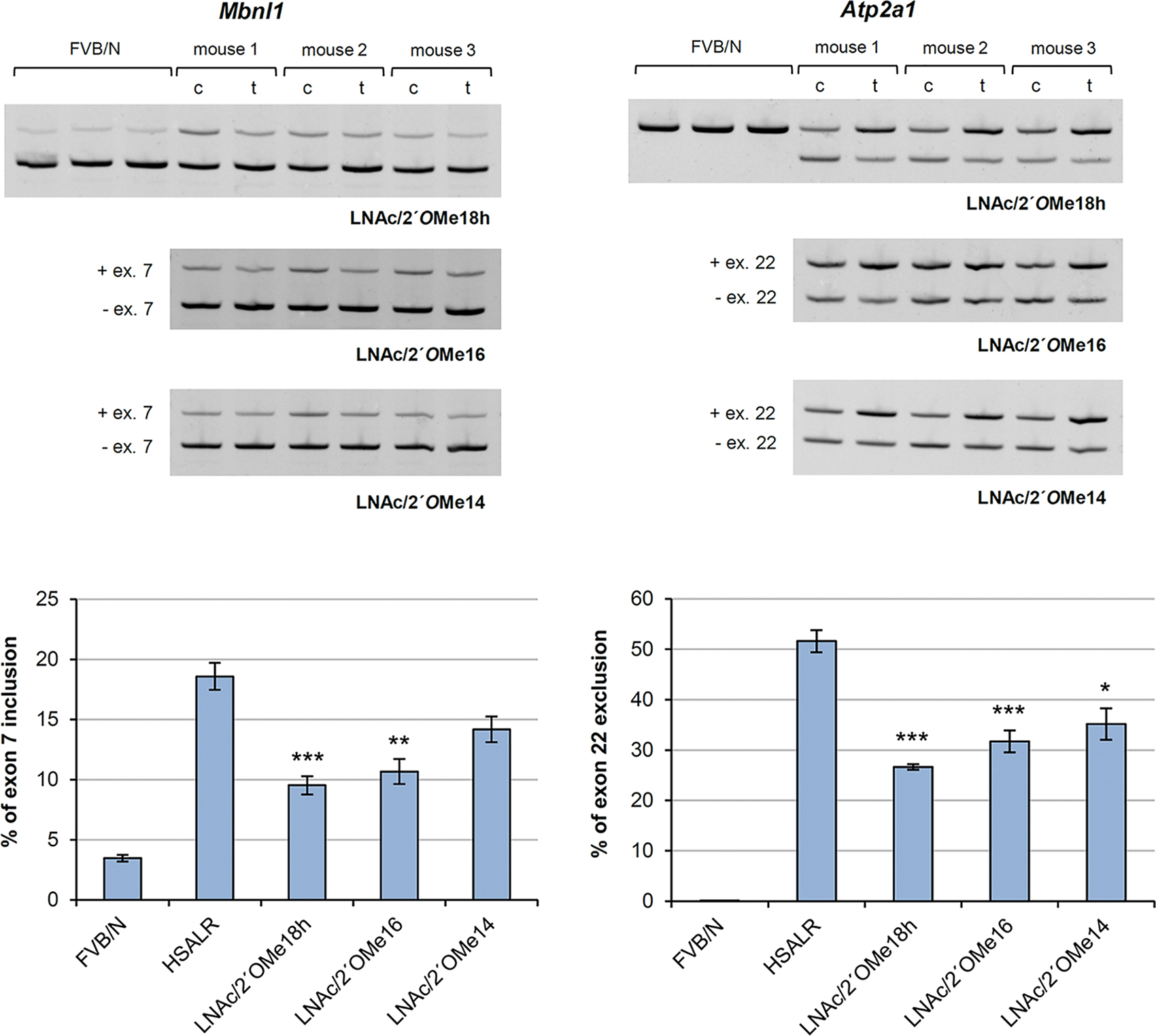
Partial reversal of misregulated alternative splicing after intramuscular delivery of chimeric LNA/2′OMe ASOs. RT-PCR analysis of alternative splicing changes for Mbnl1 and Serca1 in TA muscles of 2–3-month-old HSALR mice, sacrificed 2 weeks after treatment (t) with 4 nmol of one of the three LNA/2′OMe chimeras (n = 3 per tested ASO). The contralateral (c) TA was injected with saline only. Splice products from TA muscles of age-matched wild-type (FVB/N) mice (n = 3) are also shown. Quantification of results is expressed as the percentage of splice variant that includes or excludes the indicated exon. Values are mean ± S.E.M. *Denotes P < 0.05, **denotes P < 0.01, ***denotes P < 0.001 compared with saline controls using the two-sample t-test followed by Bonferroni correction. TA, tibialis anterior. Color images are available online.
