
Abstract
The review starts with a historical perspective of the achievements of the Gait group in synthesis of oligonucleotides (ONs) and their peptide conjugates toward the award of the 2017 Oligonucleotide Therapeutic Society Lifetime Achievement Award. This acts as a prelude to the rewarding collaborative studies in the Gait and Wood research groups aimed toward the enhanced delivery of charge neutral ON drugs and the development of a series of Arg-rich cell-penetrating peptides called Pip (peptide nucleic acid/phosphorodiamidate morpholino oligonucleotide [PNA/PMO] internalization peptides) as conjugates of such ONs. In this review we concentrate on these developments toward the treatment of the neuromuscular diseases Duchenne muscular dystrophy and spinal muscular atrophy toward a platform technology for the enhancement of cellular and in vivo delivery suitable for widespread use as neuromuscular and neurodegenerative ON drugs.
Introduction
T
However there are limitations in some cases to the effectiveness of steric blocking ONs in achieving sufficient levels of activity in vivo, especially when they are delivered systemically and need to both penetrate the required tissues efficiently, as well as thereafter to enter cell nuclei. Thus, for example, the clinical effectiveness of eteplirsen has been questioned [5]. In addition, this drug must be used at relatively high dosage, which results in it being expensive as a therapeutic agent. The poorer intracellular efficacy of steric blocking ONs, compared to the RNase H antisense types, is partly due to their intrinsic RNA target-blocking mode of action where a competition must occur for interaction with components of the RNA recognition machinery (eg, splicing complexes). Thus this process must require at least a stoichiometric quantity of ON to reach and bind to its RNA target. By contrast, the binding of a standard antisense ON triggers a rapid cleavage of the RNA target by the cellular enzyme RNase H, which has been observed to be a much more efficient cellular process and requires lower amounts of ON to achieve an effect on the RNA target [7]. Nevertheless in certain applications, particularly in interference with nuclear events such as splicing and in targeting of toxic repetitive RNA sequences, steric blocking ONs have proved to be of great therapeutic value.
Many attempts have been made over recent years to enhance the cellular and in vivo delivery of ONs to improve their effectiveness, both steric blocking and otherwise. Partly this may be achieved in some cases by judicious choices of the chemistry of the backbone of ONs. For example, the use of a phosphorothioate (PS) backbone, to replace a phosphodiester backbone, results not only in a greater stability against nuclease degradation for which it was designed but also serendipitously in enhanced serum protein binding upon systemic injection, and, therefore, increases bioavailability [8,9]. Furthermore, discovery of superior entry into certain organs and cell types in vivo, such as liver cells, has resulted accordingly in the widespread use of the PS linkage in therapeutic antisense ONs of the RNase H-directing type.
Improvements in cell and in vivo delivery, as well as further increases in nuclease stability, have been obtained by incorporation of various nucleoside sugar analogs, such as 2′-O-methyl, 2′-O-methoxyethyl, and so on, reviewed in reference [4]. In addition, there have been many studies that utilize either a covalently attached ligand as delivery agent, such as the carbohydrate cluster GalNac, which has been attached to siRNA for targeting liver [10] and which also improves the potency of ON gapmers dramatically, and these are now in clinical trials [11]. By and large lipid-packaging agents have proved of less value in the case of ONs in vivo, as opposed to siRNA, than ligand attachment.
The emergence of a new class of peptide ligand, called a cell-penetrating peptide (CPP), has offered great opportunities for exploration of the possibilities for enhancement of delivery of an attached steric blocking ON, which was clearly needed if such ONs were to become universally accepted as efficient drugs [12]. This review concentrates initially on the contributions of the Gait group in ON and conjugate synthesis toward the award of the 2017 OTS Lifetime Achievement Award as a prelude to the exciting collaborative work of the Gait and Wood groups in the development of peptide-ON conjugates as therapeutics in treatment for neuromuscular diseases.
RNAse H and Initial Steric Blocking Activities of Antisense ONs
The Gait group worked on the development of methods of oligodeoxynucleotide synthesis on solid support from 1975 [13] to 1984 [14], which was superseded by the outstanding phosphoramidite chemistry of Caruthers and colleagues [15] (OTS Lifetime Achievement Award winner of 2018) and automated by Applied Biosystems and other companies. Initial pioneering antisense experiments of Zamecnik and Stephenson were designed to be by a steric blocking mechanism [16], but later such ONs were found to utilize an RNase H directing mechanism of action to cleave a bound complementary RNA strand, reviewed in reference [3].
The field of therapeutics took a major step forward in the mid 1980s through the chemical synthesis of PS internucleotide linkages in ONs, based on the pioneering work of Fritz Eckstein (recipient of the 2015 OTS Lifetime Achievement Award [17], where a simple sulfur atom replaces an oxygen atom). PS-linked ONs are much more resistant to nuclease degradation than phosphodiesters, and thus, cellular activities were found to be much higher. PS ONs were later found to be active in in vivo models and quickly became the primary choice for therapeutics development. Initially the concept was to use ONs to block translation of mRNA, but in due course it became clear that the concentrations of such ONs needed for activity were significantly larger than for RNase-H-utilizing ONs [7]. In the years that followed most of the development of therapeutic ONs took place using gapmers, where a central core of deoxynucleotides containing PS linkages was flanked by a number of 2′-modified nucleotide analogs on each arm, such analogs being insensitive to recognition by RNase H. This also results in an increase in the binding strength of such ONs, since RNA:RNA duplexes are stronger than DNA:RNA duplexes, and also the protection of the ON ends from nuclease degradation. This configuration still allowed the cleavage by RNase H of the RNA complementary to the central core of the ON. Subsequently chemists developed many nucleotide analogs suitable for the gapmer flanks [7].
However the original concept of steric blocking started to be explored more through the use of ONs that were fully modified by the inclusion of such analogs at every position, such that the ONs are no longer able to direct RNase H cleavage. While the new therapeutics companies concentrated on development predominantly of gapmers, scientists looked to understand which are the optimal intracellular activities for steric blocking ONs [18,19].
At MRC-LMB in the Gait laboratory, in collaboration with the Karn laboratory, the protein-RNA interactions involved in trans-activation by the HIV-1 trans-activator protein Tat were elucidated as a potential anti-HIV drug target [20]. Then in collaboration with Wengel and colleagues in Denmark, ON inhibition studies were explored by use of certain 2′-modified ONs as mixmers, in particular mixmers of 2′-O-methyl (OMe) and Locked Nucleic Acids (LNAs) (Fig. 1), for the steric blocking inhibition of the trans-activation responsive RNA (TAR) stem-loop region of HIV-1 that binds the HIV-1 Tat protein. By use of a HeLa cell line containing stably integrated plasmids expressing firefly luciferase under HIV-LTR/Tat dependence, as well as a Renilla luciferase constitutive control, submicromolar, selective, dose-dependent, and sequence-dependent intracellular inhibition of Tat-TAR trans-activation by a 12-mer mixmer ON was shown [21]. No intracellular activity was observed for the corresponding OMe 12-mer. The ON length could be reduced to 12 because of the tight binding to complementary RNA of the included LNA analogs, but a 12-mer containing 11 contiguous LNA residues was less effective than a mixmer both in binding TAR and in inhibition of Tat-dependent transcription, probably because the latter is more flexible [21]. This was the first time that intranuclear cell activity had been demonstrated for a steric blocking ON.
