
Abstract
Effective in vivo delivery of small interfering (siRNA) has been a major obstacle in the development of RNA interference therapeutics. One of the first attempts to overcome this obstacle utilized intravenous injection of cholesterol-conjugated siRNA (chol-siRNA). Although studies in mice revealed target gene knockdown in the liver, delivery was relatively inefficient, requiring 3 daily injections of 50 mg/kg of chol-siRNA to obtain measurable reduction in gene expression. Here we present a new delivery approach that increases the efficacy of the chol-siRNA over 500-fold and allows over 90% reduction in target gene expression in mice and, for the first time, high levels of gene knockdown in non-human primates. This improved efficacy is achieved by the co-injection of a hepatocyte-targeted and reversibly masked endosomolytic polymer. We show that knockdown is absolutely dependent on the presence of hepatocyte-targeting ligand on the polymer, the cognate hepatocyte receptor, and the cholesterol moiety of the siRNA. Importantly, we provide evidence that this increase in efficacy is not dependent on interactions between the chol-siRNA with the polymer prior to injection or in the bloodstream. The simplicity of the formulation and efficacy of this mode of siRNA delivery should prove beneficial in the use of siRNA as a therapeutic.
Introduction
One of the early systemic siRNA delivery approaches utilized the conjugation of cholesterol to the siRNA (chol-siRNA), which improved the pharmacokinetic properties of siRNA and increased uptake by the liver (Soutschek et al., 2004). However, this delivery strategy was highly inefficient, as multiple injections of large doses of cholesterol-siRNA targeting apolipoprotein B (chol-siApoB) were required to achieve just 50% target gene knockdown in mice (Soutschek et al., 2004). Although no adverse effects were associated with such a dosing regimen, the low potency of chol-siApoB made it impractical for use in clinical settings (ROSSI, 2004).
Previously, we described the development of a targetable, polymer-based siRNA delivery platform named dynamic polyconjugate (DPC) that enables efficient siRNA delivery to liver hepatocytes after intravenous injection (Rozema et al., 2007). A central feature of this delivery system is the reversible modification of the amphipathic endosomolytic polymer composed of butyl and amino vinyl ether (PBAVE) (Wakefield et al., 2005; Rozema et al., 2007). Modification of PBAVE with carboxy dimethylmaleic anhydride (CDM) containing polyethylene glycol (CDM-PEG) and N-acetylgalactosamine (CDM-NAG) provides for surface charge masking and hepatocyte targeting via the cognate receptor for NAG, the asialoglycoprotein receptor (ASGPr) (Rozema et al., 2007). Once taken up by the hepatocyte via receptor-mediated endocytosis, delivery of siRNA to the cytoplasm is triggered by hydrolysis of the CDM bond in the acidic environment of the endosome, resulting in polymer unmasking and polymer-facilitated endosomolysis. In the prototypical DPC configuration, the siRNA is covalently attached to the polymer via a disulfide bond. Once exposed to the reducing environment of the cytoplasm, the disulfide bond is reduced and siRNA is released from the polymer, allowing the siRNA to engage the cellular RNAi machinery (Rozema et al., 2003; Rozema et al., 2007). DPC-mediated siRNA delivery is much more efficient than that relying on the injection of chol-siRNA alone and results in significantly greater levels of target gene knockdown.
We reasoned that the lack of efficient target gene knockdown using chol-siRNA alone was due at least in part to inefficient endosomal escape and subsequent degradation of the chol-siRNA in endosomal and/or lysosomal compartments. In this study, we show that the efficacy of chol-siRNA in liver hepatocytes in mice can be improved over 500-fold by simply co-injecting it with the hepatocyte-targeted, endosomolytic PBAVE polymer used in the prototypical DPC. We also demonstrate that this delivery approach is highly efficacious in non-human primates. Our data shows that the increase in efficacy is not dependent on interaction between the chol-siRNA and the polymer prior to injection or in the bloodstream prior to contact with the target cell. These results have important implications for the design and manufacture of siRNA delivery systems for clinical use.
Materials and Methods
PBAVE, CDM reagents, and formulation
Synthesis of the PBAVE polymer, CDM-PEG, and CDM-NAG were carried out as previously described (Rozema et al., 2003; Wakefield et al., 2005). NAG-DPC, in which ApoB siRNA is attached to PBAVE, was prepared as described (Rozema et al., 2007). For NAG-PBAVE preparation, CDM-PEG and CDM-NAG at a 2 to 1 wt/wt ratio were added to PBAVE at a 7 to 1 wt/wt ratio in 0.2 mL of 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffered (5 mM, pH 7.5) isotonic glucose and the reaction allowed to proceed for at least 1 hour at room temperature. Untargeted PEG-PBAVE was prepared similarly, except that CDM-NAG was omitted from the reaction and replaced with CDM-PEG. The chol-siRNA and CDM-modified PBAVE were co-injected into animals shortly after mixing, or injected separately as subsequently described in the text. For preparation of fluorescently tagged PBAVE, Cy3 NHS ester (GE Healthcare, Piscataway, NJ) or Oregon Green NHS ester (Life technologies, Grand Island, NY) was coupled to PBAVE according to the manufacturer's recommendations.
siRNAs
The siRNAs used in this study contained the following sequences:
siApoB sense: 5’-(R)-GGAAUCuuAuAuuuGAUCcA*A-3’; siApoB antisense: 5’-uuGGAUcAAAuAuAAGAuUCc*c*U-3’; siF7 sense: 5’-(R)-GGAfUfCAfUfCfUfCAAGfUfCfUfUAfCdT*dT-3’; siF7 antisense: 5’-GfUAAGAfCfUfUGAGAfUGAfUfCfCdT*dT-3’; siLuc sense: 5’-(R)-cuuAcGcuGAGuAcuucGAdT*dT-3’; and siLuc antisense: 5'-UCGAAGuACUcAGCGuAAGdT*dT-3’.
(R indicates amine or cholesterol moiety; lower case, 2’-O-methyl substitution; f, 2’-F substitution; d, 2-deoxy substitution; and *, phosphorothioate linkage.)
Factor 7 activity measurements
Plasma samples from mice were prepared by collecting blood (9 volumes) by submandibular bleeding into microcentrifuge tubes containing 0.109 M sodium citrate anticoagulant (1 volume) following standard procedures. Factor 7 activity in plasma was measured using a chromogenic assay following manufacturer's recommendations (BIOPHEN 7, Aniara, Mason, OH). Absorbance of colorimetric development was measured using a Tecan Safire 2 microplate reader at 405 nm.
Serum ApoB protein level determination
Mice were fasted for 4 hours before blood collection by submandibular bleeding. Serum ApoB levels were determined by standard sandwich enzyme-linked immunosorbent assay methods. Briefly, a polyclonal goat anti-mouse ApoB antibody, kindly provided by Dr. Kevin Williams, Thomas Jefferson University, and a rabbit anti-mouse ApoB antibody (Meridian Life Science, Inc., Saco, ME) were used as primary antibodies. A goat anti-rabbit immunoglobin G conjugated to horseradish peroxidase was used as the secondary antibody (Sigma, St. Louis, MO). Absorbance of tetramethylbenzidine (Sigma) after colorimetric development was measured using a Tecan Safire2 (Männedorf, Switzerland) microplate reader at 450 nm.
Quantitative real-time polymerase chain reaction
In preparation for quantitative polymerase chain reaction (PCR), total RNA was isolated from tissue samples homogenized in TriReagent (Molecular Research Center, Cincinnati, OH) following the manufacturer's protocol. Approximately 500 ng RNA was reverse-transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). Premanufactured TaqMan gene expression assays for mouse Factor 7 (Assay ID: Mm00487333_m1) and ß-actin (Part No.: 4352341E) were used in biplex reactions in triplicate using TaqMan Gene Expression Master Mix (Applied Biosystems). Quantitative PCR was performed by using a 7500Fast or StepOnePlus real-time PCR system (Applied Biosystems). The delta-delta cycle threshold method was used to calculate relative gene expression.