Pharmacokinetics, biodistribution, and activity of chimeric LNA/2′OMe ASOs following subcutaneous delivery
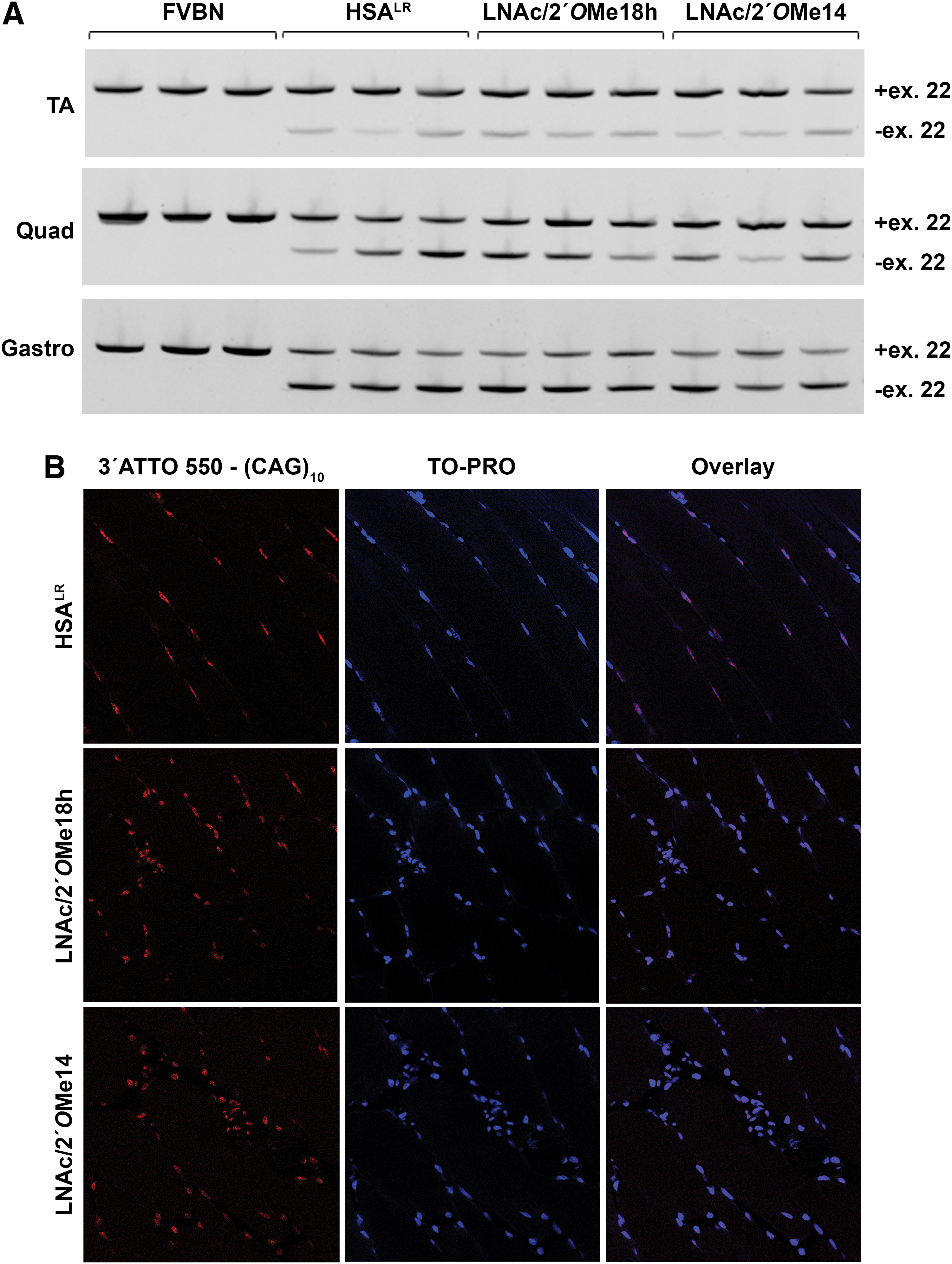
Drug pharmacokinetics was studied upon a single subcutaneous injection of 200 mg/kg of the respective ASO. Plasma levels of LNAc/2′OMe18h and LNAc/2′OMe14 rose rapidly within the first 30 min post injection, but continued to increase at a slower rate over the next 90 min (Fig. 3A). This is consistent with a relatively slow absorption rate of biomolecules administered via the subcutaneous route. To study biodistribution, 2-month-old HSALR female mice were treated with 200 mg/kg of one of the two ASOs—LNAc/2′OMe18h and LNAc/2′OMe14—or saline only, two times a week for 4 weeks. Skeletal muscles (TA, quadriceps, and gastrocnemius), diaphragm, heart, liver, and kidney tissues were isolated 2 days after the last dose and processed for LC-MS/MS analysis for the detection and quantification of the respective ASOs. Chimeric LNA/2′OMe ASOs accumulated in all tested skeletal muscles, as well as in the diaphragm and heart tissue (Fig. 3B). As expected, both ASOs accumulated to highest levels in the liver and kidneys, while size difference did not appear to affect ASO clearance. To assess