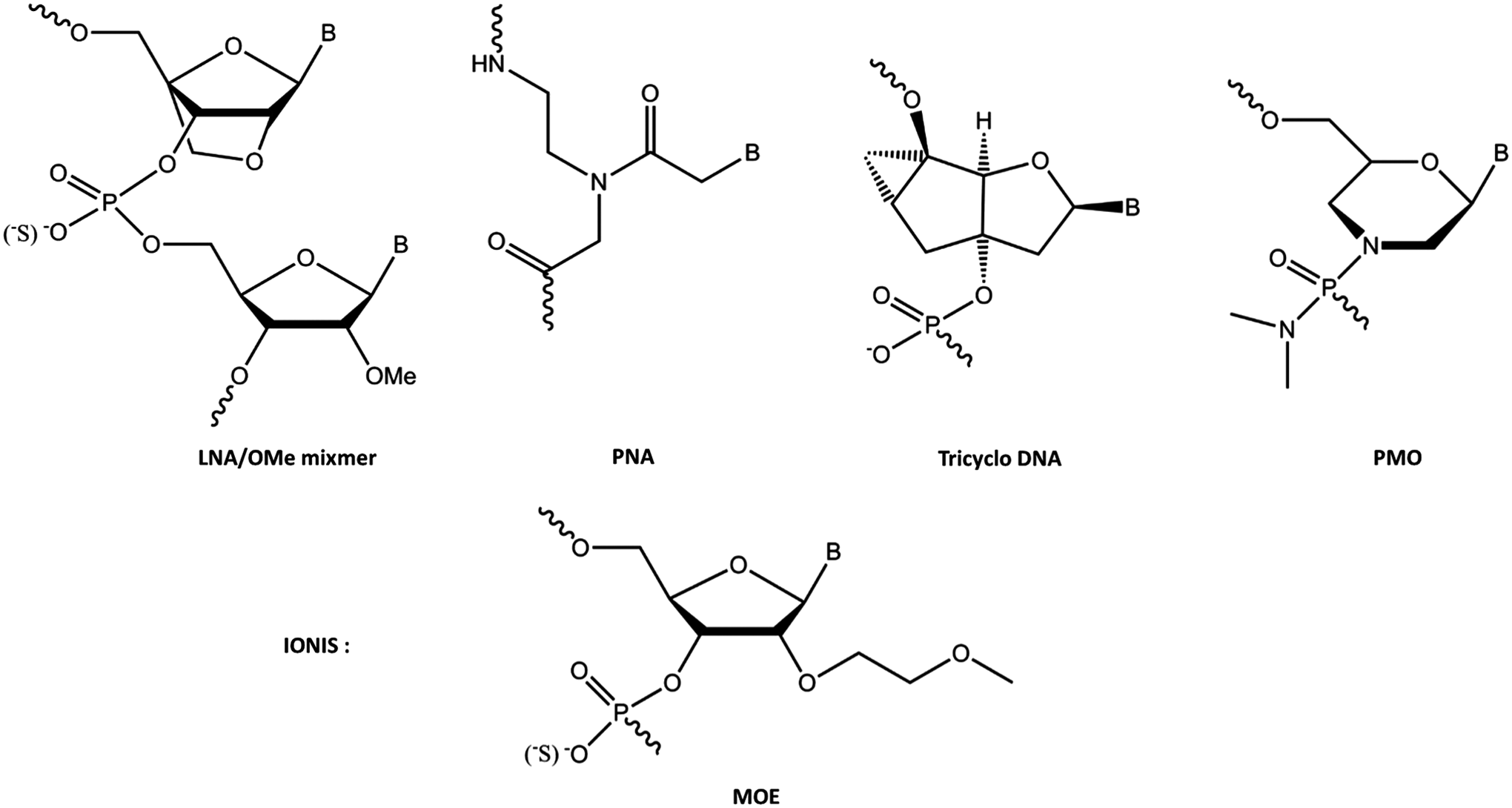
Nucleotide analogs used by the Gait group for studies of steric block ON activities in comparison to 2′-MOE nucleotides used in the Ionis research studies. ON, oligonucleotide.
In collaboration with Leumann and colleagues and Toulmé and colleagues, a 16-mer tricyclo ON (Fig. 1), although binding to TAR RNA more weakly, was found to be as good as a 16-mer OMe/LNA mixmer ON in suppressing Tat-dependent in vitro transcription in HeLa cells when delivered by cationic lipid. No inhibition was observed in the HeLa cell reporter model for tricyclo-DNA/OMe mixmers, even though their affinities to TAR RNA were strong and their cell distributions did not differ from ONs containing all or predominantly tricyclo-DNA residues. A tricyclo-DNA 16-mer showed sequence-specific inhibition of β-galactosidase expression in an anti-HIV HeLa cell reporter assay [22]. Thus intranuclear cell activity was not confined to LNA/OMe mixmers, but might be achievable with a range of ON analog types.
Cell Delivery by Conjugates of ONs with CPPs
It was clear that, to obtain nuclear activity with LNA/OMe ONs required for inhibition of Tat-dependent trans-activation, it was necessary to formulate the steric blocking ON with a cationic lipid delivery agent. As a more practical alternative to avoid the use of complex formulation strategies and which might be more appropriate for therapeutic development, certain covalently attached CPPs [23] were investigated to deliver steric blocking LNA/OMe ONs into the nucleus of cells to inhibit Tat-dependent trans-activation [24]. Although uptake into the cell nucleus had been observed with fluorescent ON-CPP conjugates previously, reviewed in reference [25], many of such results were likely to have been compromised by the use of strong cell fixatives that altered the cell distribution of CPPs during such use [26].
Thus to avoid the potential for fixation artifacts, we utilized nonfixed cells to demonstrate enhanced cellular uptake. However, disulfide-linked conjugates of a 5′-thiol substituted 12-mer LNA/OMe mixmer ON targeting the TAR RNA stem-loop with the well-known cationic CPPs, HIV-1 Tat (48–58) peptide, Penetratin, or R9F2 was only seen in cytosolic vesicles when incubated with Tat-TAR reporter HeLa cells, suggesting nuclear exclusion. No inhibition of Tat-dependent trans-activation activity was seen unless cationic lipofection was used for delivery [27]. Uptake only into human fibroblast cytosolic compartments was seen for Tat, Penetratin, R9F2, and Transportan conjugates. Large enhancements of HeLa cell uptake into cytosolic compartments were seen when free Tat peptide was added to the Tat conjugate of 12-mer OMe/LNA ON or the Penetratin peptide to the Penetratin conjugate of the same ON, suggesting the possibility that delivery activities observed by others may have been due to a packaging effect of the use of excess of these peptides [27].
