
Abstract
Advances in medicinal chemistry have produced new chemical classes of antisense oligonucleotides (ASOs) with enhanced therapeutic properties. Conjugation of the triantennary N-acetylgalactosamine (GalNAc3) moiety to the extensively characterized phosphorothioate (PS)-modified 2′-O-methoxyethyl (2′MOE) ASO exemplifies such an advance. This structure-activity optimized moiety effects receptor-mediated uptake of the ASO prodrug through the asialoglycoprotein receptor 1 to support selective targeting of RNAs expressed by hepatocytes. In this study we report the integrated assessment of data available from randomized placebo-controlled dose-ranging studies of this chemical class of ASOs administered systemically to healthy human volunteers. First, we compare the pharmacokinetic and pharmacodynamic profiles of a subset of the GalNAc3-conjugated PS-modified 2′MOE ASOs to the parent PS-modified 2′MOE ASOs for which plasma analytes are available. We then evaluate the safety profile of the full set of GalNAc3-conjugated PS-modified 2′MOE ASO conjugates by the incidence of signals in standardized laboratory tests and by the mean laboratory test results as a function of dose level over time. With hepatocyte targeted delivery, the ED50 for the GalNAc3-conjugated PS-modified 2′MOE ASO subset ranges from 4 to 10 mg/week, up to 30-fold more potent than the parent PS-modified 2′MOE ASO. No GalNAc3-conjugated PS-modified 2′MOE ASO class effects were identified from the assessment of the integrated laboratory test data across all doses tested with either single or multidose regimens. The increase in potency supports an increase in the safety margin for this new chemical class of ASOs now under broad investigation in the clinic. Although the total exposure is limited in the initial phase 1 trials, ongoing and future investigations in patient populations will support evaluation of the effects of long-term exposure.
Introduction
Today, five antisense oligonucleotides (ASOs) have been commercialized and one is under review, with indications as diverse as spinal muscular atrophy (SMA) to hereditary transthyretin amyloidosis (hATTR). Scores of other ASOs are progressing in development, representing a number of different chemical and mechanistic classes [1]. ASOs have been administered to humans by intravitreal, intrathecal, intravenous, subcutaneous (SC), and intramuscular injections, orally, and by inhalation and enema [2]. Thus, the potential of antisense technology is being realized while the technology continues to advance and produce steadily better performing agents [3].
Therapeutic oligonucleotides consist of three key elements, the hydrophobic nucleobase, a carbohydrate or modified carbohydrate, and a phosphate or phosphate analog. The nucleobase identifies target sites in RNAs through Watson–Crick hybridization. Optimal affinity and selectivity are conferred by 16–20 nucleotides [4]. In principle, 11–12 nucleotides should confer acceptable selectivity, but even using high RNA affinity oligonucleotides, the affinity is such that potency is inadequate in vivo, for review see Crooke et al. [5]. The carbohydrate is usually a deoxy ribose, a ribose, or a modified ribose. Substitutions at the 2′-position of the ribose alter the structure of the sugar and thus can be used to enhance affinity for target RNA sequences and hence potency. Modified sugars can also contribute a substantial increase in resistance to the endonucleases that perform the first step in the metabolism of oligonucleotides. The most frequently used modified sugar residues are 2′-fluoro (2′F), 2′-O-methoxyethyl (2′MOE), or constrained sugars such as “Locked nucleic acids” (LNAs) or constrained ethyl (cEt), for review see Refs. [1,3,6]. The phosphate or a modified phosphate is used to connect the nucleotides to form an oligonucleotide. The most commonly used phosphate analog is phosphorothioate (PS). Substitution of a nonbridging oxygen with a sulfur results in broader distribution of the phosphate charge and greater hydrophobicity. These changes enhance nuclease resistance and most importantly increase the promiscuity and affinity of interactions with proteins, for review see Levin et al. [7]. The neutral morpholino is also used, but has limited potency in vivo, for review see Refs. [1,3,8].
The nucleobase is critical for the potency and specificity of pharmacological effects. Carbohydrates that enhance potency and duration of effects, such as 2′MOE, cEt, or LNA, are used routinely. 2′F ribose is used extensively in small-interfering RNAs (siRNAs), but does not enhance nuclease resistance [1] and is associated with toxicities caused both by the intact oligonucleotide and one of the main degradates, 2′F ribose [9]. The PS moiety is the key determinant of distribution and the ability to cross membrane structures because of protein binding [7]. A minimum of 12 PS residues is sufficient to result in plasma protein binding, thus avoiding rapid excretion by the kidney [1]. The PS moiety also results in binding to proteins on the cell surface and in the cell, for review see Crooke et al. [10].
Double-stranded oligonucleotides, siRNAs, are highly hydrophilic and thus do not enter cells or organs without complex delivery systems, such as lipid nanoparticles, or conjugation of ligands that take advantage of high-capacity receptor systems, such as asialoglycoprotein receptors as is the case in hepatocytes for N-acetylgalactosamine (GalNAc), for review see Refs. [3,11,12]. In contrast, single-strand oligonucleotides (ASOs or single-stranded siRNAs) are amphipathic and thus can cross membranes without the need of formulations or transfection, for review see Refs. [1,2,13].
Of the chemical classes that have been studied, 2′MOE ASOs have been the most thoroughly evaluated [1,2]. Three 2′MOE ASOs have been commercialized, and one (volanesorsen) is under review in Europe, Canada, and the United States. Three, mipomersen, volanesorsen, and inotersen, are designed to exploit RNase H1-mediated RNA target reduction and are administered by weekly SC injection [14–16]. The fourth, nusinersen (Spinraza), is a fully 2′MOE-modified ASO designed to alter the splicing of a specific pre-mRNA and administered intrathecally every 4 months to treat patients with SMA [17–19].
The pharmacokinetics of PS-modified 2′MOE chimeric or “gapmer” ASOs have been extensively characterized in animals and humans. These ASOs are called chimeric or gapmer ASOs because they are designed to exploit RNAse H1-mediated cleavage of RNA and thus have a central region (usually 8–10 nucleotides) of PS deoxynucleotides (DNA like) flanked by 2′MOE PS residues on the 5′ and 3′ poles, for review see Refs. [1,10,20]. Absorption and distribution are defined by the PS moieties and thus are similar for PS gapmers and fully 2′-modified PS ASO used to alter splicing or effect translation inhibition or polyadenylate site masking or upstream open-reading frame or translation inhibitory structure-enhanced translation [5,21,22]. After SC dosing, absorption of these ASOs is nearly 100%. The absorption phase half-life is ∼60 min in humans and the distribution half-life is 30–60 min, for review see Refs. [1,23]. The pharmacokinetics of these ASOs are remarkably consistent in all species tested [mice, rats, dogs, nonhuman primates (NHPs), and humans].
Plasma protein binding is critical to distribution and these drugs bind to a number of plasma proteins, but the main plasma repository is albumin. The dissociation constant (Kd) for albumin varies from species to species, the degree of lipidation of albumin, the method used to measure binding and, of course, the number of PSs, but for an ASO that has 19 PS residues, the Kd is 140–150 μM. This binding is sufficient to prevent rapid renal clearance and yet the affinity is weak enough to support disproportionation between albumin and capillary, interstitial, and cell surface proteins, for review see Refs. [1,2]. These ASOs are widely distributed at lower doses to liver, kidney, fat cells, the spleen, and bone marrow [7,24]. At higher doses, the primary tissues saturate and these ASOs can then accumulate in secondary tissues [3,23]. Suborgan pharmacokinetics are well characterized in animals for the liver and kidney [25–27]. The liver accumulates ∼20% of a total systemic dose achieving ∼200–400 μg/g liver in PS 2′MOE ASO concentration at therapeutic doses [28–30]. These ASOs distribute to all the major cell types in the liver with nonparenchymal cells accumulating most of the liver's ASO levels.