Agarose gel electrophoresis
Chol-siRNA (2 μg) was mixed with Oregon Green–labeled PBAVE (10 μg) with or without CDM-NAG modification for 10 minutes in 20 μl of gel running buffer (1×Tris-acetate-ethylenediaminetetraacetic acid [TAE]) at ambient temperature. Control samples containing individual components were similarly incubated. Samples were then applied to a 2% agarose gel (Agarose 1000, Invitrogen). Electrophoresis was carried out under volt-limiting conditions at 100 V for 45 minutes using 1×TAE buffer. All samples containing PBAVE contained a mixture of 4 to 1 w/w of unlabeled PBAVE and Oregon Green–labeled PBAVE. The agarose gel was stained in TAE buffer containing 0.5 μg/mL of ethidium bromide to visualize chol-siRNA after electrophoresis. The sample migration pattern was analyzed using a Kodak Gel Logic 200 Imaging System (Kodak, Rochester, NY).
Confocal microscopy
Dy547-labeled cholesterol-conjugated small interfering Factor 7 (siF7) and Oregon green-labeled NAG-PBAVE were injected into the tail vein of mice. Liver tissue harvest and preparation of tissue sections were performed as previously described (Rozema et al., 2007). Tissue sections were counterstained with Alexa-633-conjugated phalloidin (20 nM, Invitrogen, Carlsbad, CA) and To-pro-3 (40 nM, Invitrogen) in PBS as previously described (Rozema et al., 2007). Mounted slides were analyzed using a Zeiss LSM710 confocal microscope (Carl Zeiss Microimaging, Inc., Thornwood, NY). Co-localization analyses of fluorescence signals were performed using the Zeiss confocal ZEN software.
Mice and injection procedures
Female mice (strain ICR), 6 to 8 weeks old, were obtained from Harlan Sprague-Dawley (Indianapolis, IN). Six- to eight-week-old male or female ASGPr (129-Asgr2tm1her) and female low-density lipoprotein receptor (LDLr; B6.129S7-Ldlrtm1Her/J) knockout mice were obtained from Jackson Laboratory (Bar Harbor, ME). Wild type (WT) female C57B/6J mice (Harlan Sprague-Dawley), 6 to 8 weeks old, were used as controls for studies using knockout mice. All mice were handled in accordance with animal use protocols approved by the Animal Care and Use Committee at Arrowhead Madison Inc. (Madison, WI). Mice were maintained on a 12 hours light/dark cycle with free access to water and food (Harlan Teklad Rodent Diet, Harlan, Madison, WI). For co-injections in mice, CDM-modified PBAVE and chol-siRNA were mixed together and injected as a bolus into the tail vein in a total volume of 0.2 mL of HEPES-buffered (5 mM, pH 7.5) isotonic glucose under standard conditions. In some studies, in which chol-siRNA and CDM-modified PBAVE were injected separately, a total volume of 0.3 mL was injected.
Non-human primate study
Male rhesus monkeys were housed at the University of Wisconsin–Madison. Prior to injection, animals were sedated with ketamine (10 mg/kg, injected intramuscularly). One animal received a single injection of 7.8 mL (2 mL/kg) containing chol-siApoB (2 mg/kg) and NAG-PBAVE (15 mg/kg). Sample was injected into the small saphenous vein slowly over 5 minutes. The control animal received 7.8 mL (2 mL/kg) of delivery vehicle only. Blood samples were collected every 3–4 days for gene silencing and clinical chemistry analysis of serum glucose, blood urea nitrogen, creatinine, creatine kinase, aspartate transaminase, lactate dehydrogenase, bilirubin, gamma-glutamyl transpeptidase, alanine transaminase, total protein, albumin, alkaline phosphatase, calcium, phosphorus, iron, sodium, potassium, and chloride levels (Meriter Laboratories, Madison, WI). All procedures were carried out in accordance with the National Research Council's Guide for the Care and Use of Laboratory Animals (National Research Council [U.S.], 2011) and were approved by the University of Wisconsin–Madison and Arrowhead Madison, Inc. animal care and use committees.
Results
Co-injection of a hepatocyte-targeted endosomolytic polymer and cholesterol-conjugated siRNA results in efficient target gene knockdown in mice
We first evaluated whether providing a means for endosome escape increased the efficacy of chol-siRNA after intravenous injection in mice. The endosomolytic polymer PBAVE, without covalent siRNA attachment, was modified with CDM-PEG and CDM-NAG as previously described for hepatocyte-targeted delivery (Rozema et al., 2007). This modified PBAVE is referred to as NAG-PBAVE. In these experiments, we used a chol-siRNA targeting coagulation Factor 7 (chol-siF7). F7 is expressed exclusively in hepatocytes and secreted into the bloodstream. The level of F7 activity in plasma is easily monitored by a simple chromogenic assay.
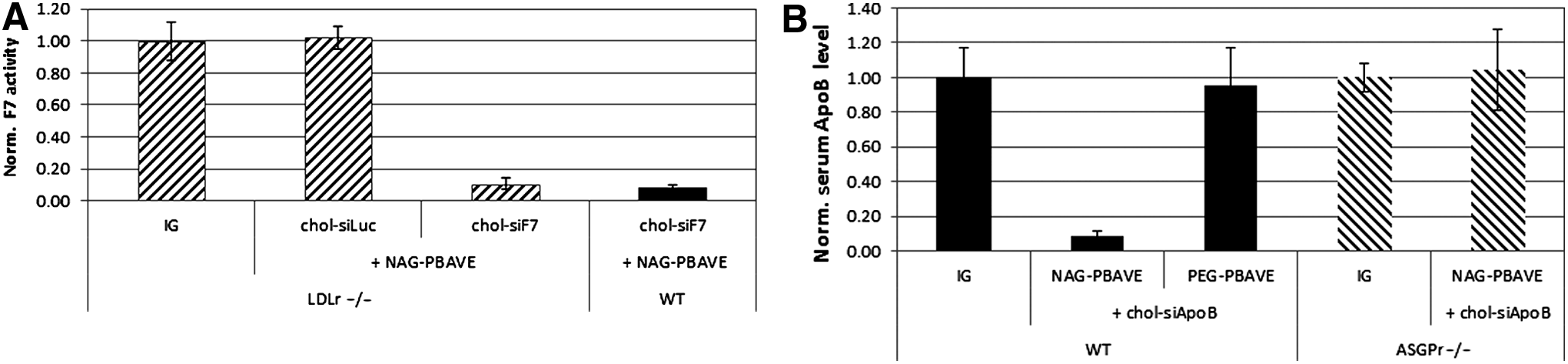
Injection of a 5 mg/kg dose of chol-siF7 alone did not lead to F7 gene silencing (Fig. 1A). This was not surprising, as this dose is more than 10-fold below that required for gene knockdown using chol-siRNA, as shown previously (Soutschek et al., 2004). In contrast, co-injection of NAG-PBAVE with just a 1 mg/kg dose of chol-siF7–enabled robust F7 gene silencing. Plasma F7 activity levels were reduced by 95%±3% with a corresponding reduction in F7 messenger RNA (mRNA) level in the liver of 93%±3% relative to mice that received isotonic glucose only. This level of gene silencing was similar to that obtained with the prototypical hepatocyte-targeted DPC (NAG-DPC) containing 1 mg/kg F7 siRNA covalently attached to the PBAVE polymer. Further reduction in F7 was observed in mice receiving a 5 mg/kg dose of chol-siF7 co-injected with NAG-PBAVE. No significant F7 reduction was observed when a control cholesterol-conjugated siRNA against firefly luciferase was co-injected with NAG-PBAVE or when F7 siRNA without cholesterol conjugation (siF7) was co-injected. In addition, when chol-siF7 was injected with untargeted PBAVE (PEG-PBAVE) no gene silencing was observed. These results indicate that both cholesterol conjugation of the siRNA and the hepatocyte-targeted NAG-PBAVE are required for target gene knockdown.