CPP Conjugates of Charge Neutral PNA ONs
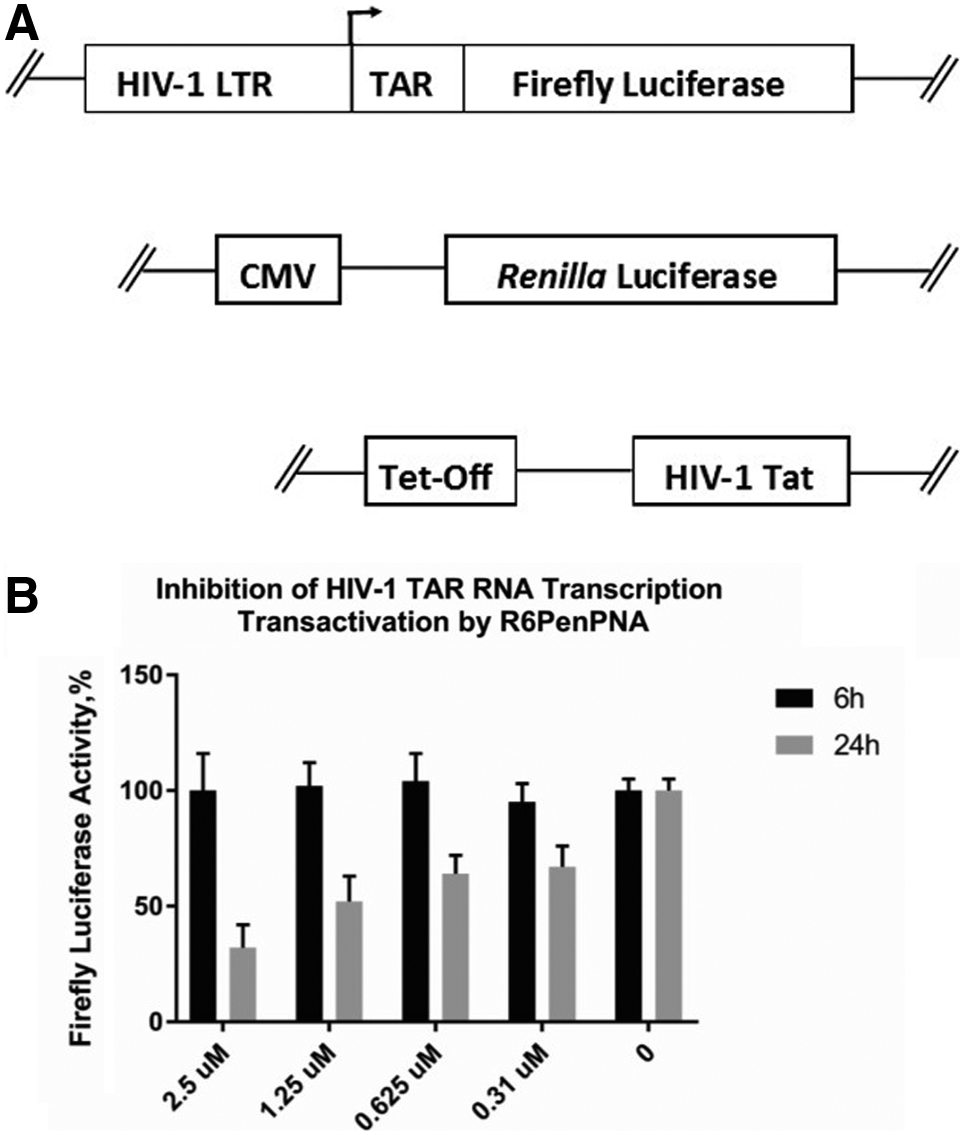
To find ON analogs that may enter cells and be delivered to the nucleus more easily, attention was therefore turned to the use of CPP conjugates of charge-neutral ONs and, in particular, PNA ONs (Fig. 1). In this study, by contrast, much greater success in nuclear delivery was observed when certain CPPs were disulfide linked to a 16-mer charge neutral PNA targeting the TAR RNA stem loop. Disulfide-linked PNA conjugates of two types of CPP [Transportan or a novel chimeric peptide R6-Penetratin, where six Arg residues were added to the N-terminus of the well-known Penetratin peptide (Fig. 2)] exhibited dose-dependent inhibition of Tat-dependent trans-activation in the Tat-TAR dependent reporter HeLa cell assay when incubated for 24 h (Fig. 3). Activity was reached within 6 h if chloroquine, an endosomal release agent, was coadministered (data not shown). While some nuclear uptake was observed in nonfixed HeLa cells following incubation of fluorescein-labelled Transportan or R6-Penetratin conjugated PNA, the majority of fluorescent signal was observed in cytosolic vesicles. Interestingly, stably linked conjugates of Tat and Transportan (or TP10, a shorter Transportan analog) with PNA were inactive when delivered alone, but attained trans-activation inhibition only in the presence of chloroquine [28]. Thus although it was possible to obtain nuclear activity for CPP-PNA conjugates, the major part of such conjugates was seen to be located in cytosolic vesicles.
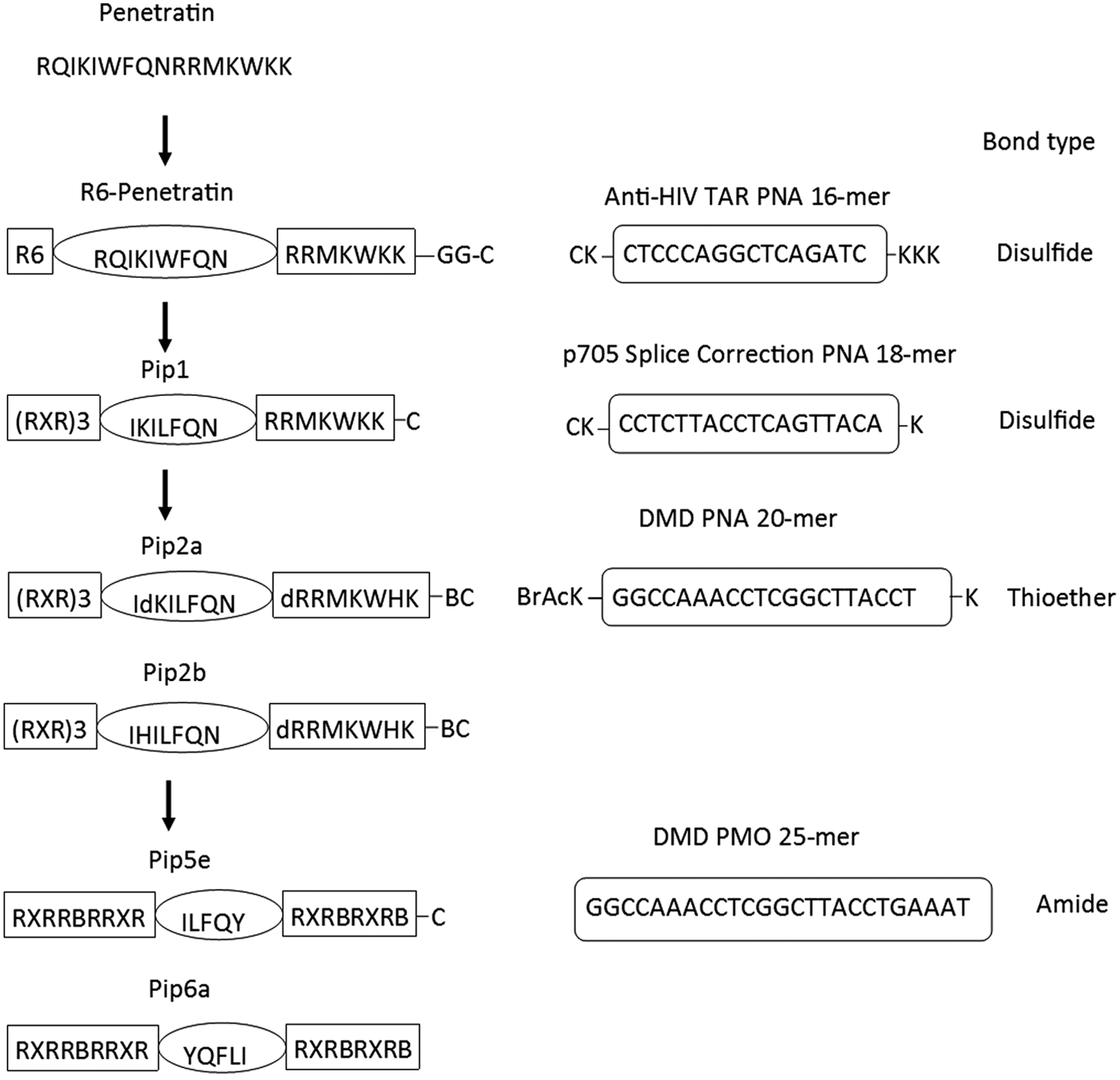
Development of novel Pip sequences starting from the CPP Penetratin. These include on the left side R6-Penetratin (R6-Pen), Pip1, which was the initial peptide disulfide linked to PNA 705 for splicing redirection, Pip2a and Pip2b, the initially protease stabilized peptides, as well as the more recent Pip5e and Pip6a. PMO sequences and their linkage functionalities are shown on the right side. R6-Pen and early Pip were conjugated to an anti-TAR PNA or a splice-correcting PNA705 through a disulfide linkage, whereas later Pip were either stably thioether linked to PNA705 or amide linked to PMO for exon skipping in DMD mdx cells. CPP, cell-penetrating peptide; DMD, duchenne muscular dystrophy.