the effect of ASO treatment on Mbnl1-dependent splicing misregulation, all three skeletal muscles were processed for RNA extraction and RT-PCR analysis of Serca1 splice variants. Unfortunately, even at this high-dosage regimen, our chimeric LNA/2′OMe ASOs did not correct the misregulated splicing of Serca1 exon 22 (Fig. 4A). Consistent with these data, FISH analysis of quadriceps muscle tissue from ASO-treated HSALR mice revealed no decrease in the number or intensity of nuclear-retained CUGexp-Mbnl1 foci compared with saline-treated controls (Fig. 4B).
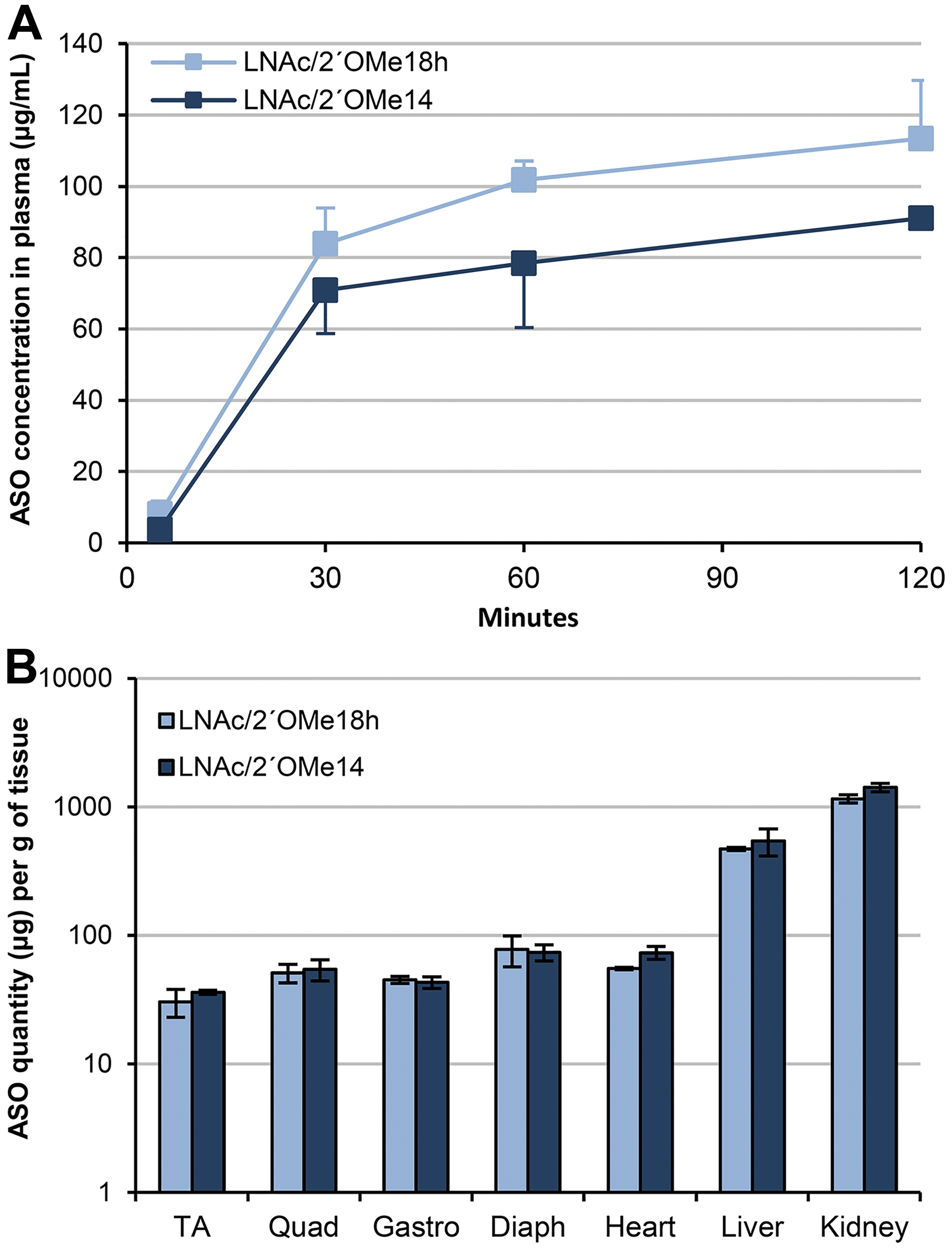
Pharmacokinetics and biodistribution of chimeric LNA/2′OMe ASOs.
Toxicity status of chimeric LNA/2′OMe ASOs in vivo
To determine the safety of chimeric LNA/2′OMe ASOs in vivo, body and tissue weights, as well as plasma markers of hepatic injury or renal function, were measured after treatment with 200 mg/kg of the indicated ASO, twice a week for 4 weeks. Body weights, as well as the weights of spleen, liver and kidney tissues, were similar between ASO- and saline-treated HSALR mice (Fig. 5A). Studies of hepatic cytotoxic markers showed slightly elevated levels of AST in LNAc/2′OMe18h- and LNAc/2′OMe14-treated HSALR mice compared