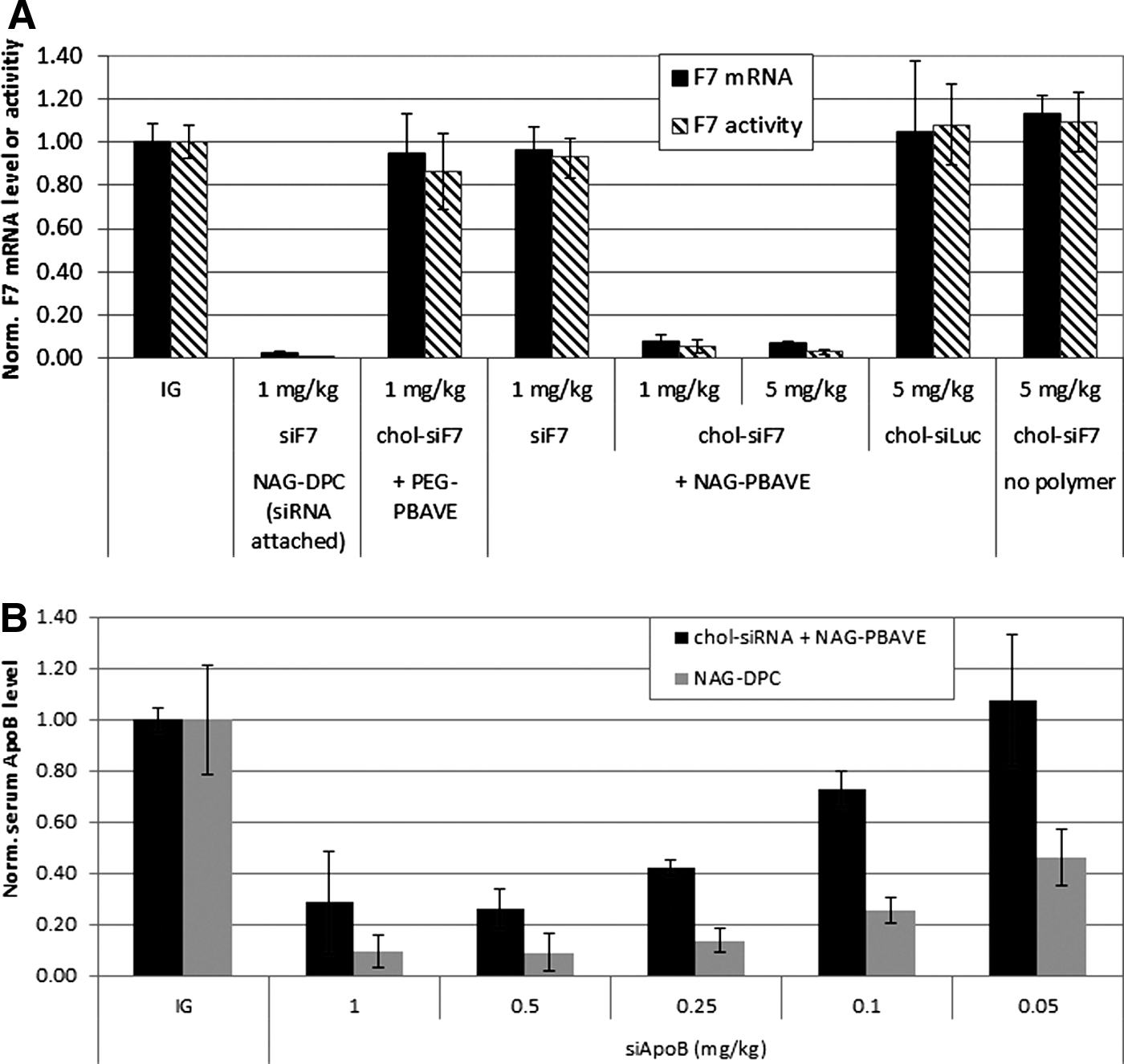
Co-injection with polymer composed of butyl and amino vinyl ether modified with carboxy dimethylmaleic anhydride containing polyethylene glycol and carboxy dimethylmallieic anhydride containing N-acetylgalatosamine (NAG-PBAVE) improves efficacy of cholesterol-conjugated small interfering RNA (chol-siRNA) in mice.
We next compared the relative efficacy of the co-delivery approach with delivery using the NAG-DPC in which the siRNA is covalently linked to PBAVE in a dose titration experiment. In this experiment, we used a siRNA sequence targeting apolipoprotein B (siApoB). Apolipoprotein B (ApoB) is a gene expressed in hepatocytes that is involved in cholesterol transport and metabolism (Rhainds and Brissette, 1999). In both delivery configurations, the amount of PBAVE was held constant and the amount of co-delivered cholesterol-conjugated siApoB (chol-siApoB) or siApoB directly attached to PBAVE was varied. Results indicate that approximately 0.25 mg/kg of chol-siApoB was required to achieve a 50% reduction in serum ApoB levels of mice when co-delivered with NAG-PBAVE (Fig. 1B). Serum clinical chemistry parameters indicated no adverse effects resulting from the polymer doses injected in these studies (data not shown). This represents a greater than 500-fold improvement in the efficacy of chol-siApoB compared with previously reported results using the same chol-siApoB alone (Soutschek et al., 2004). This level of efficacy is somewhat less than that achieved with NAG-DPC, in which siApoB is covalently attached to PBAVE. Using NAG-DPC, a 0.05 mg/kg siApoB dose was required to achieve a 50% reduction in ApoB protein levels. It is remarkable that the efficiency of the co-injection method can approach that of NAG-DPC, considering that in the co-injection method NAG-PBAVE and chol-siApoB are not covalently attached. We believe that the benefits gained from the simplified formulation and manufacturing procedures required for the co-injection method will outweigh the slight decrease in efficacy.
Simultaneous knockdown of two genes after co-injection of NAG-PBAVE and chol-siRNAs in mice
There are diseases, such as cancer, that might benefit from therapeutic strategies that target multiple genes. In addition, it could be beneficial to use multiple siRNAs to target different sequences within the same transcript, for example, when treating infections caused by mutable viruses in which nucleotide substitutions could lead to resistance to a single siRNA. The relative simplicity of the formulation procedure required for co-injection would facilitate the inclusion of multiple chol-siRNAs. We tested whether we could target multiple sequences by co-injecting NAG-PBAVE with a mixture of chol-siApoB and chol-siF7 in mice and determining the effects on ApoB and F7 levels. After a single co-injection of NAG-PBAVE and a 1 mg/kg dose of each chol-siRNA, a 90%±6% reduction in serum ApoB protein and an 88%±1% reduction in plasma F7 activity were observed (Fig. 2). The degree of silencing of each gene was similar to that observed when each chol-siRNA was injected separately, suggesting that there was minimal impact on the efficacy of either chol-siRNA when both were co-injected with NAG-PBAVE.
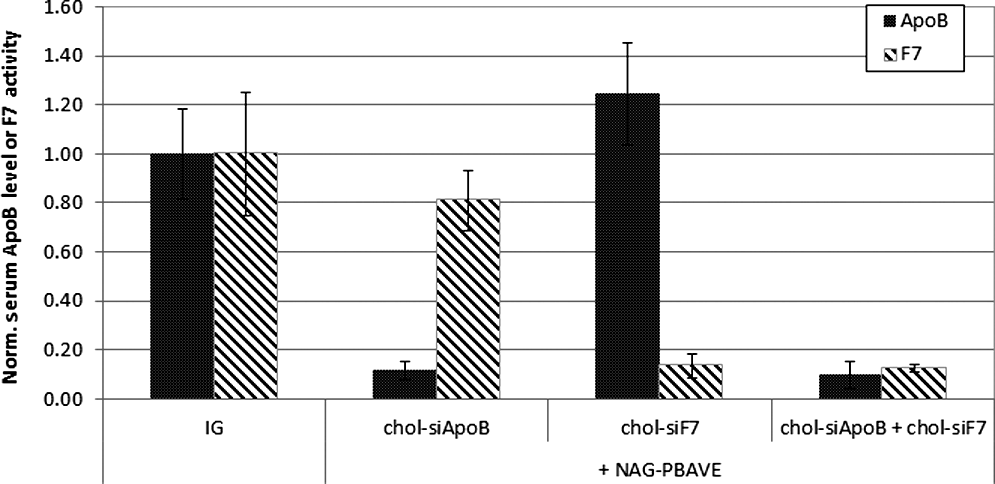
Simultaneous knockdown of 2 liver genes in mice. Serum ApoB protein levels (solid bars) or F7 activity (diagonal shaded bars) in ICR mice 2 days after a single i.v. injection of either isotonic glucose alone, chol-siApoB with NAG-PBAVE (10 mg/kg), chol-siF7 (1 mg/kg) with NAG-PBAVE (10 mg/kg), or both chol-siApoB and chol-siF7 at 1 mg/kg each with NAG-PBAVE (10 mg/kg). ApoB protein and F7 activity levels were normalized to those measured in mice receiving isotonic glucose only. Bars indicate group mean values, error bars indicate SD, n=4.