A Splicing Redirection Assay
The R6-Penetratin peptide was clearly a very useful lead for potential therapeutic development toward steric blocking activity with a PNA cargo. To better assess the potential of this approach for nuclear delivery, in collaboration with Lebleu and colleagues [29], we adopted the HeLa 705 splicing system that was developed by Kole and colleagues based on a mutation discovered in a β-thalassemia patient that resulted in the introduction of a functionally deleterious cryptic splice site [30]. Such activities in the nuclei of cells were particularly suited to fully modified ONs since cleavage, and hence destruction, of the pre-mRNA was not appropriate and instead the ON needed to be bound tightly to block and compete with the action of proteins involved in splicing. The main requirement was the optimal positioning of the ON to hybridize with the pre-mRNA for maximal splicing redirection depending on the particular application. The HeLa 705 system utilizes a steric blocking ON targeted toward a cryptic splice resulting in the removal of the aberrant intron from the luciferase mRNA and subsequent correct protein synthesis that allows for a sensitive readout with a large dynamic range [30] (Fig. 4).
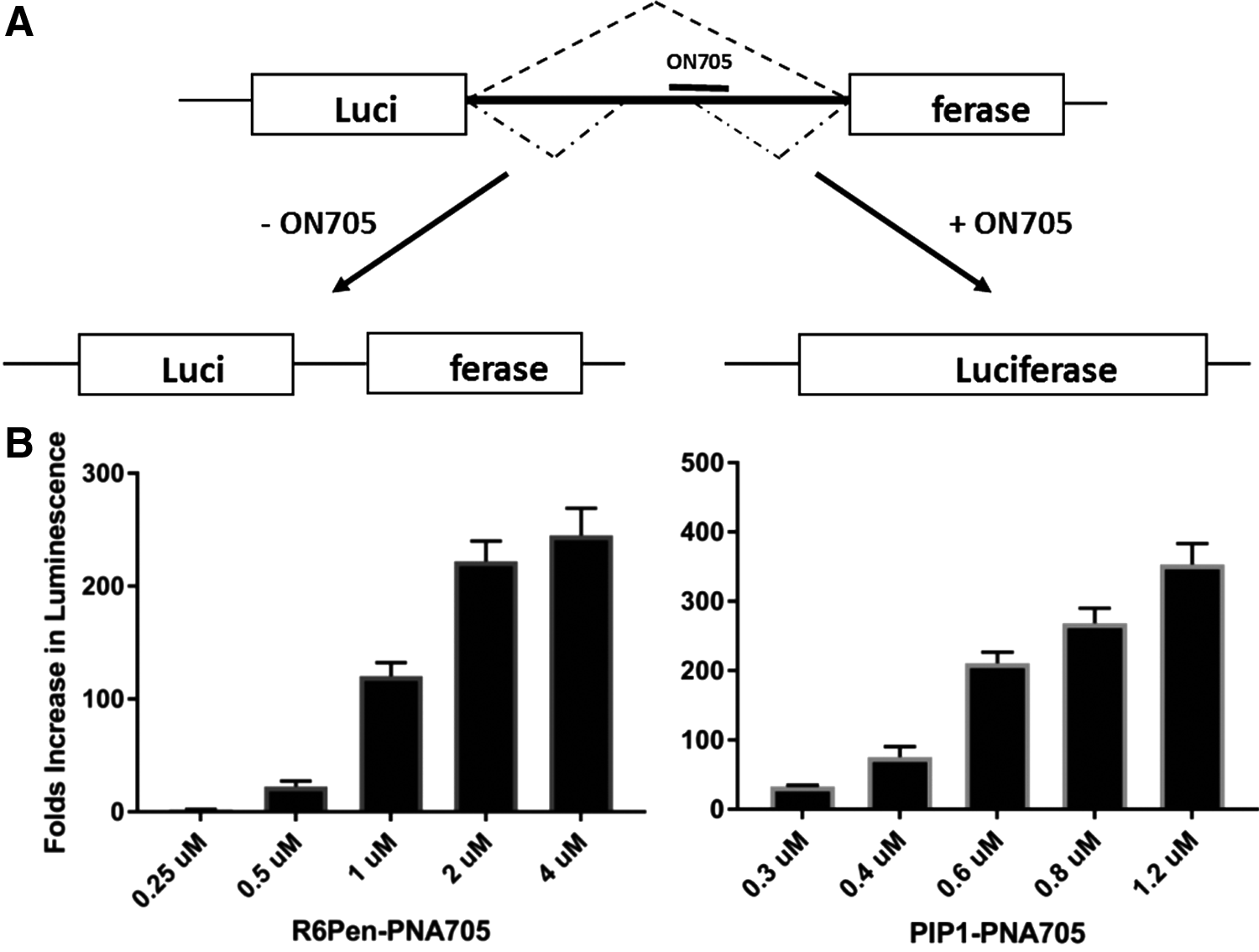
Utilization of a (Lys)8-PNA-(Lys) 18-mer targeted to the 705 splice site did not demonstrate splicing correction unless the endosome-disrupting agent chloroquine was added. Subsequent analysis with a N-terminal fluorescently labelled derivative of the (Lys)8-PNA-(Lys) suggested that lack of activity was due to ON becoming trapped in endosomal compartments [31]. By contrast, under identical conditions, the use of R6-Penetratin (R6-Pen) peptide conjugated to the same 18-mer PNA demonstrated upregulation of luciferase in a luminescence assay of 105-fold higher compared to unconjugated PNA705 [29]. The addition of the endosomal release agent chloroquine to R6Pen-PNA705 increased splicing in the HeLa705 cells two to threefold, suggesting that even with this better CPP most of the R6Pen-PNA705 was still trapped in endosomal compartments. However, the activity level in the absence of chloroquine was higher than that seen for an 8-Arg peptide (R-Ahx-R)4-conjugate of PMO and a conjugate of PNA with the same peptide (Arzumanov, Gait, et al. unpublished results).
These results led to the use of R6-Penetratin as the starting point for design of peptides, which might be suitable as a cell and in vivo tissue delivery agent of PNA and later PMO ONs.
CPP Conjugates of PNA for Exon Skipping in DMD Cells and as In Vivo Delivery Agents
In 2007 the Gait laboratory in Cambridge and the Wood laboratory in Oxford began what turned out to be an extremely fruitful collaboration. The HeLa pLuc705 system was used to develop a series of modified R6-Penetratin peptides that were more stable to protease digestion in mouse serum, called PNA internalization peptides (Pip) (Fig. 2), which were conjugated to an 18-mer PNA705 model ON. First we showed that introduction of an R-X-R motif (X = aminohexanoic acid, as had been used previously [29]) and shortening of the hydrophobic region resulted in a peptide (Pip1) that conferred threefold higher splicing higher activity compared to R6-Penetratin (Fig. 4) and greater than 10-fold higher than both the previously reported peptide [R-Ahx-R]4, as well as unconjugated 18-mer PNA705 (data not shown). We showed that Pip1–PNA705 and other Pip conjugates are internalized in HeLa cells by an energy-dependent mechanism and that the predominant pathway of cell uptake of biologically active conjugate is via clathrin-dependent endocytosis [32].