Elimination half-lives from all tissues including the liver are long, 2–4 weeks. Elimination is effected by endonuclease cleavage in the DNA gap for gapmers resulting in half molecules that may be further degraded by exonucleases. As these degradates have only eight to nine PS moieties, they are rapidly cleared by glomerular filtration [7,31,32]. These drugs do not interact with cytochrome p450 enzymes [33]. They do not bind to drug binding sites on albumin, so there are limited drug–drug interactions [34].
Because members of a chemical class share similar properties, we have constructed databases that integrate all safety and tolerability observations from toxicity studies in NHPs and all randomized double-blind placebo-controlled trials for each chemical class of ASOs we are developing. Although the potency of 2′MOE ASOs has increased compared with mipomersen because of more effective screening, the potency of more recently discovered 2′MOE ASOs are consistent with ED50s of ∼150–200 mg/week for targets in the liver [3,35]. Of course, the pharmacokinetic properties of this chemical class are also similar as discussed previously. Similarly, the safety and tolerability profiles have been characterized.
Three publications that summarize the analyses of the integrated safety database that include results from >2,600 subjects treated with 16 2′MOE ASOs systemically in 52 completed clinical trials have been published [36–38]. These publications established the clinical safety of the 2′MOE ASO and highlighted two adverse events, thrombocytopenia and exacerbation of renal dysfunction in patients with hATTR and thrombocytopenia in patients with familial chylomicronemia syndrome that seem to be the result of unique interactions between two different ASOs at doses of 300 mg/week in two unique patient populations. It is important to emphasize that only members of the same chemical class share similar properties and that even what may seem minor chemical differences, for example, 2′MOE versus 2′-O-methyl (2′OMe), can greatly influence the behavior of ASOs. Therefore, the comments above refer strictly to the properties of members of the 2′MOE chemical class.
More recently, conjugation of triantennary N-acetylgalactosamine (GalNAc3) to 2′MOE ASOs has been shown to increase productive delivery of ASOs to the liver resulting in substantial increases in potency for hepatocyte produced target RNAs [1,39–44]. Today ten 2′MOE ASOs conjugated with a GalNAc3 moiety are in development and 9 have completed at least a 4-week clinical trial in normal volunteers. In total, >600 subjects have been exposed to this chemical class with >200 subjects exposed for 6 months or longer. The purpose, therefore, of this publication is to provide an initial assessment of the performance GalNAc3-conjugated 2′MOE ASOs after weekly or monthly SC dosing in healthy human volunteers. The experience in NHPs with this chemical class will be the subject of a separate report.
Materials and Methods
Clinical trial protocols were approved by the respective institutional review boards, or independent ethics committees. All studies complied with the guidelines of the Declaration of Helsinki and the International Conference on Harmonization Guidelines on Good Clinical Practice. Written informed consent was obtained from all participants before participation in the study. All studies were dose ranging and included a placebo-control group. The route of study drug administration was by SC injection in all protocols.
Bioanalytical methods
Human plasma drug concentrations were determined for each ASO using hybridization-based bioanalytical methods [45]. For GalNAc3-conjugated 2′MOE ASOs, the assay quantitated full-length ASOs (including fully conjugated, partially conjugated with 1, 2, or 3-sugar deletions, and unconjugated ASO). All plasma sample analyses were performed based on the principles and requirements described in 21 Code of Federal Regulations Part 58.
Pharmacokinetic analysis
Noncompartmental analysis methods were used for pharmacokinetic characterization of the plasma concentration data (Phoenix WinNonlin v.6 or higher; Certara, L.P., Cary, NC). Plasma pharmacokinetic parameters reported include peak plasma concentration (Cmax) and time to Cmax (Tmax), AUC0–24h, clearance at steady state (CLss/F), and apparent terminal elimination half-life (t1/2λz).
Dose–response analysis
The relationship between each pharmacodynamic biomarker response (ie, the individual plasma target protein levels) and dose in human following multiple dose treatments (typically 2 weeks after the last treatment) was analyzed with an inhibitory Emax model using GraphPad Prism version 5 (GraphPad Software, Inc., La Jolla, CA) by the following equation: E = 100/(1 + 10^(LogX − LogED50)), where E is the measured response (% baseline), X is the weekly dose, and ED50 is the weekly dose that produced 50% of maximum drug-induced effect.
Safety assessments
Samples were collected before dosing for standard clinical laboratory tests and collected at several time points within a 24-h period after SC dose administration for coagulation tests and complement split products. Data were imported from individual study data sets into one SAS data set for each laboratory test [46].
Injection site reactions (ISRs) were defined as injection site erythema, injection site swelling, injection site pruritus, injection site pain or tenderness that started the day of SC injection, and persisted (start to stop) for 2 days or more. Flu-like symptoms were defined as either (1) influenza-like illness or (2) pyrexia, feeling hot or body temperature increased, plus at least two of the following: chills, myalgia, or arthralgia that started on the day of injection or the next day.
Statistics
Evaluable subjects were those who received at least one dose of study drug. Data are presented by the incidence of events and descriptive summary statistics of laboratory test results. All study data were included for analysis of the incidence of events, except where noted. The baseline was defined as the last nonmissing value before the first dose. An event was defined as data falling outside the normal range or reaching the specified threshold, as defined by protocol stopping rules, standard reporting, or Grade 3 criteria provided by the Food and Drug Administration in Guidance to Industry for healthy adults and adolescents [47].
The over-time analysis of laboratory test results included all study data up to last visit in the single dose regimen cohort, and up to 10 days after the last dose in the multidose regimen cohort. The lower-limit-of-normal and upper-limit-of-normal (ULN) displayed in the mean results over time figures represent the median values (Supplementary Table S1). If there was no reference range available, then the range was calculated as the (mean ±2 × SD) of the baseline values in the data set.
A meta-analysis using subject-level data was performed to compare ASO-treated dose groups to placebo group on the multidose regimen test results during the treatment period, defined as the period from first dose to up to 10 days after last dose. The endpoints evaluated were the absolute change from baseline. The data were compared between GalNAc3-conjugated 2′MOE ASO dose groups and placebo using an analysis of covariance model with the dose group and trial as factors and baseline value as a covariate.
Results
To compare the behaviors of the parent PS 2′MOE ASOs with the GalNAc3-conjugated drugs, we performed similar randomized placebo-controlled dose-ranging phase 1 studies in normal volunteers. Single doses of 5–120 mg and multiple doses of 10–120 mg were evaluated in these studies. The multiple dose regimens included six weekly SC doses (Supplementary Table S2), and we also evaluated monthly doses for the GalNAc3-conjugated 2′MOE ASOs. Because GalNAc3-conjugated PS-modified 2′MOE ASOs are substantially more potent for liver targets, they were administered at lower doses than the parent PS-modified 2′MOE ASOs.
Comparison of the pharmacokinetic properties of PS 2′MOE parents with the GalNAc3 conjugates
Figure 1 gives a schematic of the primary mechanism of entry into hepatocytes for GalNAc3-conjugated 2′MOE ASOs and the estimated exposure compared with the parent. We have shown that liver concentrations can be estimated by plasma trough concentrations by combining these calculations with liver subfractionate studies conducted in animals to estimate total liver and hepatocyte/nonhepatocyte concentrations [39,48,49]. The lower doses used for GalNAc3-conjugated PS-modified 2′MOE ASOs result in lower peak plasma concentrations (Cmax) and plasma concentration area under the curves (AUCs). The fraction of dose distributed to the liver of GalNAc3-conjugated PS 2′MOE ASOs is similar to the parent PS-modified 2′MOE ASOs; however, ∼80% of the total drug in the liver is delivered to hepatocytes for the GalNAc3 conjugates, in contrast to ∼12% with the parent ASOs. This is the reason GalNAc3-conjugated ASOs are so much more potent for hepatocyte targets than the parent ASOs. In addition, GalNAc3-conjugated 2′MOE ASOs achieve an equivalent potency at hepatocyte concentrations that are twofold to threefold lower than that of the parent. Therefore, in addition to enhancing total delivery to hepatocytes, more of the ASO is internalized in hepatocyte through “productive” pathways, that is, pathways that deliver ASOs to sites within the cell where target RNAs can be hybridized with the ASO. This has been shown conclusively in vitro and in animals [39,44,48,49].