Efficacy in non-human primates
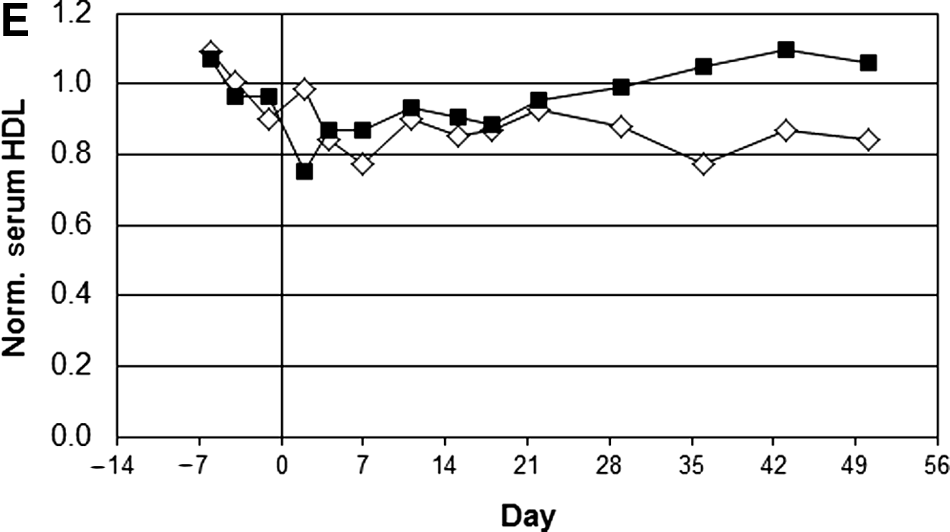
To evaluate the efficacy of this co-delivery approach in larger animals, a 2 mg/kg dose of chol-siApoB was co-injected with NAG-PBAVE into a rhesus monkey. After a single dose, a steady reduction in serum ApoB protein was observed, reaching a maximum of 78% reduction relative to pre-dose levels on day 14 after injection (Fig. 3A). Maximal ApoB protein reduction was sustained until day 30 and then serum ApoB protein level slowly recovered to the pre-dose level by day 50. A similar slow onset and long duration of gene silencing was also observed using NAG-DPC to deliver covalently attached siApoB (data not shown). Serum ApoB reduction was accompanied by reductions in serum cholesterol (39%) and triglyceride (60%) levels (Fig. 3B, C), as would be expected in animals with lowered ApoB levels. A corresponding reduction in LDL was also observed, again consistent with ApoB gene silencing (Fig. 3D). No significant changes in high-density lipoprotein (HDL) levels (Fig. 3E) or in toxicity-indicating serum clinical chemistry parameters were observed (data not shown). The maximal level of ApoB knockdown in non-human primates was similar to that observed in mice, indicating that this delivery system is proportionally effective in larger animals. The single-dose efficacy of this approach and the long duration of gene silencing observed in non-human primates could be a beneficial feature for clinical applications.
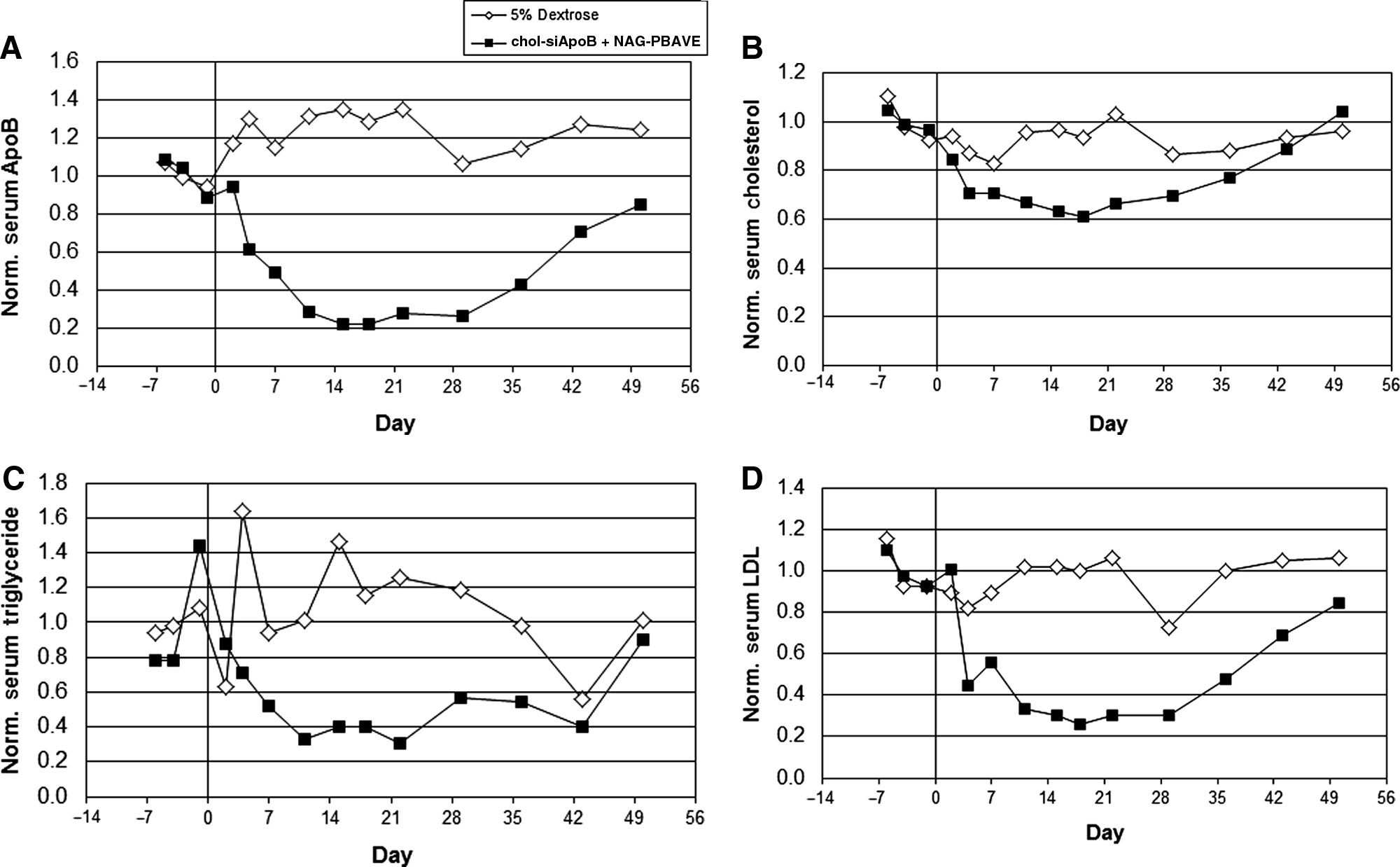
Co-injection of chol-siRNA and NAG-PBAVE is efficacious in rhesus monkey. Levels of serum ApoB protein
A functional ASGPr but not LDLr is required for gene knockdown activity of cholesterol-conjugated siRNA when co-injected with NAG-PBAVE
A previous study showed that delivery of chol-siRNA to mouse liver was at least partially dependent on the presence of the LDLr (Wolfrum et al., 2007). We utilized LDLr –/– mice to determine whether the LDLr was required for target gene knockdown when chol-siApoB was co-injected with NAG-PBAVE. No difference in the extent of target gene knockdown was detected between LDL –/– mice and wild type mice (Fig. 4A). These results indicate that the presence of the LDLr is not required for effective target gene knockdown when chol-siRNA is co-injected with NAG-PBAVE.