Exon skipping has been established for some years as a therapeutic opportunity for the treatment of patients with DMD. Most DMD patients have out-of-frame mutations in the DMD gene that results in a truncated and nonfunctional dystrophin protein. In some cases, especially within the repeating units of the spectrin-like rod domain, it is possible to induce in-frame deletions using a synthetic ON targeted to redirect splicing around the genomic deletion, so as to produce a semifunctional protein similar to that produced by less severely affected Becker muscular dystrophy patients [33]. In the well-known mdx mouse model of DMD, targeted removal of the in-frame exon 23, which contains a premature termination codon, has become a well-established model to demonstrate the therapeutic potential of splice switching. Serum-stabilized Pip2a or Pip2b (Fig. 2) conjugated to a 20-mer PNA (PNADMD) targeting exon 23 demonstrated enhanced exon-skipping activity in mdx myotube cultures in the absence of an added transfection agent at concentrations where the naked PNADMD was inactive [24]
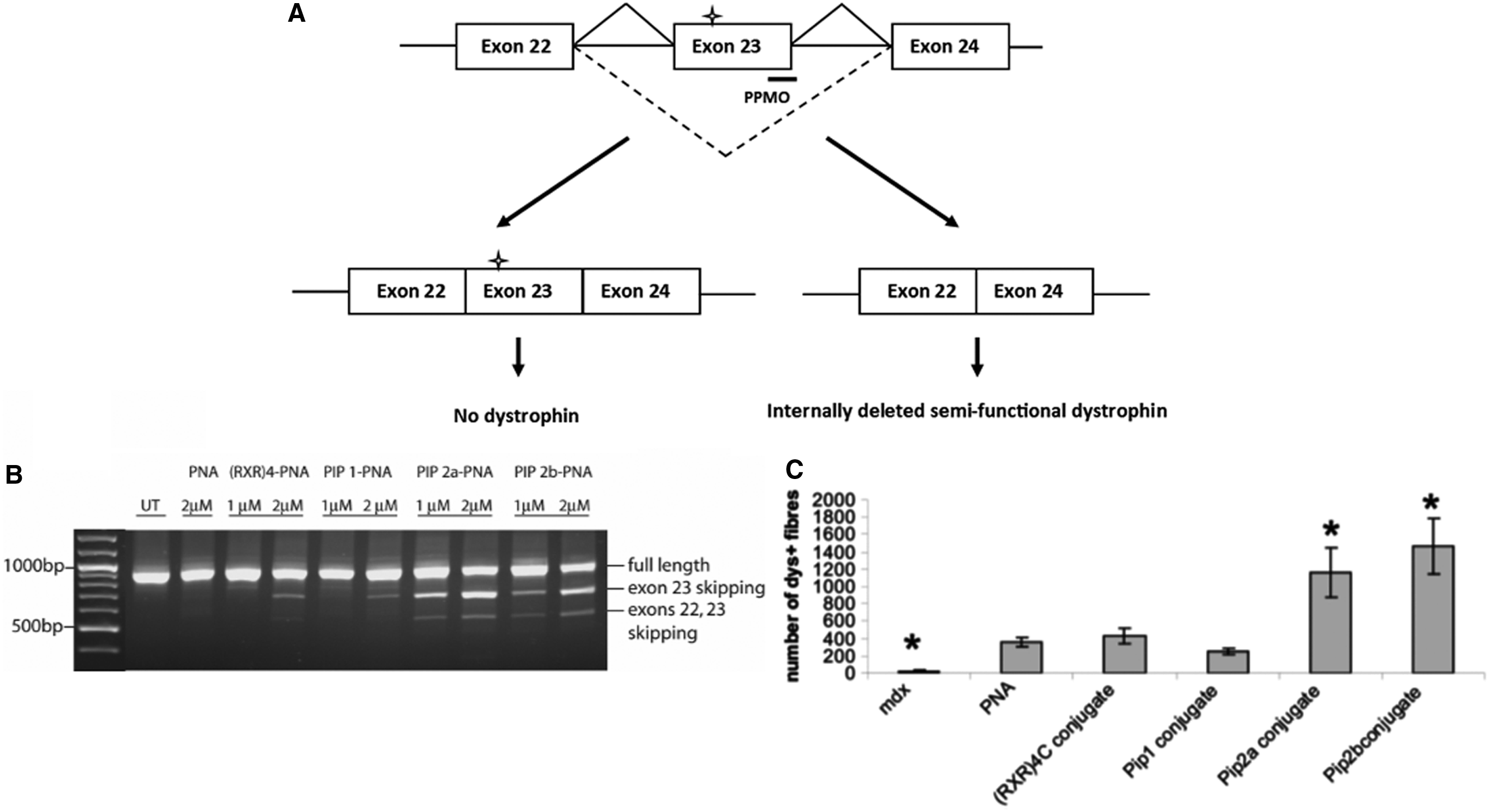
In further Pip-PNA synthesis and activity studies, Pip2a and Pip2b conjugates to PNA705 were each joined through a stable thioether linkage, and the peptide part was predominantly stable for 1 h in 20% mouse serum (data not shown). Thus these peptides were considered to be sufficiently stable for use in vivo. Therefore in vivo experiments were carried out such that injection into the tibialis anterior muscles of dystrophic mdx mice of Pip2a-PNADMD or Pip2b-PNADMD resulted in greater exon skipping as determined by RT-PCR and approximately threefold higher numbers of dystrophin-positive fibers compared to naked PNADMD or the then standard active control peptide-PNADMD conjugate, (R-Ahx-R)4-PNADMD
Systemic Delivery of Pip-PMO for Treatment of DMD
Quite surprisingly however, when Peptide-PNA was injected systemically by intravenous tail vein injection into mdx mice, the levels of dystrophin production were found to be no higher than naked PNA alone (data not shown). No simple reason could be deduced for this failure, but for further systemic studies we decided instead to switch the ON type to PMO (Fig. 1) that was utilized also by Sarepta.
Further iterations of Pip design for therapeutic development focused on shortening and simplification of the hydrophobic core region and modification of the cationic arms such that the combination of arginine (R), aminohexanoic acid (Ahx, X), and beta-alanine (B) had a more uniform composition. This Pip5 series of peptides, exemplified by Pip5e (Fig. 2), demonstrated enhanced splice-switching activity in vitro compared to previous Pip-PNA iterations (Fig. 6A). A single dose systemic comparator study with the arginine-rich B-PMO (B peptide = RXRRBRRXRRBRXB) demonstrated that Pip5e-PMO had similar efficacy in skeletal muscle, but remarkably enhanced activity in the heart with 25% wild-type dystrophin restoration compared to ∼3% for the B-PMO (Fig. 6B) [34].