with saline controls, and similar levels of ALT in all ASO-treated and untreated mice (Fig. 5B). Plasma CPK, a marker of muscle damage, was reduced in all ASO-treated HSALR mice, particularly with ASO LNAc/2′OMe14 administration. Studies of renal function showed similar levels of creatinine in all ASO-treated and untreated mice, and slightly elevated levels of BUN in LNAc/2′OMe18h-treated HSALR mice, compared with saline controls.
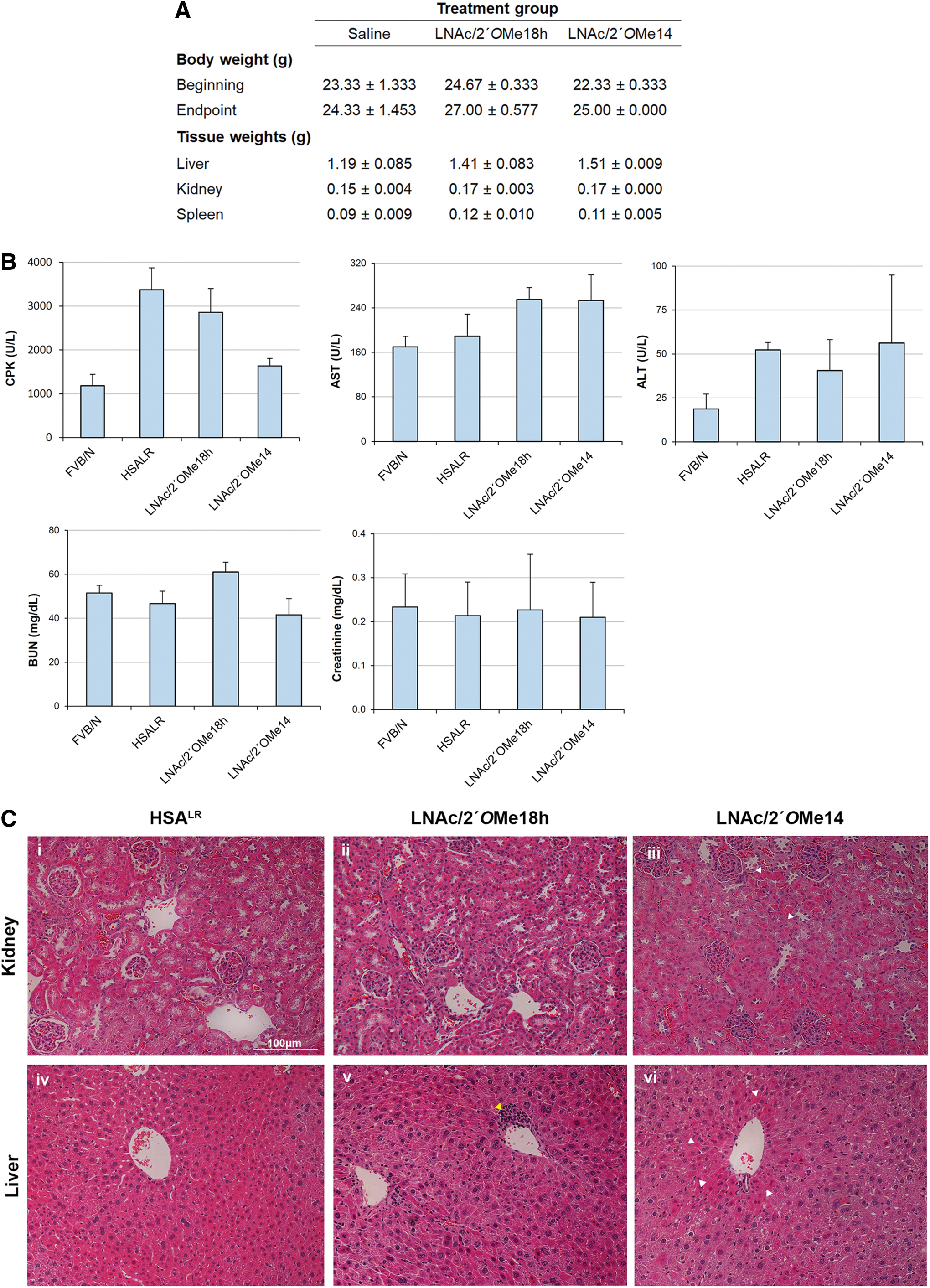
ASO safety was further assessed by histological analysis of liver and kidney sections stained with hematoxylin and eosin (Fig. 5C). The majority of sections examined, from both control and ASO-treated HSALR mice, exhibited mild hyperemia probably due to the fact that no animal perfusion had been carried out before tissue isolation. Assessment of renal tissue sections revealed no histological abnormalities in saline- and LNAc/2′OMe18h-treated HSALR mice, while in two mice injected with LNAc/2′OMe14, we observed acute mild necrosis affecting ∼10% of the proximal convoluted tubules. Assessment of liver sections revealed randomly distributed necrosis in ∼10% of hepatocytes in two mice injected with LNAc/2′OMe18h. A single lesion of perivascular lymphocytic infiltration was also observed in one of the two mice, an event that could also be random. Importantly, there was one LNAc/2′OMe18h-treated mouse that showed no histological abnormality, similar to HSALR controls. Liver sections of LNAc/2′OMe14-injected mice exhibited diffuse, mild-to-moderate vacuolation and mild hepatocellular necrosis.
Discussion
DM1 is a highly complex disease that affects multiple organs and systems in the body. The primary pathologic mechanism is the sequestration of MBNL1 protein by the CUGexp repeat tract, leading to loss of nuclear MBNL1-dependent splicing regulation. Therapeutic research in the last decade has mainly been concentrating on the use of chemically modified ASOs that inhibit protein sequestration to the repeat site and thus restore MBNL1 function in the nucleus. Chemical alterations to the 2′-hydroxyl position of the ribose sugar—2′OMe, MOE, LNA, and cEt—have been particularly appealing for antisense applications as they increase target binding affinity, stability, and pharmacokinetics in vivo. In this study, we combined two of the most commonly used sugar modifications in a chimeric design to help offset potential negative effects, while retaining a high target binding affinity.