Efficacy of co-injected chol-siRNA and NAG-PBAVE is dependent on asialoglycoprotein receptor (ASGPr) but not LDL receptor (LDLr) expression.
We have observed that the cognate receptor for NAG, ASGPr, is necessary for target gene knockdown in liver hepatocytes when siRNAs are attached to the PBAVE as in the prototypical DPC formulation (unpublished data). ASGPr is a heterodimeric membrane protein consisting of 2 different hepatic lectins (HL-1 and HL-2) and is highly expressed on hepatocytes. Binding of ligand by ASGPr induces rapid internalization by receptor-mediated endocytosis. The expression of both subunits is required for normal receptor function, including receptor-mediated endosomal uptake (Ishibashi et al., 1994). To evaluate the role of ASGPr for delivery of chol-siApoB with NAG-PBAVE, we utilized HL-2 –/– mice and monitored ApoB levels in the serum after co-injection. Results indicate that effective gene silencing was completely abolished in HL-2 –/– mice (Fig. 4B). Gene silencing was also abolished in WT mice when untargeted PEG-PBAVE was co-injected with chol-siApoB in place of NAG-PBAVE. These results indicate that specific ASGPr-mediated uptake of NAG-PBAVE is a critical requirement for enhancing the efficacy of chol-siRNA.
NAG-PBAVE co-localizes with cholesterol-conjugated siRNA in mouse liver hepatocytes
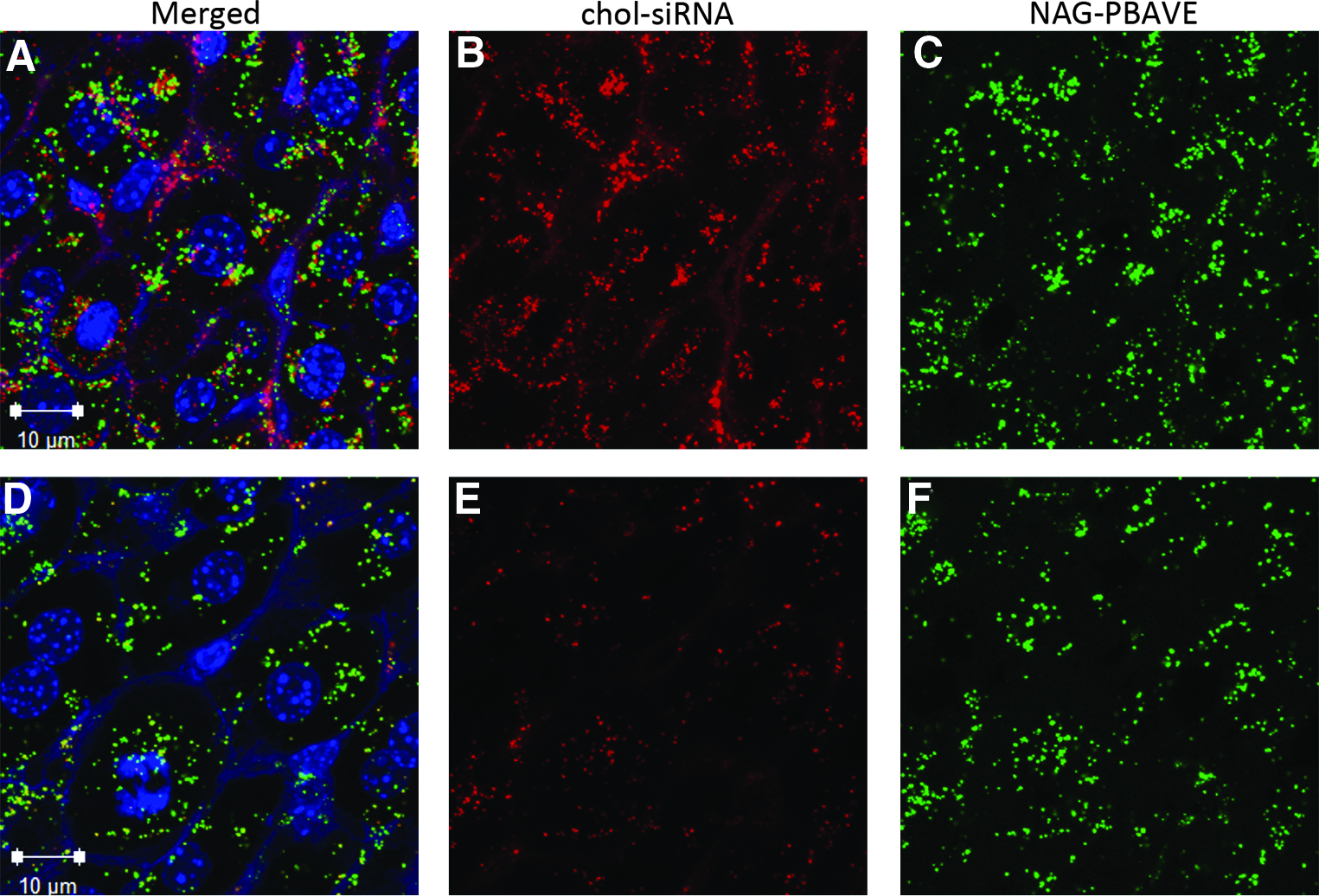
The lack of gene silencing in ASGPr deficient mice points to an essential role of ASGPr-mediated endocytosis of NAG-PBAVE for effective chol-siRNA delivery. As previously described, a decrease in pH, such as that which occurs during endosome maturation, can trigger the unmasking of NAG-PBAVE, thus revealing PBAVE's endosomolytic activity (Rozema et al., 2003; Rozema et al., 2007). We hypothesized that effective delivery of chol-siRNA with NAG-PBAVE would require that both components be localized to the same endosome in order that lysis of the endosome by unmasked PBAVE could facilitate escape of the chol-siRNA and allow its entry into the cytoplasm. Confocal microscopy analyses of liver tissue sections from mice co-injected with Dy547-labeled chol-siRNA and Oregon Green-labeled NAG-PBAVE revealed that chol-siRNA and NAG-PBAVE signals overlap, especially within liver hepatocytes. (Fig. 5A–F). At 15 minutes after co-injection (Fig. 5A–C), 99% of the signal from the NAG-PBAVE co-localized with signal from the chol-siRNA (Table 1). The majority of these co-localized signals was punctate, small in size, and mostly scattered throughout the cytoplasm of hepatocytes. The size and location of these signals are reminiscent of early endosomes (Skjeldal et al., 2012). At 2 hours after injection, many of the NAG-PBAVE signals appear brighter and larger than at 15 minutes (Fig. 5D–F). It is possible these signals originate from NAG-PBAVE residing in late endosomes and lysosomes, which are typically larger than early endosomes (Skjeldal et al., 2012; Marbet et al., 2006). Immunostaining with the late endosome/lysosome marker LAMP1 confirmed that NAG-PBAVE was found in this type of subcellular compartment at 2 hours post-injection (data not shown). Furthermore, most co-localized signals were found either in close proximity to the nucleus or along hepatic bile canaliculi tracks, both known to be lysosome-rich areas in the liver (CRAWFORD, 1996; Marbet et al., 2006). These data indicate that co-injection of NAG-PBAVE and chol-siRNA results in their co-localization to the same intracellular vesicles, most likely endosomes. This co-localization remains visible at later time points, when endosomes would be expected to have matured into late endosomes/lysosomes.