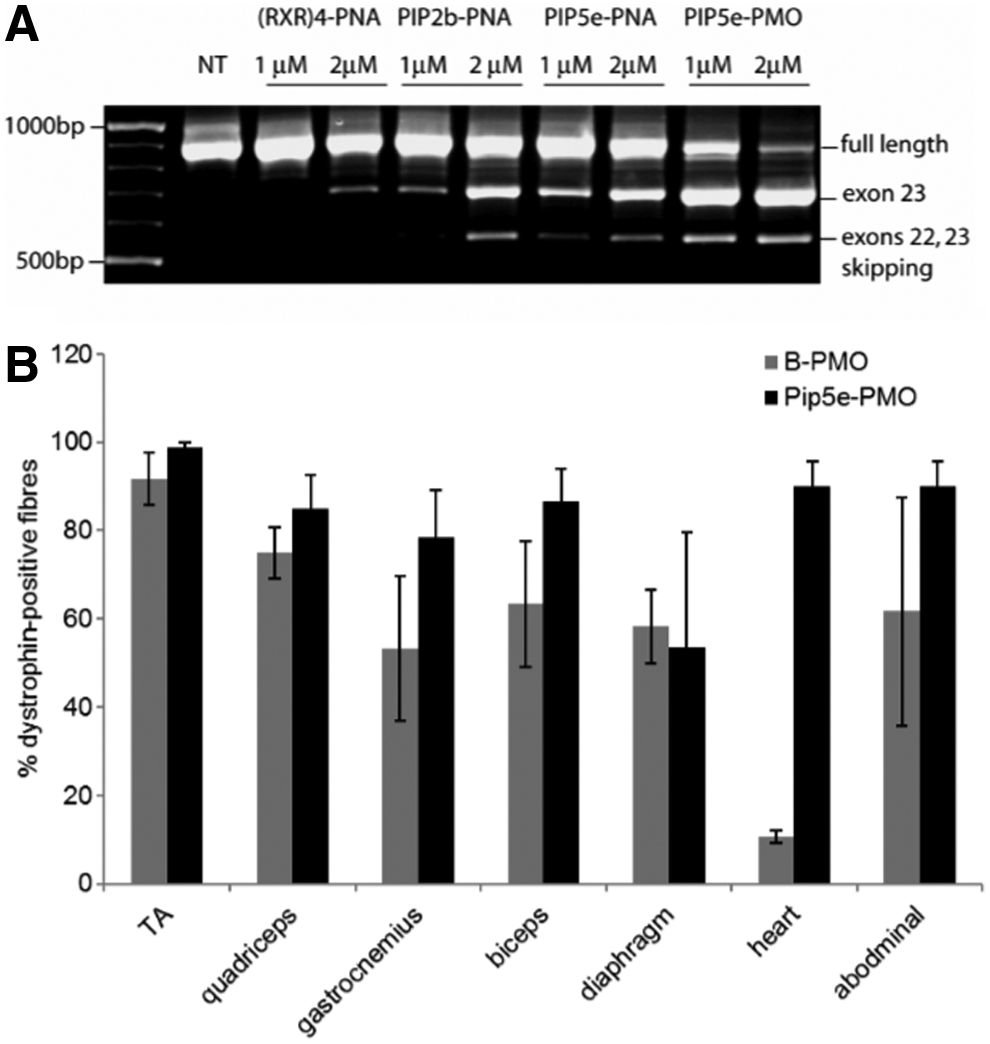
This result distinguished the Pip series of peptides from other arginine-rich CPPs and encouraged further series of Pip to be chemically synthesized for use as P-PMO conjugates. First we synthesized derivatives of Pip5e-PMO, consecutively assigned as Pip6-PMOs, and carried out single i.v. dose administration (12.5 mg/kg) into mdx mice [35]. These peptide-PMOs comprised alterations to the central 5-amino acid hydrophobic core of the Pip5e peptide
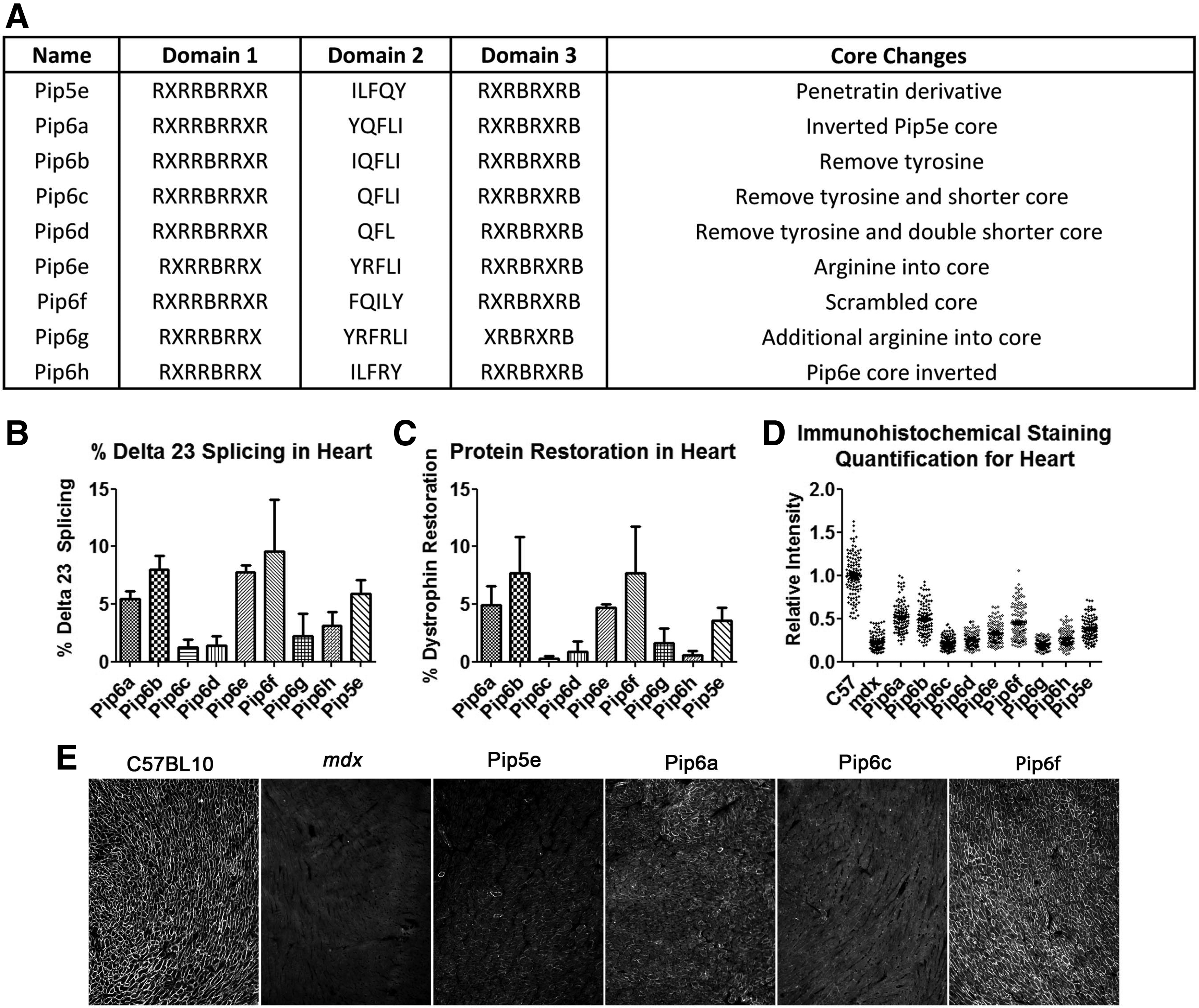
Sequences of Pip6-PMO conjugates, cardiac muscle splicing, and protein restoration following systemic administration of conjugates. A single 12.5 mg/kg systemic injection of peptide-PMO was administered to mdx mice, and tissues were harvested 2 weeks later.