Our chimeric LNA/2′OMe ASOs act by directly competing with MBNL1 for CUG binding to restore protein function. At the cellular level, we have proven that the combination of LNA and 2′OMe nucleotides in an 18mer ASO construct (LNAc/2′OMe18h, LNAa/2′OMe18h) is more potent than an LNA/DNA mixmer of the same length and LNA composition (LNAc/DNA18). However, when introduced in short (11mer) antisense constructs, the combination of LNA and 2′OMe nucleotides was ineffective in correcting Mbnl1 exon 7 missplicing in primary HSALR myoblasts. By contrast, a 10mer ASO, with a fully modified LNA sugar core, has previously been shown to significantly correct MBNL1-sensitive splicing defects in proliferating human DM1 cells and TA muscle of HSALR mice [32]. Considering that LNA monomers have superior binding affinity compared with 2′OMe nucleotides and that reduced ASO lengths compromise hybridization stability, a fully modified LNA sugar core would theoretically be more beneficial than an LNA/2′OMe combination when designing short antisense constructs. However, the LNA/2′OMe combination can be used to make longer (14mer–18mer), fully modified ASOs with high binding affinities and potentially reduced off-target annealing. The impact of LNA nucleotide inclusion on the efficacy of truncated exon-skipping oligonucleotides has previously been demonstrated in an in vitro study by Le et al. While an 18mer 2′OMe-PS ASO with a two-nucleotide truncation was able to maintain exon 23 skipping efficiency in vitro, further reductions in ASO size significantly reduced the efficacy, with 12mer and 14mer variants showing no activity at all [41]. Importantly, the introduction of four LNA nucleotides in the 14mer, 16mer, and 18mer 2′OMe-PS ASOs significantly improved the yield of exon 23 skipped RNA product, thus demonstrating that shorter ASOs with high steric-blocking activities can be constructed by combining LNA and 2′OMe-PS nucleotides [41].
When delivered locally into the TA muscle of HSALR mice, LNAc/2′OMe18h, LNAc/2′OMe16, and LNAc/2′OMe14 ASOs were able to partially reverse the misregulated alternative splicing of Serca1 exon 22 and Mbnl1 exon 7. As expected, LNAc/2′OMe18h was the most potent ASO, since a single 18mer molecule can occupy and block a larger (CUG)n target region in the mutant mRNA (it is worth rementioning that we injected equimolar doses of the three ASOs into the TA muscle). Although our results clearly demonstrate partial reversal of misregulated spliced exons, the efficacy of our chimeric ASOs appears to be lower than that reported for other steric-blocking ASOs—25mer morpholino and 10mer all-LNA—at the intramuscular level [32,42]. This is probably due to suboptimal electroporation conditions caused by the use of an in ovo, rather than in vivo, electroporator.
The dosage selection for systemic administration was based on the disease pathogenesis and the mode of action of our LNA/2′OMe chimeras. For RNA-degrading gapmer ASOs against Dmpk mRNA, dosages as low as 25 mg/kg/week have been reported [37]. Given that a single gapmer ASO molecule can target and degrade many mRNA molecules, lower drug amounts would be required to achieve an effect. By contrast, a steric-blocking ASO will remain bound to a single target site, thus increasing the number of ASO molecules needed to elicit an effect. The situation is further amplified in the case of DM1, where dozens of steric-blocking ASO molecules would be required to inhibit MBNL1 sequestration to a level that restores alternative splicing regulation. To our knowledge, the highest reported dosage of a naked, steric-blocking ASO delivered systemically was 200 mg/kg/week, to induce dystrophin (Dmd) exon 23 skipping in the mdx mouse model of Duchenne muscular dystrophy (DMD) [43]. We therefore decided to proceed with a dosage regimen of 200 mg/kg per injection, administered subcutaneously two times a week for 4 weeks.