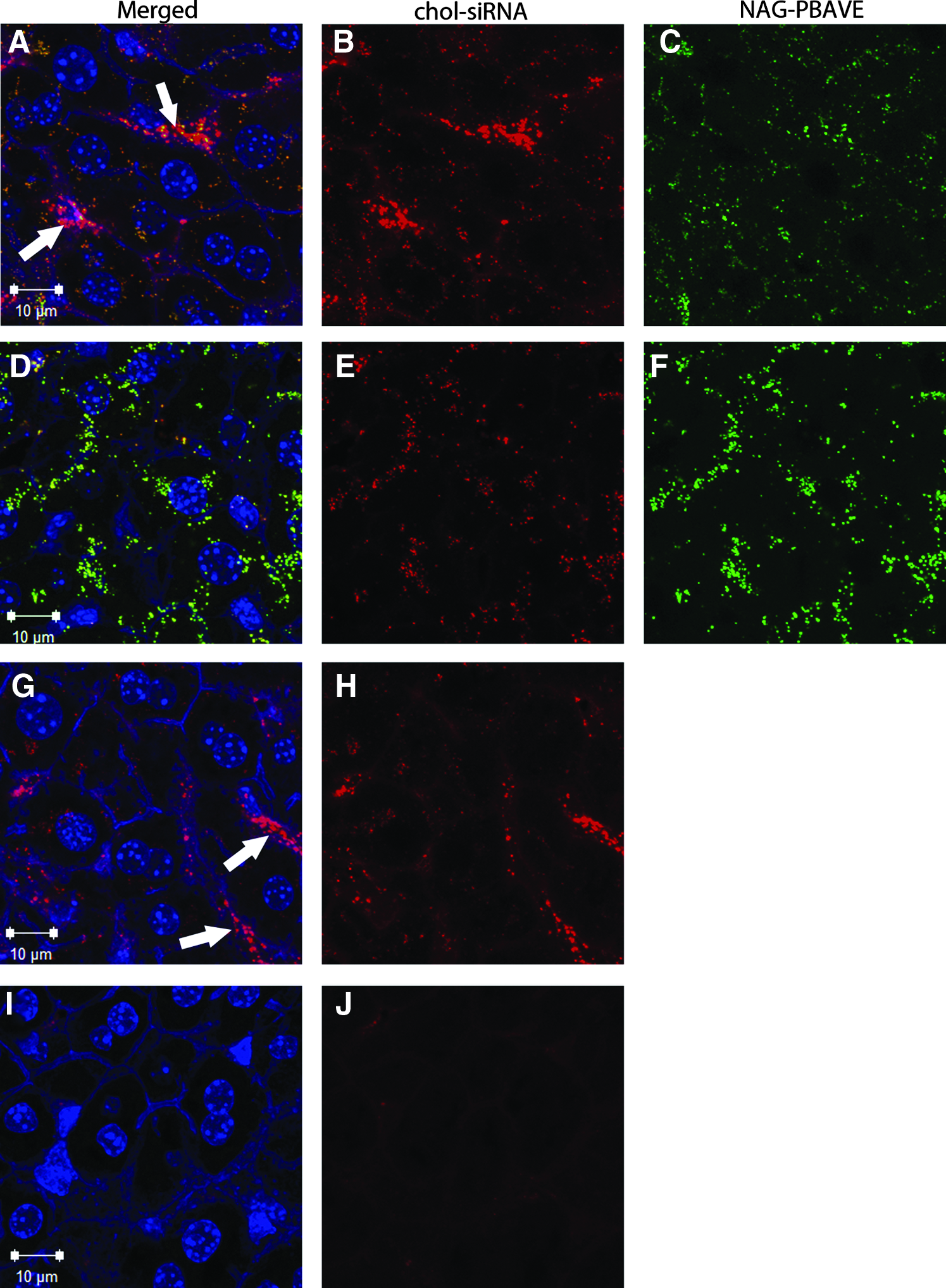
Confocal micrographs of chol-siRNA and NAG-PBAVE in mouse liver sections. Confocal images of co-injected Dy547-labeled chol-siF7 (2 mg/kg) and Oregon Green-labeled NAG-PBAVE (10 mg/kg) at 15 minutes
Values represent percentage of pixels fluorescence signal from chol-siRNA and from NAG-PBAVE at 15 minutes and 2 hours (based on chol-siRNA injection time) that are co-localized with the other using the co-localization application provided with the Zeiss LSM710 confocal software.
chol-siRNA, cholesterol-conjugated small interfering RNA; NAG-PBAVE, polymer composed of butyl and amino vinyl ether modified with carboxy dimethylmaleic anhydride containing polyethylene glycol and carboxy dimethylmaleic anhydride containing N-acetylgalactosamine.
While nearly all the visible NAG-PBAVE was co-localized with chol-siRNA in hepatocytes 15 minutes after co-injection, only 7% of the total chol-siRNA signals co-localized with NAG-PBAVE signals (Table 1). A large proportion of the chol-siRNA signal that did not co-localize with NAG-PBAVE appeared to be present in sinusoids (white arrows in Fig. 5) and not in hepatocytes. By 2 hours after injection, the proportion of chol-siRNA that co-localized with NAG-PBAVE increased to 87% and was found mainly within the hepatocytes. This increase in co-localization is mainly due to a decrease of chol-siRNA that is not co-localized with NAG-PBAVE in the liver. This decrease could be due to nuclease degradation of the chol-siRNA and its elimination when not co-localized with NAG-PBAVE. This is supported by the qualitative decrease in overall chol-siRNA signal intensity in the liver over time in animals injected with chol-siRNA alone (Fig. 5G–J). At 15 minutes after injection, the pattern and intensity of chol-siRNA signal in these animals was similar to that observed when chol-siRNA was co-injected with NAG-PBAVE (Fig. 5G–H). However, by 2 hours after injection, the amount of chol-siRNA signal detectable in liver was much reduced compare to that detectable when NAG-PBAVE was co-injected (Fig. 5I–J). These results suggest that co-injection of NAG-PBAVE with chol-siRNA alters how chol-siRNA is processed in hepatocytes, leading to preservation of chol-siRNA in endosomes containing NAG-PBAVE. In addition to its primary function as an endosomolytic agent, this highlights a possible second functional role of unmasked NAG-PBAVE polymer, the protection from nuclease degradation of chol-siRNA co-localized in the same endosome.
NAG-PBAVE and chol-siRNA do not interact prior to injection
For NAG-PBAVE and chol-siRNA to be localized in the same endosome, they would need to be either (1) taken up into the same endosome during a single endosomal uptake event or (2) co-localized as a result of fusion of endosomes from different uptake events. One scenario by which NAG-polymer and chol-siRNA could be taken up into the same endosome would be if NAG-PBAVE and chol-siRNA formed a complex that could bind to the ASGPr and trigger receptor-mediated endocytosis. We tested whether NAG-PBAVE and chol-siRNA could interact during formulation by assessing potential complex formation by gel electrophoresis. Results indicate that NAG-PBAVE does not form complexes with chol-siRNA, as the migration of pre-incubated NAG-PBAVE and chol-siRNA was unchanged compared with each incubated alone (Fig. 6). This was not unexpected, as the overall negative charge of the NAG-PBAVE would inhibit electrostatic interactions with the negatively charged phosphate groups on the chol-siRNA. These results suggest that intracellular co-localization of NAG-PBAVE and chol-siRNA as observed by confocal microscopy is not a result of NAG-PBAVE/chol-siRNA complex formation during formulation.