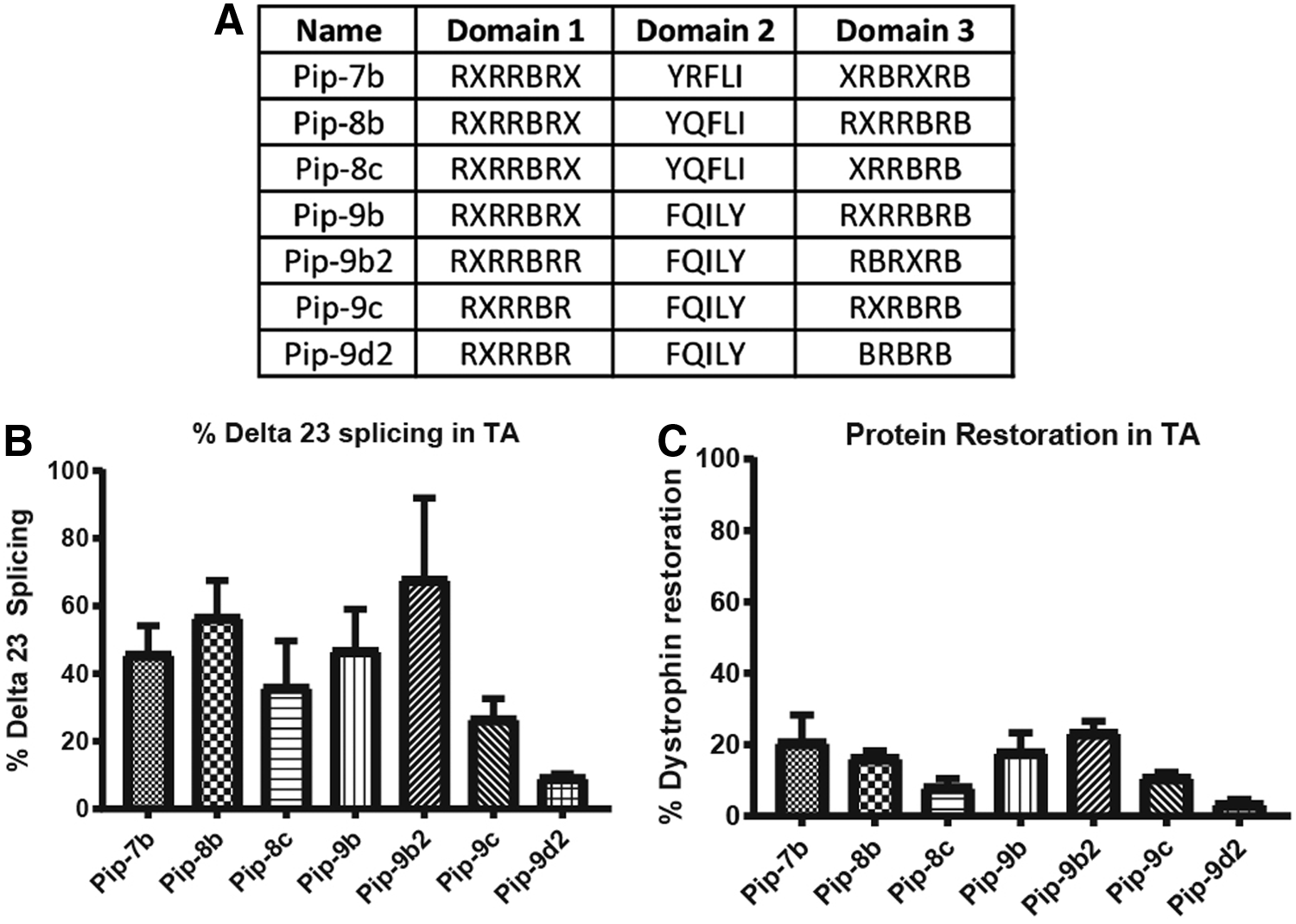
One of the key challenges for therapeutic success of this class of CPP-PMOs remains the toxicity associated with these peptides. Lethargy in rats following high dose administration of (R-X-R)4-XB-PMO has been previously noted [36], and studies in nonhuman primates suggested that the toxicology profile would need to be further improved for clinical development. Our hypothesis was that reduction in the number of arginine residues could confer improved toxicology outcomes, and thus, we sought to reduce the number of Arg residues in the Pip6 series, which contains 10 Arg residues, to between 6 and 9. Thus we developed the Pip 7, 8, and 9 series that were based on the hydrophobic core sequences used in three of the potent Pip6 series, namely Pip6e, Pip6a, and Pip6f, respectively. Carrying out single intravenous dose screening studies in mdx mice demonstrated a general pattern of reduced splice-switching activity as Arg residues were removed from the CPP. However the composition of the core region and the precise sequences of the cationic arms still remained the determinant of splicing activity (Fig. 8). Studies are currently underway focusing on the in vivo toxicological profile of these compounds and understanding the minimal requirements of CPP composition to maintain robust splice-switching activity while minimizing organ toxicity, especially nephrotoxicity.
Systemic Delivery of Pip-PMO for Treatment of SMA
Nusinersen (commercialized as spinraza), a wholly 2′-O-MOE PS ON (Fig. 1), has been approved for treatment of SMA by direct spinal delivery [6]. SMA is primarily a neurodegenerative disease of the lower motor neurons, and so the spinal cord is the primary tissue for therapeutic disease modification. SMA is caused by the deletion of survival motor neuron 1 gene, SMN1. Humans have a second paralog gene of SMN, called SMN2, which, through a single base change in an exon recognition motif, results in ∼90% of transcripts lacking exon 7 and which results in nonfunctional SMN protein [37]. Thus the number of copies of SMN2 and hence the cumulative levels of the 10% of transcripts that do produce a functional protein is the key determinant of SMA clinical outcome.
In SMA, multiple tissue and organs are affected by the low levels of SMN protein (reviewed in [38]). Heterogeneity of phenotype between patients is directly linked to copy numbers of SMN2 and thereby the levels of SMN protein [39]. Antisense ONs are used to sterically block a splicing repressor motif within intron 7 and thereby increase the exon 7 included transcripts and hence SMN protein expression (Fig. 9A) [40,41]. Unlike the dystrophin gene, a single ON targeted to a single site in the gene for SMN is able to provide treatment for nearly 100% of patients. While direct injection into the central nervous system by lumber puncture is the clinical route of administration for spinraza, seminal work from Adrian Krainer's group showed increased efficacy and survival when the drug was delivered systemically to neonatal SMA mice rather than by direct intracerebroventricular treatment of the central nervous system (CNS) [42]. This suggests that treatment with an effective systemic delivery agent for an ON may have an improved benefit for patients.