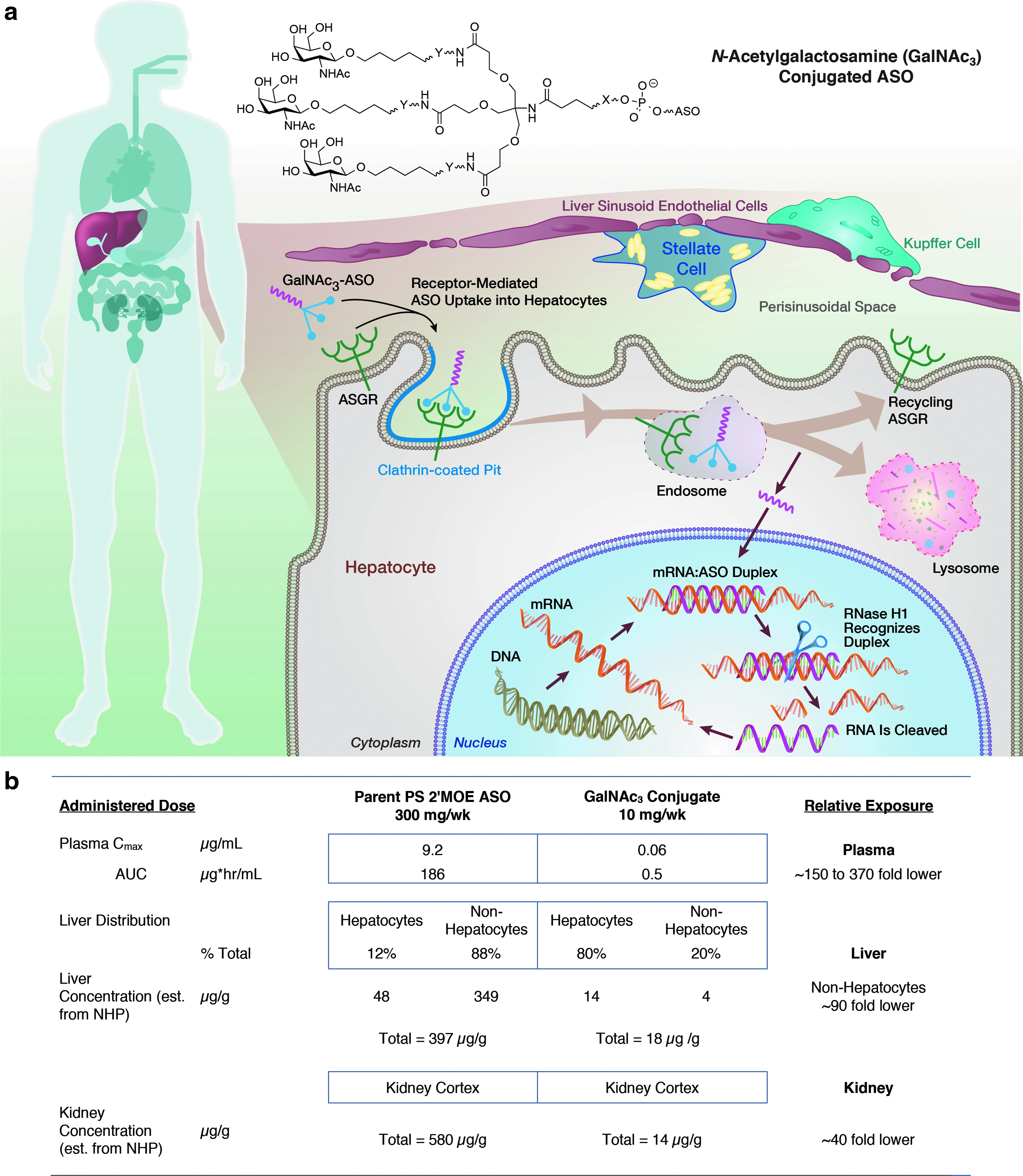
GalNAc3-conjugated PS 2′MOE ASOs demonstrate an increased fraction of total liver distribution to hepatocytes compared with parent 2′MOE ASOs.
Table 1 gives the GalNAc3-conjugated PS-modified 2′MOE ASOs for which phase 1 data were available for safety and tolerability assessments. Directly assayable plasma analytes were available to assess pharmacodynamic effects for four GalNAc3 conjugates, ANGPTL3-L, Apo(a)-L, ApoCIII-L, and FB-L, with data available to support a direct comparison of the properties of the GalNAc3 versions with the parent molecules for three conjugates, ANGPTL3, Apo(a), and ApoCIII (volanesorsen). Thus, we focused on these ASOs for pharmacokinetic and pharmacodynamic comparisons.
Evaluation of Eight Unique GalNAc3-Conjugated Phosphorothioate 2′-O-Methoxyethyl Antisense Oligonucleotides
Molecular target indicates the RNA target, e.g. mRNA or pre-mRNA. Symbols and nomenclature are based on HUGO standards.
Data not included in primary integrated analyses (monthly dose ranging pharmacokinetics and pharmacodynamics only).
AGT, angiotensinogen; ANGPTL3, angiopoietin-like 3; Apo, apolipoprotein; APOC3, apolipoprotein C3 (apoCIII); CFB, complement factor B (FB); FB, factor B; GHR, growth hormone receptor; HBV S, hepatitis B virus surface antigen; HV-RCT, healthy volunteer randomized controlled trial; KLKB1, kallikrein B1 (PKK, prekallikrein); L, GalNAc3; LPA, lipoprotein (a) [apo(a)]; PO, phosphodiester; PS, phosphorothioate.
As given in Table 2, the elimination half-lives of a representative parent PS-modified 2′MOE ASOs and the respective GalNAc3 conjugate were similar. Of course, the Cmax and AUC values were lower for the GalNAc3 conjugates, reflecting the lower dose, and the times to peak plasma concentrations (Tmax) were similar (as given in Fig. 2). However, distribution from plasma for the GalNAc3 version was more rapid as shown by the approximately fivefold higher rate of clearance from plasma (CLss/F). The more rapid distribution phase may suggest more rapid extraction from plasma by tissues, but this explanation remains speculative. The extent of plasma protein binding by the GalNAc3 conjugates was similar to the parent 2′MOE ASOs in all species tested (mouse, 94%–99%; monkey, 97%–99%; humans, 96%–99%). The other pairs of PS-modified 2′MOE ASOs studied behaved similarly (data not shown).