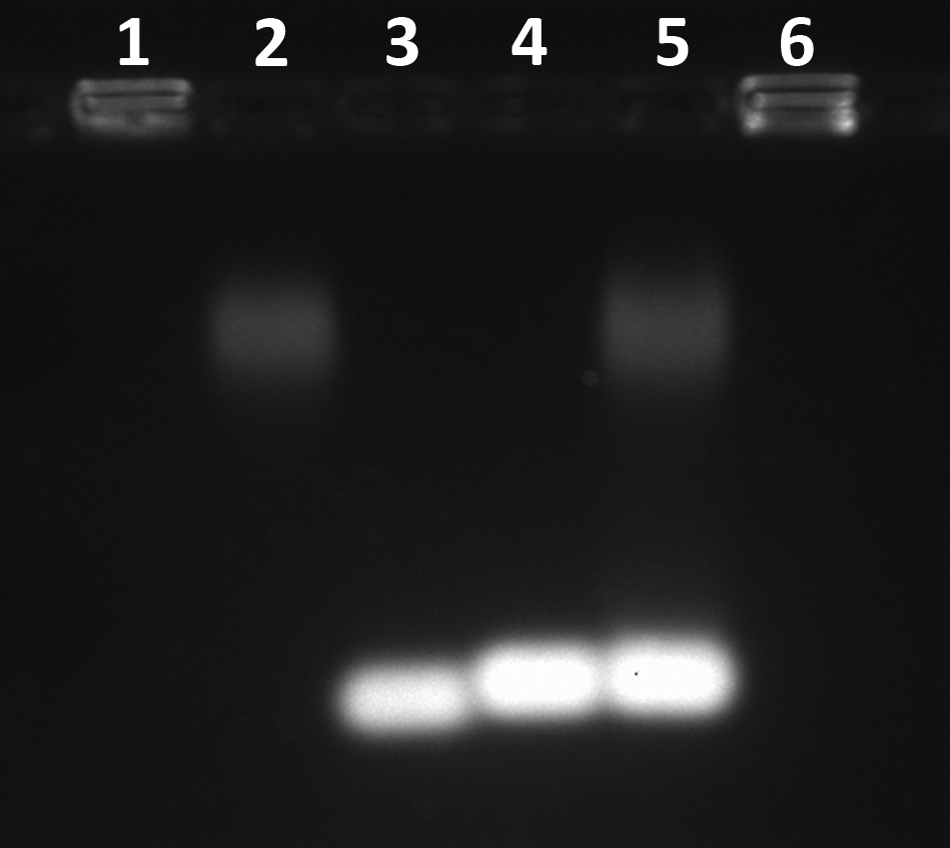
NAG-PBAVE and chol-siRNA do not interact in solution. Potential interaction between chol-siRNA and NAG-PBAVE was evaluated by a gel retardation assay. Samples were incubated at room temperature for 10 minutes prior to loading onto the gel. Lane 1, unmodified PBAVE; lane 2, NAG-PBAVE alone; lane 3, siRNA alone (no cholesterol); lane 4, chol-siRNA alone; lane 5, chol-siRNA+NAG-PBAVE; lane 6, chol-siRNA+unmodified PBAVE. PBAVE used in all samples was fluorescently tagged with Oregon Green. The gel was stained in gel running buffer containing ethidium bromide (0.5 μg/mL) to visualize the chol-siRNA and analyzed using a Kodak Gel Logic 200 Imaging System.
However, once the PBAVE is unmasked, as would be expected to occur in the endosome, the charge would become net positive and a complex with the chol-siRNA would be expected to form through electrostatic interactions. Indeed, mixing chol-siRNA with unmasked PBAVE prior to electrophoresis resulted in the formation of a complex that could not enter into the gel and remained in the well (Fig. 6, lane 6). It is possible that such a complex occurs in the endosome and may help to explain the increased stability of chol-siRNA in the presence of co-localized NAG-PBAVE in hepatocytes as visualized by confocal microscopy.
NAG-PBAVE and chol-siRNA can be injected separately without loss of gene knockdown activity
Data presented in the previous section indicated that NAG-PBAVE and chol-siRNA do not form complexes prior to injection. However, this experiment did not allow us to rule out the possibility that NAG-PBAVE and chol-siRNA might interact in the bloodstream or on the cell membrane just prior to cell entry. This could conceivably occur if NAG-PBAVE were to become partially unmasked prior to cell uptake and interact electrostatically with chol-siRNA. In order to test this, we performed separate injections of NAG-PBAVE and chol-sRNA and monitored gene knockdown activity. We reasoned that if gene knockdown activity required that NAG-PBAVE and chol-siRNA interact prior to cell uptake, injecting them individually and separated by time would lead to a significant diminution of activity. We have previously shown that >90% of NAG-PBAVE is cleared from the blood 1 hour after injection (Mudd et al., 2010). A large majority of the NAG-PBAVE present in the liver at this time point is internalized by hepatocytes as shown in Fig. 5. In the following experiment, we first injected the NAG-PBAVE alone followed by injection of chol-siApoB 1, 2, 4, or 6 hours later. Injection of the compounds in the reverse order, specifically, injection of chol-siApoB first followed by NAG-PBAVE, was also performed. We found that injection of either compound by up to 2 hours after injection of the first compound had only a minor effect on efficacy (Fig. 7). Most of the material injected would have been cleared from the bloodstream and taken up by cells at this time point, suggesting that complex formation prior to contact with hepatocytes is likely not required for activity. However, activity was completely abolished if either compound was injected 6 hours after injection of the first, indicating a finite time window exists between injections before activity is lost.
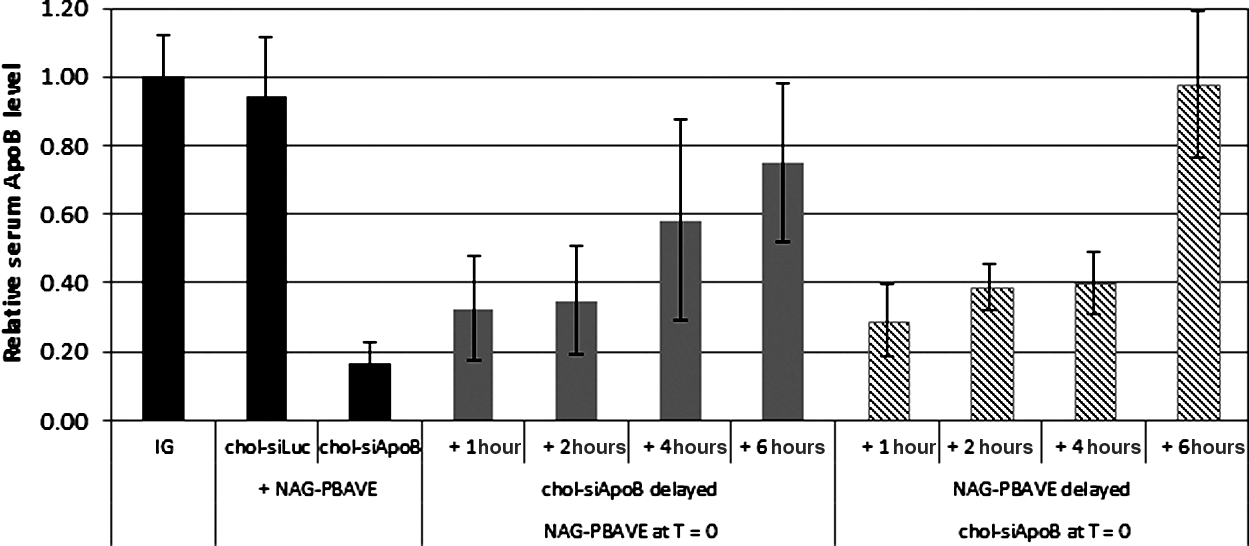
NAG-PBAVE and chol-siRNA can be injected separately. Relative serum ApoB protein levels in ICR mice 2 days after co-injection of control chol-siLuc (1 mg/kg ) or chol-siApoB (1 mg/kg ) with 20 mg/kg NAG-PBAVE (dark solid bars); 20 mg/kg NAG-PBAVE injected at time (T)=0, followed by injection of 1 mg/kg chol-siApoB at T=1, 2, 4, or 6 hours (light solid bars); 1 mg/kg chol-siApoB injected at T=0, followed by injection of 20 mg/kg NAG-PBAVE at T=1, 2, 4, or 6 hours (diagonal shaded bars). Serum ApoB levels were normalized to those measured in mice receiving IG only. Bars indicate group mean values; error bars indicate SD, n=5.
Chol-siRNA co-localizes with PBAVE in liver hepatocytes when injected separately
In order to determine if separately injected NAG-PBAVE and chol-siRNA could still co-localized to the same hepatocyte endosomes, confocal microscopy studies were performed. In these studies, Oregon Green-labeled NAG-PBAVE was injected first followed by injection of Dy547-labeled chol-siRNA 1 hour later. Liver samples were collected at 15 minutes or 2 hours after chol-siRNA injection and sections were prepared for microscopy. Remarkably, despite being injected 1 hour after NAG-PBAVE when more than 90% of the injected NAG-PBAVE has been cleared from the blood and is found within the liver almost entirely inside hepatocytes, 22% of the chol-siRNA signal co-localized with NAG-PBAVE 15 minutes after chol-siRNA injection (Table 1). Nearly all of the co-localization occurred in hepatocytes (Fig. 8A–C). This suggests that intracellular vesicles/endosomes containing chol-siRNA had rapidly fused with pre-existing endosomes in hepatocytes containing NAG-PBAVE. Interestingly, the percentage of chol-siRNA that co-localized with NAG-PBAVE was approximately 3 times greater than that which co-localized when it was co-injected with NAG-PBAVE (Table 1). One possible explanation for this observation is that there are a larger number of pre-existing, fusion competent endosomes containing NAG-PBAVE present 1 hour after injection than at 15 minutes after co-injection of NAG-PBAVE and chol-siRNA. If chol-siRNA and NAG-PBAVE are taken up independently by the hepatocyte, as our data suggest, then vesicles/endosomes containing chol-siRNA would have a greater opportunity to fuse with endosomes containing NAG-PBAVE simply because of their sheer number at this time point. This would result in a greater percentage of chol-siRNA signals that overlap with NAG-PBAVE signals.