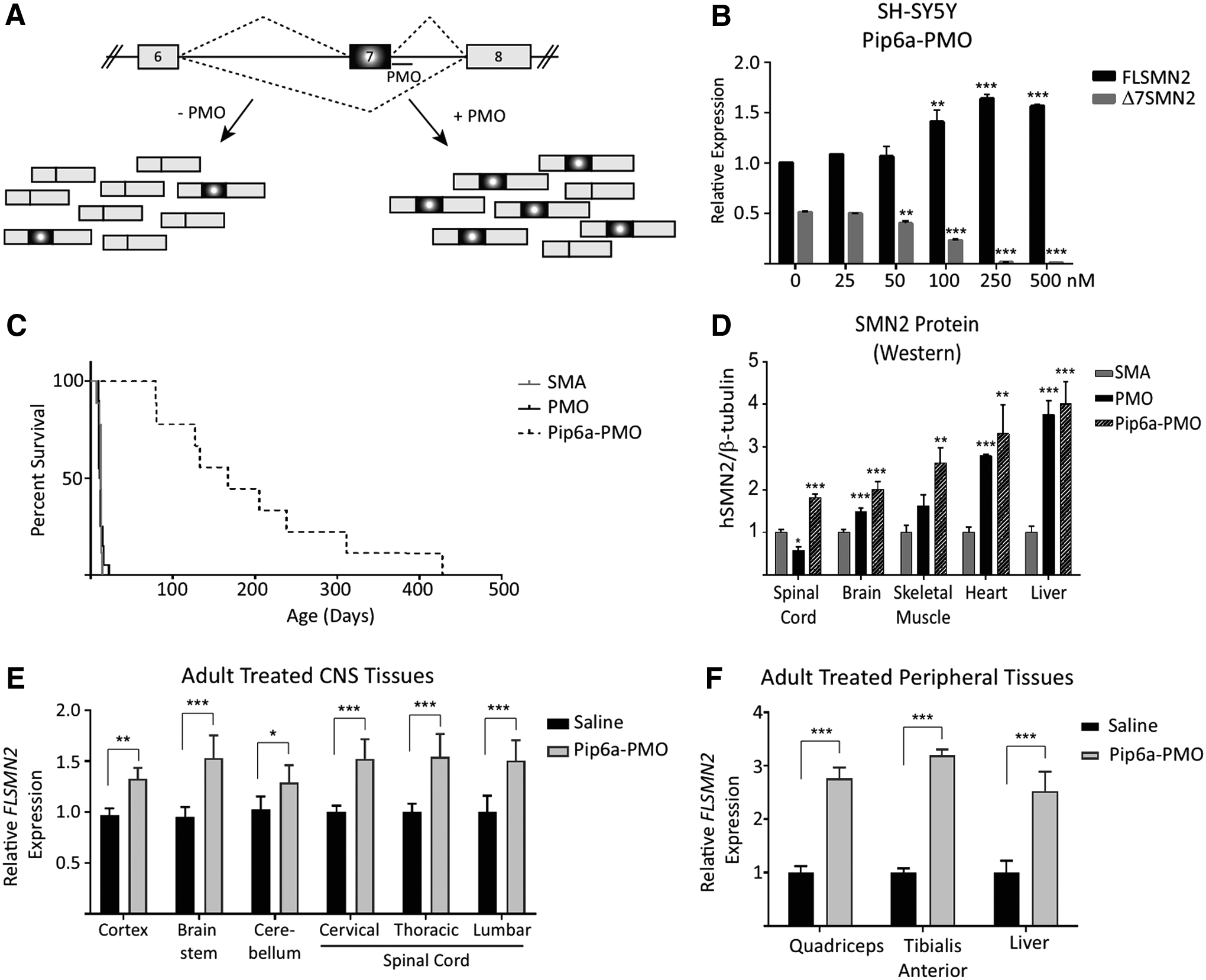
(
Following demonstration that Pip6a-PMO targeted to the repressor sequence at the end of intron 7 increased exon inclusion dose-dependently in cells (Fig. 9B), we therefore started treatment of severe SMA pups and generated biodistribution studies in adult mice using a Pip6A conjugated PMO as a potential efficacious systemic therapeutic [43]. Type I SMA pups normally survive between 7 and 12 days of age. Dosing with Pip6a-PMO at 10 μg/g yielded a median survival of 167 days, a similar survival length to animals given two 80 μg/g doses of nusinersen [42], whereas PMO alone resulted in a median survival of only 12 days (Fig. 9C). SMN protein as judged by western blotting was increased in all tissues assayed 7 days postadministration (Fig. 9D). In unaffected adult mice harboring the human SMN2 allele, both exon inclusion (Fig. 9E) and SMN protein (Fig. 9F) were increased significantly over saline treated mice when injected with 18 mg/kg Pip6a-PMO and tissues harvested 7 days postadministration.
During observation of the biodistribution of Pip6a-PMO in treated adult mice we were pleased to discover that the Pip6a delivered PMO not only to the skeletal muscle and heart but also to the brain and spinal cord. This was evidenced through upregulation of SMN expression in brain and spinal cord (Fig. 9D), as well as through labelled Pip6a-PMO expression being observed in the brain (data not shown). We have also found that other peptide-PMOs (P-PMOs) are capable of reaching the brain and spinal cord [44]. Now that we know that the Pip series is capable of transporting PMO into the CNS, in mice at least, we have continued with other P-PMOs to further increase this effect while simultaneously work is proceeding to enhance the safety and tolerability to develop clinically relevant peptides (patent application submitted).
The Future of P-PMO for Treatment of Neuromuscular and Neurodegenerative Diseases
Our extensive studies over several years of some 40 or 50 Pip as covalent PMO conjugates have demonstrated how substantial increases in splice redirecting activity (exon skipping and exon inclusion) are obtained when such Arg-rich peptides are used as cell and in vivo delivery agents. Furthermore, by use of the mdx mouse model of DMD, we have been able to obtain some insights into how Pip sequence variation affects activities. This has led us into exploration of the use of CPP-PMO conjugates in other neuromuscular diseases through use of alternative mouse models, as well as development of further series of peptides as PMO carriers.
For example, we have recently been studying the effects of Pip6a-PMO in HSA-LR mice, a model of the DM1 disease type in type 1 myotonic dystrophy. In this study we have found that Pip6a-PMO targeting the CUG repeats at the 3′ end of the gene for dystrophin myotonica protein kinase (DMPK), when injected intravenously into HSA-LR mice, corrects splicing defects in the mice, normalizes the global transcriptome at both expression and splicing levels, normalizes the DM1 specific phenotype, namely myotonia, and corrects DM1-specific molecular symptoms in DM1 muscle cells (data not shown). Work is now under way in development of newer CPPs that as conjugates of PMO retain efficacy, but with reduced toxicological measures, which may be suitable drug candidates for treatment of DM1 disease.
In the case of DMD, we have been exploring the use of several new series of Arg-rich CPPs based on the original Pip series. In this study we have not only studied how further sequence modulation affects the exon skipping activity and dystrophin production in the mdx mouse model of disease but also measured parameters associated with nephrotoxicity, which is the primary toxicological outcome of this class of compounds. New peptide leads as PMO conjugates are now emerging, and it is hoped that these will lead in due course to a clinical trial for DMD treatment.
A second exciting development is the discovery that Pip6a and another Arg-rich peptide, upon systemic injection, are able to carry a PMO across the blood-brain barrier (BBB) and into brain and spinal cord of mouse models of SMA and lead to both exon inclusion within SMN2, as well as phenotypic enhancements [43,44]. This opens up future opportunities with other neuromuscular diseases and many neurodegenerative disorders that could benefit from P-PMO systemic treatment. The promise of therapeutic ON technology envisaged in the 1970s and 1980s is truly coming to fruition, not only in the already well-advanced RNase-H-dependent antisense strategies but also now in the use of steric blocking ONs and their delivery by new classes of Arg-rich CPPs. The power of such platform technologies to enhance systemic delivery of these potent effectors is yet to be realized, but remains an exciting prospect for the antisense therapeutic field and patients alike.
Footnotes
Acknowledgments
M.J.G. thanks the many members of his research group (MRC-LMB) and M.J.A.W.'s group (University of Oxford) over many years that have contributed to the work described above. The authors thank Bernard Lebleu and his colleagues (University of Montpellier II) for collaboration and important contributions to the understanding of cell delivery of Pip conjugates of ONs.
Author Disclosure Statement
M.J.G., M.J.A.W., and C.G. are cofounder shareholders of PepGen, a company dedicated to the peptide-based enhancement of delivery of therapeutic oligonucleotide analogs. All other authors declare no conflicts of interest.