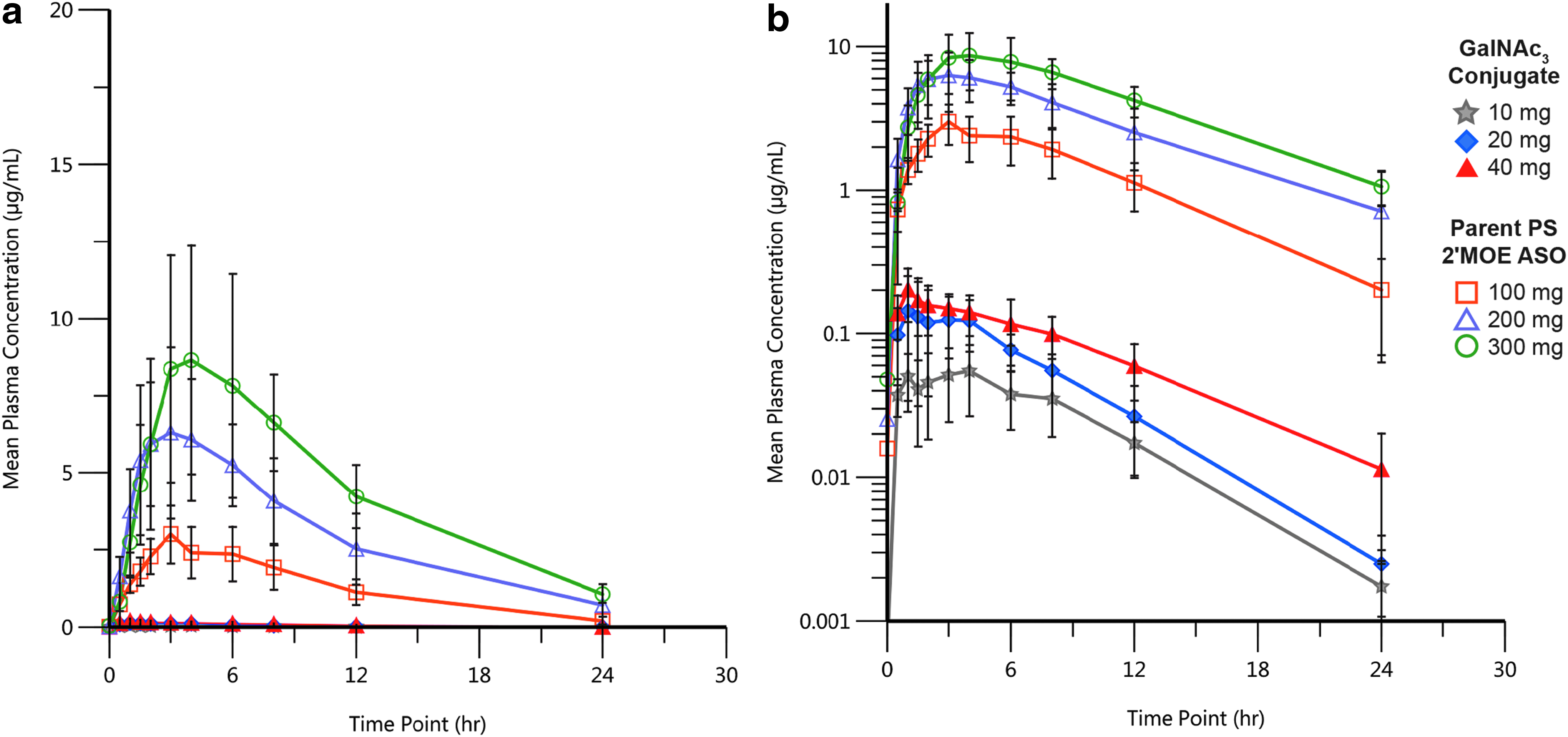
Pharmacokinetic profile after multiple SC doses of GalNAc3 conjugate versus parent PS 2′MOE ASO. Comparison of 0- to 24-h plasma concentration–time profiles,
Pharmacokinetic Properties of a Parent and GalNAc3-Conjugated Phosphorothioate 2′-O-Methoxyethyl Antisense Oligonucleotide Targeting Apo(a)
Values are presented as mean ± SD, except Tmax, which is presented as median (minimum, maximum). CLss/F is the plasma clearance at steady state after subcutaneous administration. CLss/F was calculated by actual dose/AUC0-τ, where AUC0-τ refers to the area under the plasma concentration-time curve (AUC) from time 0 to dosing interval τ following the last dose and τ = 168 hours.
2′MOE, 2′-O-methoxyethyl; ASO, antisense oligonucleotide; AUC, area under the curve; CLss/F, clearance at steady state; Cmax, peak plasma concentration; Tmax, time to Cmax; SD, standard deviation; t1/2λz, terminal elimination half-life.
Pharmacodynamics
The most striking difference between the parent and GalNAc3 versions is potency (Fig. 3). The ED50s for the parents range from 120 to 210 mg/week, whereas the ED50s for the GalNAc3 versions range from 4 to 10 mg/week. The ED50s for target reduction for nearly all the PS 2′MOE ASOs are in this range. The consistency of performance of PS 2′MOE ASOs and the GalNAc3 versions is remarkable and consistent in species ranging from mouse, NHPs, to humans. This consistency in pharmacokinetics and pharmacodynamics supports many drug development efficiencies. To compare the pharmacokinetic properties of the parent and GalNAc3 versions at doses resulting in equivalent target reductions, Supplementary Fig. S1 compares the pharmacokinetics of the parent and GalNAc3-conjugated 2′MOE ASO for apolipoprotein(a) at 300 mg versus 10 mg, respectively. These doses result in ∼60% reduction of plasma Apo(a) levels. With the exception of a significantly lower Cmax, the GalNAc3 version behaved similarly to the parent. This was true for the other pairs of parent and GalNAc3-conjugated 2′MOE ASOs (data not given).
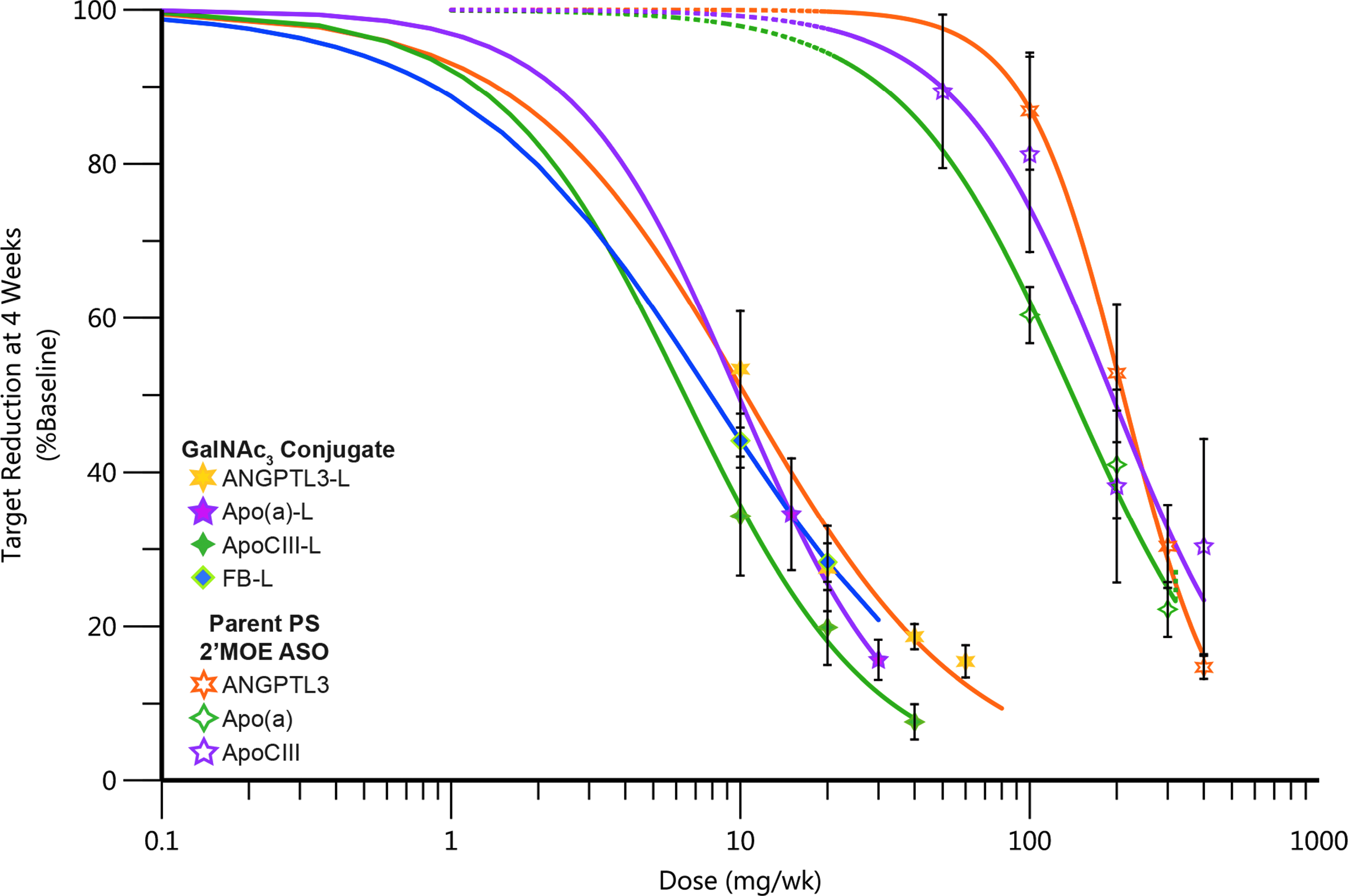
Increased potency observed with GalNAc3-conjugated PS 2′MOE ASOs in phase 1 healthy volunteer studies. Data shown are the mean, error bars represent the standard error, and the lines are fit to the mean data.