Confocal micrographs of liver sections from mice receiving chol-siRNA and NAG-PBAVE injected 1 hour apart. Oregon Green-labeled NAG-PBAVE (10 mg/kg) was injected at T=0, followed by injection of Dy547-labeled chol-siF7 (2 mgkg−1) at T=1 hour. The liver was harvested 15 minutes
Although the absolute number of fusion competent endosomes containing NAG-PBAVE may be greater at 1 hour after injection than 15 minutes after injection, not all endosomes containing NAG-PBAVE may be fusion-competent at this stage. The data indicates that only 63% of the NAG-PBAVE signal co-localized with the chol-siRNA signal 15 minutes after chol-siRNA injection versus 99% when NAG-PBAVE and chol-siRNA were co-injected (Table 1). This may be because a portion of NAG-PBAVE containing endosomes exist as late endosomes/lysosomes at this time point. These would be expected to be less competent to fuse with endosomes containing chol-sRNA than early endosomes (Gruenberg et al., 1989; Luzio et al., 2009).
At 2 hours after chol-siRNA injection, the overall chol-siRNA signal in the liver was qualitatively lower than at 15 minutes, but the proportion of chol-siRNA signal that co-localized with NAG-PBAVE increased to 84% (Fig. 8D–F; Table 1). This increase in chol-siRNA co-localization and decrease in overall chol-siRNA signal over time is similar to that observed when chol-siRNA and NAG-PBAVE are co-injected. This may be at least partially explained by degradation and/or elimination of chol-siRNA that was not co-localized with NAG-PBAVE in hepatocytes, as described previously. Together, the confocal and knockdown data are consistent with the view that NAG-PBAVE and chol-siRNA can be taken up by hepatocytes independently, co-localize to the same endosome by vesicle fusion, and elicit gene knockdown via PBAVE-mediated endosomolysis and chol-siRNA release to the cytoplasm.
Discussion
In this study, we have shown that the in vivo efficacy of cholesterol conjugated-siRNA in hepatocytes can be dramatically improved by the co-injection of a targeted endosomolytic agent. This delivery strategy also enabled, for the first time to our knowledge, the demonstration of chol-siRNA-mediated gene knockdown in non-human primates. The endosomolytic agent utilized, PBAVE, was previously used in the DPC delivery system in which the siRNA is covalently attached (Rozema et al., 2007). By eliminating the siRNA attachment step and instead co-injecting NAG-PBAVE and chol-siRNA, constraints associated with conjugation chemistry and siRNA/polyplex formulation are removed. This greatly simplifies the formulation procedure and opens up the possibility to explore the use of alternative endosomolytic agents to effectively delivery siRNA to liver hepatocytes.
As in our original DPC formulation, the co-delivery strategy is dependent on the use of hepatocyte-targeted NAG-PBAVE. The lack of functional delivery in ASGPr knockout mice or in the absence of the NAG ligand on PBAVE confirmed that hepatocyte targeting of the PBAVE via the ASGPr is necessary. Receptor-mediated endosomal uptake of ligands including NAG by the ASGPr is well established, and it is likely that NAG-PBAVE is taken up into hepatocyte endosomes by the same mechanism.
We also showed that it is necessary that the siRNA is conjugated to cholesterol. Precisely how the chol-siRNA is and taken up by hepatocytes is not entirely known. Our observations that NAG-PBAVE and chol-siRNA do not form complexes when mixed together, and that target gene knockdown can occur even if the two are injected up to 2 hours after the first, makes it unlikely that uptake in the same endocytotic event is necessary for activity. This implies that the ASGPr required for uptake of NAG-PBAVE is not directly involved in chol-siRNA uptake. Other receptors/mechanisms for chol-siRNA uptake are likely responsible.
The mechanism of liver uptake of chol-siRNA injected alone has been examined by others (Wolfrum et al., 2007). In that study, chol-siRNA was shown to bind LDL and HDL, and liver uptake was reduced in mice lacking LDLr and in mice lacking the HDL scavenger receptor SR-B1. A more recent study using reconstituted HDL-like particles containing recombinant Apolipoprotein E and dimyristoylphosphatidylcholine as a delivery vehicle for chol-siApoB showed that gene silencing was somewhat less effective in LDLr –/– mice, suggesting LDLr may be partly involved in the uptake of these particles as well (Nakayama et al., 2012). In contrast, our data using LDLr –/– mice indicated that the LDLr expression is not required for target gene knockdown when chol-siRNA is co-delivered with NAG-PBAVE. Another candidate receptor for chol-siRNA uptake is the LDLr family member low density lipoprotein receptor-related protein 1 (LRP1). We found that gene silencing was not affected in LDLr and LRP1 double-knockout mice when co-injected with chol-siF7 and NAG-PBAVE (data not shown), suggesting that LRP1 is also not required for this delivery approach. We have not examined the role that HDL or the HDL receptor SR-B1 may play. Further investigation will be necessary to define the mechanism of functional chol-siRNA uptake by liver shown in our studies.
Regardless of how chol-siRNA is taken up by the cell, our confocal data indicate that chol-siRNA and NAG-PBAVE come to reside in the same endosome. This would be required for NAG-PBAVE-mediated endosomal escape of chol-siRNA from a conceptual standpoint and would account for the increased efficacy of the co-injection approach. One possibility is that NAG-PBAVE and chol-siRNA are taken up by separate endosomes which fuse once inside the cell. Endosomal fusion is a well-documented phenomenon (Gruenberg et al., 1989; Skjeldal et al., 2012). Although we cannot completely rule out the possibility that functional delivery was the result of a small number of endosomes in which chol-siRNA and NAG-PBAVE were taken up together, our data strongly suggest that endosomal fusion is a requirement for the relatively efficient delivery of chol-siRNA by NAG-PBAVE.
Previously it was shown that other lipophilic siRNA conjugates besides chol-siRNA can be utilized to elicit target gene knockdown in mouse liver, albeit at high doses (Wolfrum et al., 2007). The potency of these siRNAs depended on the length of the alkyl chain, with only long chain fatty acid conjugates showing gene knockdown activity. Our own preliminary experiments indicate that the co-delivery approach described here can also greatly improve the efficacy of these long chain fatty acid-siRNA conjugates (data not shown). In addition, we have found that siRNAs that are conjugated to non-lipophilic hepatocyte targeting ligands such as NAG are also effective for target gene knockdown in hepatocytes using the co-delivery approach (data not shown). This suggests that a wide variety of liver-tropic targeting ligands can be used. Given the range of ligands that could potentially be used on the polymer and siRNA, it will be of interest to determine if the co-injection approach can also be used to target non-liver tissues.
Chol-siRNA was first demonstrated to be effective in mice almost a decade ago. However, the low systemic efficacy of chol-siRNA alone hindered its use in large animals, including humans. By combining chol-siRNA with a reversibly masked endosomolytic targeted polymer in a simple co-injection approach, its use in clinical applications should be facilitated.
Footnotes
Acknowledgments
We thank the members of Axolabs GmbH (formerly Roche Kulmbach RNA Therapeutics) for siRNA synthesis and helpful discussions. We also thank the Arrowhead Madison Laboratory Animal Resources (Linda Goth, Sheryl Ferger Tracie Milarch, and Rachael Schmidt,) and Assay (Stephanie Bertin, Jacob Griffin, Jessica Montez, and Tom Reppen) Groups for their outstanding technical assistance. The authors are grateful to Dr. Christine Wooddell for critically reading the manuscript.
Author Disclosure Statement
No competing financing interests exist.