Despite high accumulation levels in skeletal muscle tissues, comparable with those reported in Goyenvalle et al. for tricyclo-DNA oligomer against Dmd, our chimeric LNA/2′OMe ASOs were unable to reduce the number of CUGexp-Mbnl1 foci and reverse Serca1 exon 22 missplicing in skeletal muscle tissues. Before this study, Leger et al. were the only group to demonstrate complete reversal of misregulated Serca1 alternative splicing after systemic administration of a peptide-linked, steric-blocking morpholino ASO in HSALR mice [39]. Importantly, when administered naked, the ASO failed to show any improvement in Serca1 missplicing in all tested skeletal muscles. These results imply that in the absence of a tissue-targeting conjugate, ASO uptake is not sufficient to enable intracellular efficacy.
The covalent attachment of cationic cell-penetrating peptides (CPPs) to charge-neutral steric-blocking ASOs—for example, morpholino—has shown to considerably enhance ASO uptake and activity in DM1 and DMD mouse models [39,44,45]. However, this is very much restricted to charge-neutral chemistries, as conjugation of cationic CPPs to anionic PS-modified ASOs would lead to electrostatic interactions, causing self-aggregation and disrupted binding to the target region. Consequently, in an attempt to increase ASO delivery and Dmpk mRNA degradation in DM1 muscle tissues, Ionis Pharmaceuticals and collaborators proceeded with the introduction of a lipid-based motif (5′-N-Palmitoylhexylamino) to the 5′ end of their PS-modified gapmer [46]. Conjugation of a hydrophobic cell-penetrating ligand to our chimeric LNA/2′OMe ASOs could therefore enhance uptake in skeletal muscle tissues to a level that elicits a corrective effect. However, it is still questionable whether this approach would be more effective than a ligand-conjugated RNA-degrading ASO.
A critical step in the success of oligonucleotide therapies is the implementation of early screening strategies to address potential toxicologic effects. The mechanisms by which ASOs can cause cellular toxicity can be classified in two categories: (1) hybridization-dependent toxicity caused by ASO binding to a partially complementary, off-target sequence and (2) hybridization-independent toxicity, which includes dose-related hyperaccumulation, and immunostimulatory and aptameric binding effects [47]. All types of toxicities are highly dependent on the sequence and the modification pattern of the individual ASO. Despite the high-dosage regimen used, corresponding to a cumulative dose of 3.2 g/kg, our 18mer and 14mer LNA/2′OMe ASOs showed minimal hepatotoxicity based on minor histological lesions and slightly elevated AST levels. Previous studies of LNA-modified gapmer ASOs have shown significantly elevated levels of hepatic transaminases (ALT/AST), profound histological lesions, and, in some cases, early morbidity, at cumulative doses of 100 mg/kg (ie, a more than 30-fold lower dose than the one administered in our study) [33–35]. Considering the different modification pattern and mode of action of LNA/DNA gapmer ASOs versus LNA/2′OMe chimeras, a direct toxicologic comparison would not be justified. Nevertheless, we can safely argue that hyperaccumulation of steric-blocking chimeric LNA/2′OMe ASOs does not appear to cause any profound hybridization-independent toxicity in liver and kidney tissues.
To conclude, our study demonstrates that direct inhibition of Mbnl1 sequestration by systemic delivery of chimeric LNA/2′OMe ASOs is an ineffective approach for DM1 therapy. Nevertheless, the data obtained open prospects for the application of LNA/2′OMe chimeras as steric-blocking agents for the treatment of other diseases. The combinatory introduction of LNA and 2′OMe chemistries to a fully modified PS backbone did not cause profound histological toxicity and resulted in relatively high levels of ASO accumulation in skeletal muscles, diaphragm, and heart tissue. Therefore, in the concept of a much simpler pathogenic model that requires inhibition of a single target site per RNA molecule, chimeric LNA/2′OMe ASOs could prove to be highly beneficial and safe.
Footnotes
Acknowledgments
The authors thank Professor Charles A. Thornton for providing us with the HSALR mouse model. Special thanks go to Mrs. Revecca Papacharalambous for the tissue embedding and Ms. Chrystalla Kamma for the sectioning and H&E staining.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by grants to L.A.P. and N.P.M. from the A.G. Leventis Foundation and AFM, respectively.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.