Because of the substantial increase in potency conferred by GalNAc3 conjugation for reducing RNA targets expressed in the hepatocyte, and the elimination half-lives from tissues for these agents range from 3 to 4 weeks, we also evaluated monthly administration of the GalNAc3-conjugated 2′MOE ASOs. The main purposes of this exercise were to define monthly dose–response curves and to assess if the mid-dose trough level of analytes were reasonably maintained. The monthly doses behaved similarly to the comparable doses in the single-dose and multiple-dose weekly cohorts, and the target reductions were maintained between doses as predicted by the pharmacokinetic properties of these drugs (Fig. 4 and Table 3).
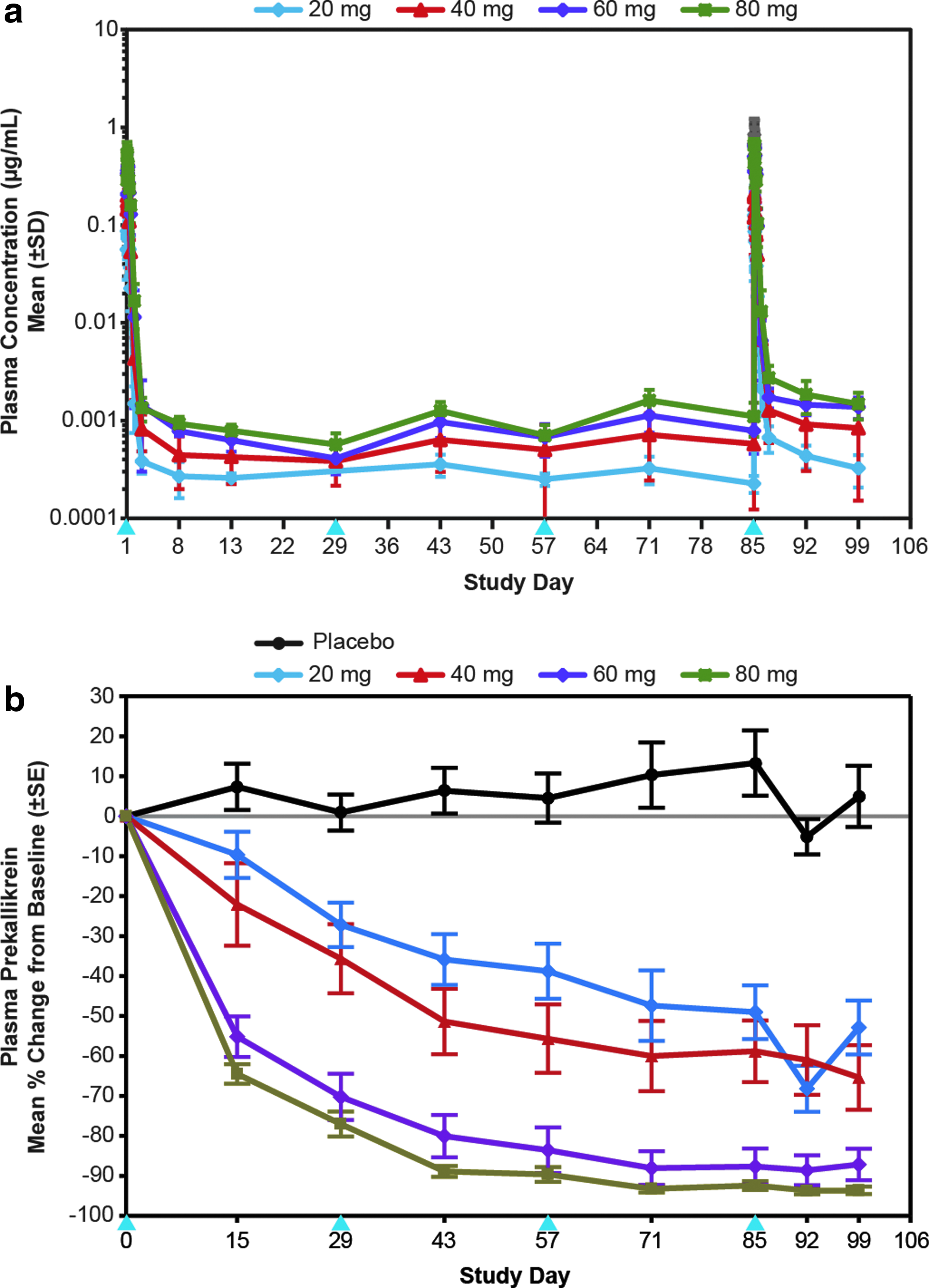
Pharmacokinetic and pharmacodynamic profile of GalNAc3-conjugated PS 2′MOE ASO in humans after monthly SC doses on study days 1, 29, 57, and 85.
Pharmacokinetic Properties of a GalNAc3-Conjugated Phosphorothioate 2′-O-Methoxyethyl Antisense Oligonucleotide Targeting Prekallikrein
Values are presented as the mean ± SD, except Tmax, which is presented as median (minimum, maximum). CLss/F is the plasma clearance at steady state after subcutaneous administration. CLss/F was calculated by actual dose/AUC0-τ, where AUC0-τ refers to the area under the plasma concentration-time curve (AUC) from time 0 to dosing interval τ following the last dose and τ = 672 hours.
NA, not available.
Safety
Integrated analysis of the safety data for the GalNAc3-conjugated 2′MOE ASOs included laboratory tests for liver (alanine transaminase, aspartate transaminase, total bilirubin, alkaline phosphatase, and albumin), kidney (serum creatinine, blood urea nitrogen, calculated glomerular filtration rate, and urine total protein), hematology (platelets, absolute neutrophil count, lymphocytes, hemoglobin, and hematocrit), coagulation (activated partial thromboplastin time and prothrombin time) and complement activation (complement split products Bb and C5a).
The results from the multiple ascending weekly doses are given in the primary figures and tables (Figs. 5–9 and Tables 4 and 5) with the respective protocol-specified dose schedules given in Supplementary Table S2; and the results of single ascending doses in the supplementary material (Supplementary Figs. S2–S5 and Supplementary Tables S8 and S9).
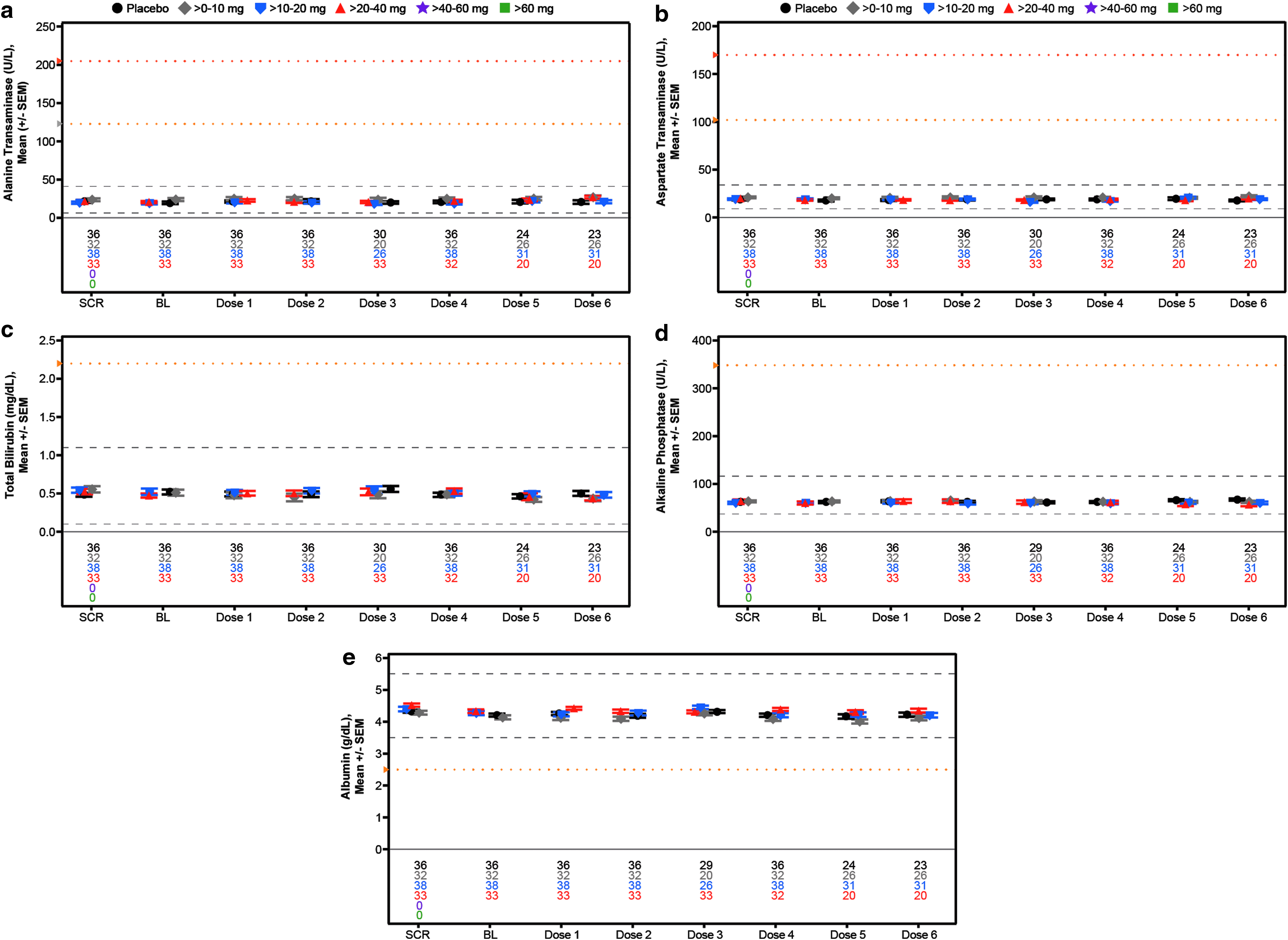
Mean laboratory measurements over time for assessment of liver function in the weekly multidose regimen cohort,
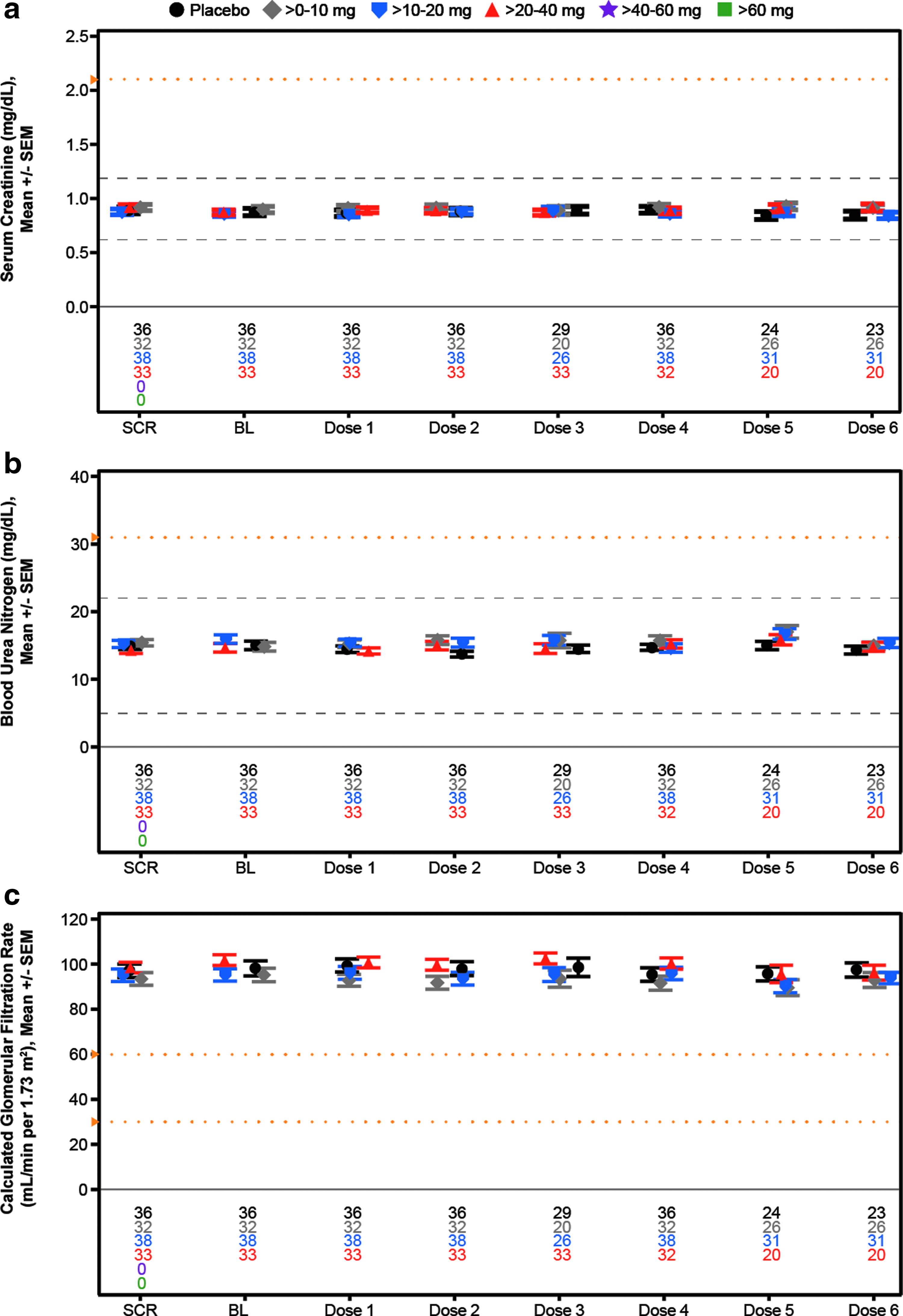
Mean laboratory measurements over time for assessment of kidney function in the weekly multidose regimen cohort,
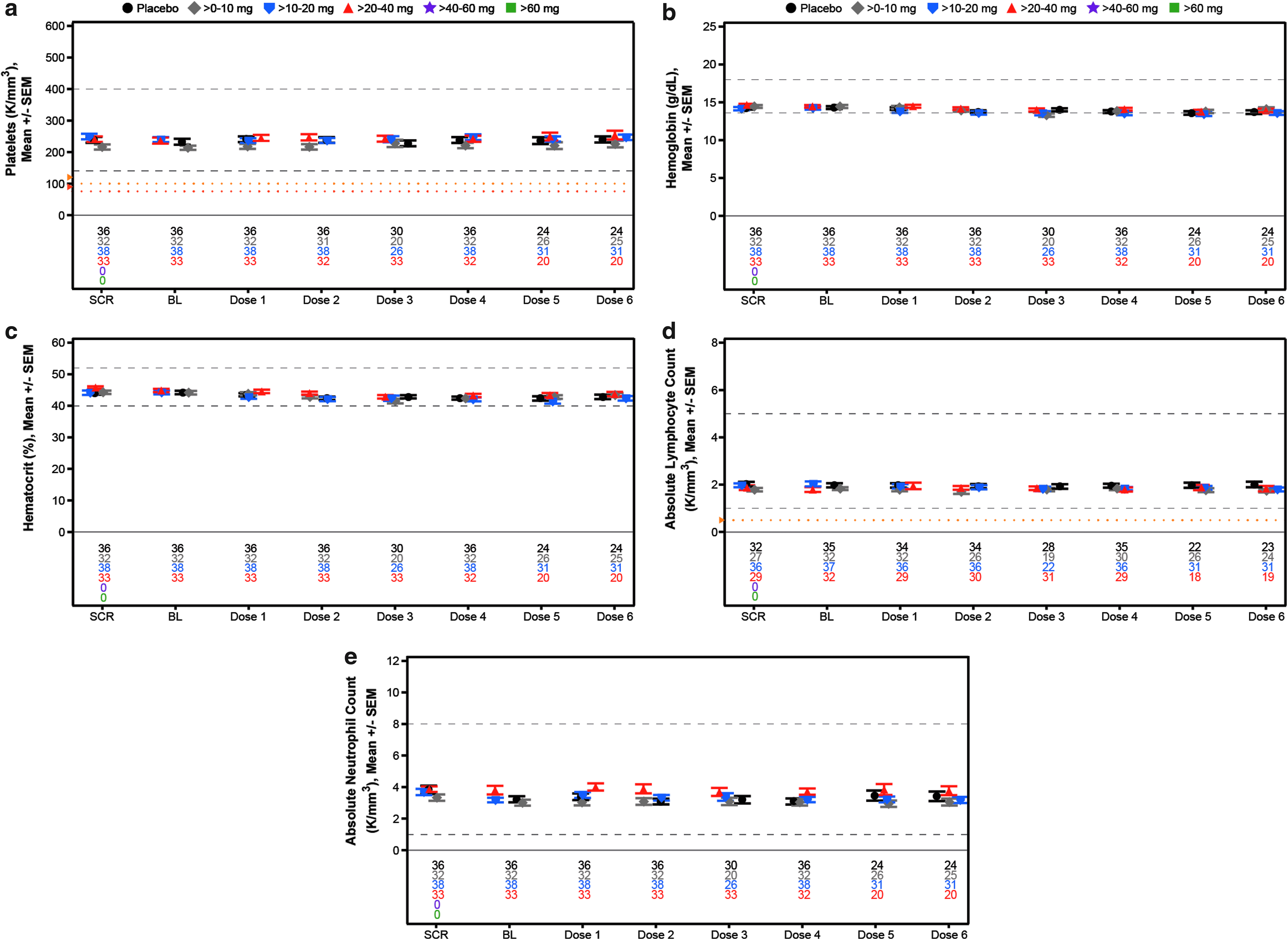
Mean laboratory measurements over time for assessment of hematology in the weekly multidose regimen cohort,
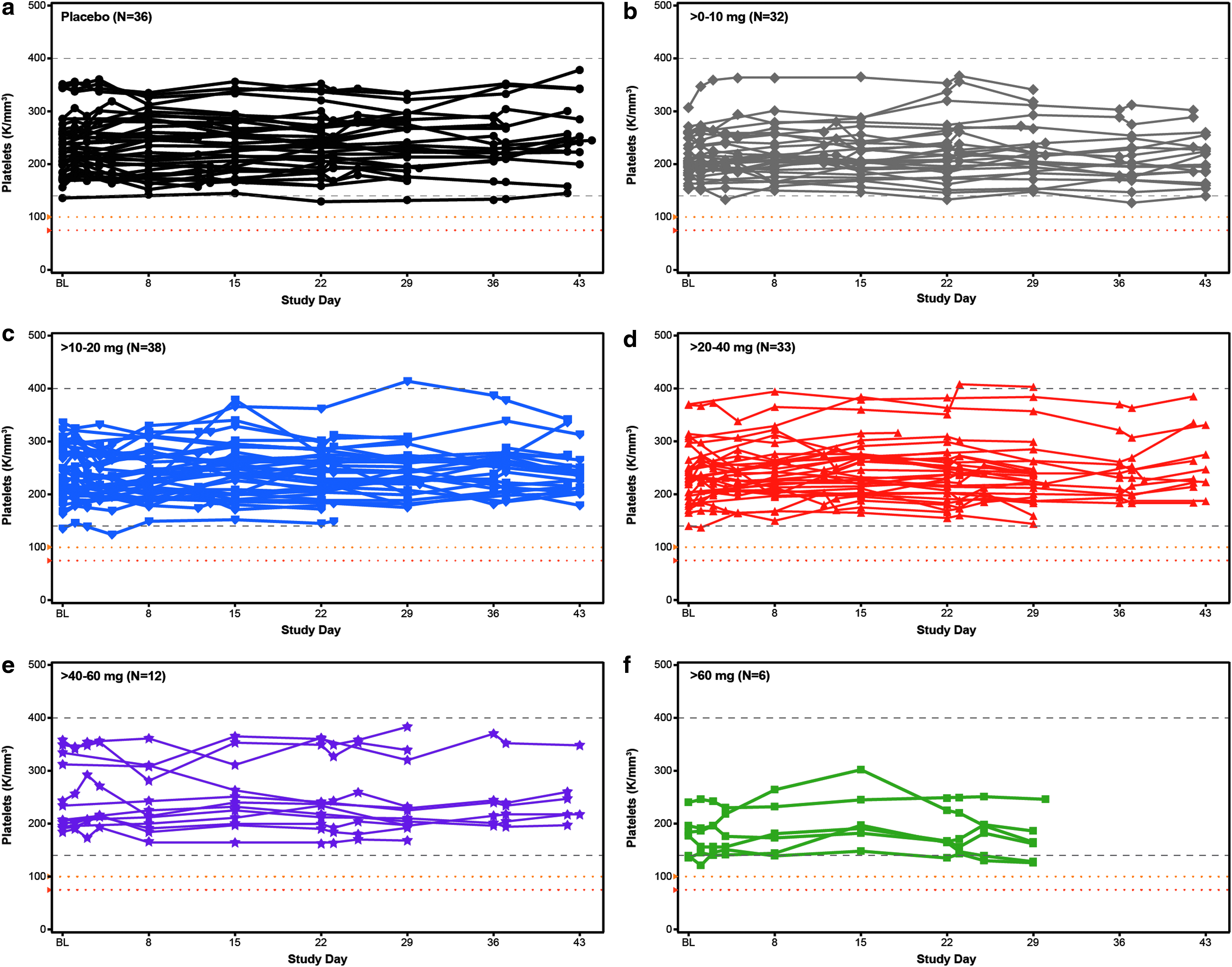
Individual subject assessment of platelet counts over time in the weekly multidose regimen cohort by dose level,
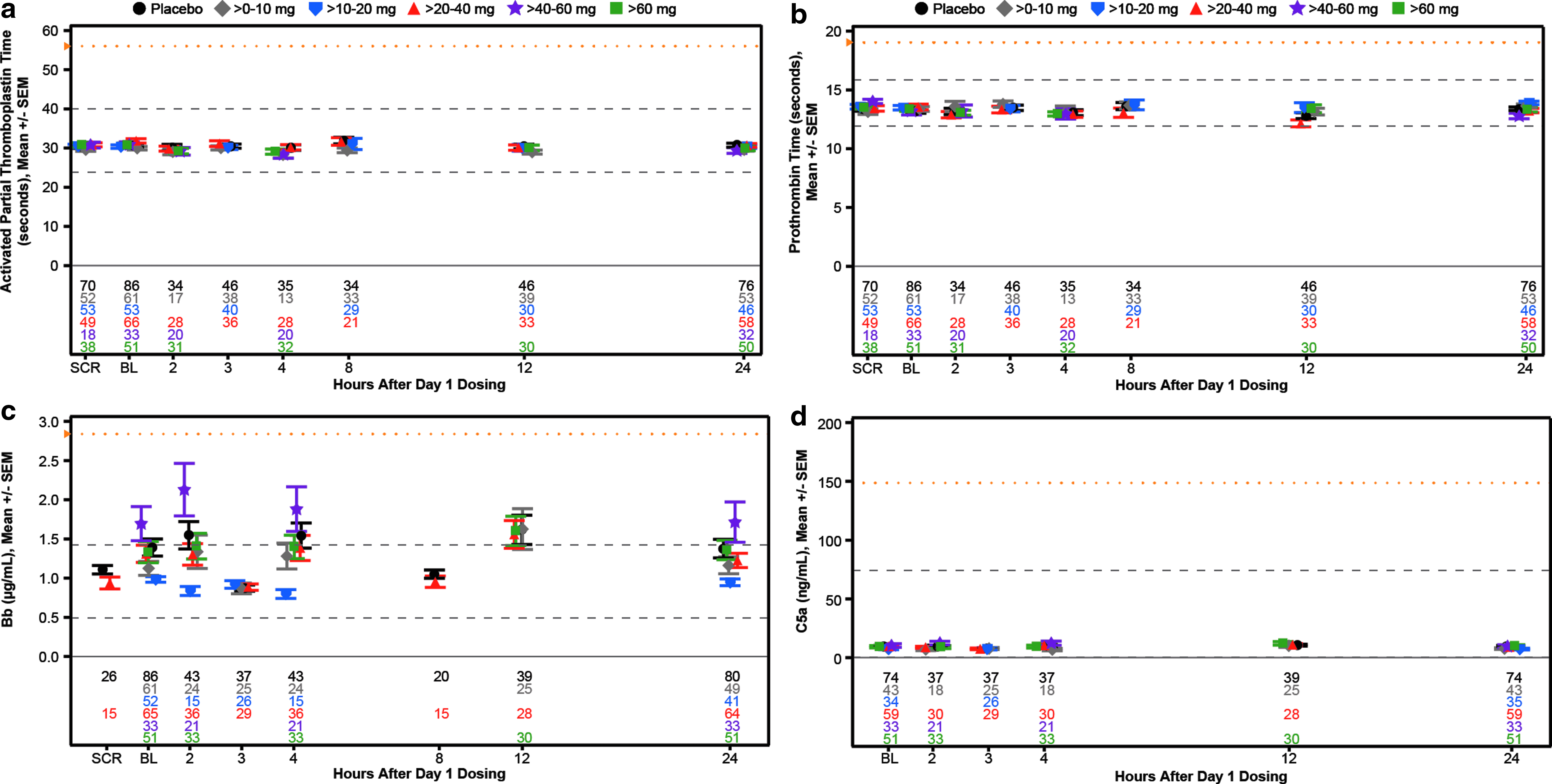
Mean laboratory measurements of coagulation and complement activation over the 24-h period postdose 1 in single and weekly multidose regimen cohorts,
Demographics and Baseline Characteristics of the Weekly Multi-Dose Regimen Cohort (N = 157)
BMI, body mass index; P25, 25th percentile; P75, 75th percentile; SD, standard deviation; SEM, standard error from the mean.
Incidence of Abnormal Laboratory Test Results in Weekly Multidose Regimen Cohort (N = 157)
Incidence of events is based on confirmed test results for liver, kidney, and hematology parameters during the study period. The incidence of coagulation and complement activation events was determined by a single observation. A confirmed event was defined as a consecutive abnormal laboratory value on a different day. If there was no consecutive test to confirm, then the initial observation was presumed confirmed. If there were multiple values on the same day but different time, the worst value was used. ALT and AST events were confirmed by two consecutive measurements at least 7 days apart with all values between the initial and subsequent test also above the specified threshold.
Urine protein was determined by dipstick.
ALT, alanine transaminase; APTT, activated partial thromboplastin time; AST, aspartate transaminase; BSLN, baseline; BUN, blood urea nitrogen; eGFR, estimated glomerular filtration rate; ULN, upper-limit-of-normal.
The demographics of the subjects in the multidose cohort were as expected for normal volunteers (Table 4). As can be seen, there were no effects observed at any dose level on the mean values of any of the analytes measured (Figs. 5–7). To identify individuals who may have had excursions in any analyte, we show the incidences of data falling outside the normal range or reaching a specified threshold. Any abnormal value during treatment or follow-up is reported for liver, kidney, and hematology analytes (Table 5). No abnormal liver analytes above three times the ULN were observed in the 157 subjects studied. Two subjects displayed creatinine clearances <60 mL/min, one in the placebo group and one in the >10–20 mg/week group. The placebo subject entered the study with a creatinine clearance of 57 mL/min, received each of the four planned doses and the creatinine increased during treatment, but was still low and then returned to approximately the entry level at last observation, meeting the event criteria. The ASO-treated subject was a 57-year-old woman who at entry had a creatinine clearance of 63 mL/min and received each of the eight planned doses of study drug. The creatinine clearance decreased to 58 mL/min in week 1, meeting the event criteria, and was stable at that range. A single 55-year-old male subject in the >10–20 mg/week dose group, who received each of six planned doses, entered with a hemoglobin of 11.2 g/dL and declined to 10.2 g/dL in week 4.
To evaluate potential effects on platelets more closely, we constructed individual-subject spaghetti plots of platelet values for all drugs and all doses tested. No abnormal values were observed as given in Fig. 8 for each subject in the multidose regimen cohort (N = 157) and Supplementary Fig. S5 for each subject in the single-dose regimen cohort (N = 193). In addition to these results there was no effect on coagulation or evidence of complement activation relative to baseline during the 24-h period after the first dose in both the single- and multidose cohorts (Table 5 and Fig. 9). In short, there were no dose-related abnormalities in any parameter tested.
The tolerability of the GalNAc3 2′MOE ASOs was excellent. There were no discontinuations because of adverse events. No flu-like symptoms were reported, and the incidence of mild injection site reactions was only 3%. There were no moderate or severe ISRs. With the PS 2′MOE type drugs, the incidence of ISRs ranged from 8% to 48% in similar phase 1 studies [36].
Discussion
GalNAc3 conjugation is the first example of targeted delivery of PS 2′MOE ASOs and represents a significant advance for targets expressed in the liver. The mechanism of targeting does not increase total liver concentrations of the ASOs. Rather, the ligand results in a greater fraction of total liver ASO to be delivered to the hepatocyte without significant changes in the elimination of the ASO [29,39,49]. Since at the low dose employed for GalNAc3 ASOs the total hepatocyte concentration does not exceed the concentrations observed for the parent drugs [39], two important conclusions were obtained. First in humans, as has been demonstrated in animal model studies [39], the GalNAc3 moiety must result in a higher fraction of 2′MOE ASO delivered to hepatocytes being delivered productively. The second conclusion is also important: For PS 2′MOE ASOs, GalNAc3 conjugation does not increase the total hepatocyte exposure at therapeutic doses arguing that hepatocyte safety should be as attractive as the safety of the unconjugated ASOs. This is supported by the absence of liver signals. The reduced doses that can be used for GalNAc3-conjugated 2′MOE ASOs means that peak plasma concentration-related adverse events are likely to be reduced. Lower doses also enhance tolerability and support less frequent than weekly dosing.
The success of GalNAc3 conjugation has stimulated broader efforts to identify ligands that enhance delivery of ASOs to other organs. We and our colleagues at Astra-Zeneca have reported that conjugation of Glp1 to PS ASOs dramatically increased delivery to the pancreatic islet cells resulting for the first time in robust activity against pancreatic targets [50]. Broader research on targeting ligands may enhance delivery to other organs in due course.
Footnotes
Acknowledgments
The authors thank Tracy Reigle for graphics support; Ekaette Mbong, PhD and Lisa Hannan, PhD for technical support; Eugene Schneider, MD, Michael L. McCaleb, PhD, Brett P. Monia, PhD, Sanjay Bhanot, MD, PhD, and the clinical development project teams for the individual phase 1 trials; John Su, PhD, and the biometrics team for support on the integrated safety database.
Author Disclosure Statement
All authors are employees of Ionis Pharmaceuticals.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.