
Abstract
Catalytic oligonucleotides, known as DNAzymes, are a new class of nucleic acid-based gene therapy that have recently been used in preclinical animal studies to treat various cancers. In this study the systemic distribution, pharmacokinetics, and safety of intravenously administered anti-MMP (matrix metalloproteinase)-9 DNAzyme (AM9D) were determined in healthy FVB and in MMTV-polyoma virus middle T (PyMT) transgenic mice bearing mammary tumors. MMP-9 is known to be involved in tumor cell development, angiogenesis, invasion, and metastasis. Sulfur-35 (35S) labeled ([35S]-AM9D) administered intravenously, without the use of carrier molecules, to healthy and mammary tumor bearing MMTV-PyMT transgenic mice distributed to all major organs. The order of percentages of [35S]-AM9D accumulation in different organs of healthy and MMTV-PyMT mice were blood>liver>kidney>lung>spleen>heart and mammary tumor>blood≈liver>kidney>spleen>lung>heart, respectively. The amount of AM9D accumulated in mammary tumors 2 hours post injection was 0.6% and 0.2% higher than in either blood or liver, respectively, and its rate of initial clearance from mammary tissue was at least 50% slower than the other organs. Approximately 43% of the delivered dosage of [35S]-AM9D was cleared from the system via feces and urine over a period of 72 hours. No evidence of acute or chronic cytotoxicity, local or widespread, associated with AM9D treatment (up to 75 mg AM9D /kg of body weight) was observed in the organs examined. These data suggest that DNAzyme in general and AM9D in particular can be used systemically as a therapeutic agent to treat patients with breast cancer or other metastatic and surgically inaccessible tumors.
Introduction
N
We and other investigators have recently used DNAzyme molecules in cell-based assays and preclinical animal studies to treat various cancers. Qu and colleagues (Qu et al., 2008), used DNAzyme targeting Aurora kinase A mRNA to inhibit growth of human prostate cancer in vivo. Aurora kinase A upregulation is involved in the malignant progression of prostatic and many other types of cancers (Zhao et al., 2007; Qu et al., 2008). Zhang and colleagues (Zhang et al., 2002), showed that intratumoral treatment with DNAzyme targeting vascular endothelial growth factor receptor 2 (VEGF2) mRNA led to a 75% reduction in tumor size compared with saline treated control. In addition, treatment of MDA-MB-231 xenograft solid tumor model with an EGR-1 mRNA–specific DNAzyme (DzF) resulted in a profound sequence-specific inhibition of tumor growth compared to saline treated control (Mitchell et al., 2004). Further, DNAzyme targeting c-jun (DZ13), a prototypic member of the AP-1 transcription factor involved in cellular proliferation and cell death, is shown to inhibit growth of basal cell and squamous cell carcinomas when injected intratumorally in animal models of skin cancer (Cai et al., 2012). DZ13 inhibition of tumor growth, however, was partially due to the induction of tumor immunity (Cai et al., 2012). We have recently shown that down regulation of matrix metalloproteinase-9 (MMP-9) production with anti-MMP-9 DNAzyme (AM9D) in vivo leads to the inhibition of tumor growth and angiogenesis and induction of apoptosis in both animal models of glioma (Batson et al. unpublished data) and in a MMTV-polyoma virus middle T (PyMT) transgenic mouse model of breast cancer (Hallett et al., 2013). AM9D selectively cleaves MMP-9 mRNA without affecting the expression of other related MMP members (Hallett et al., 2013). MMPs are a family of enzymes that play a crucial role in both normal connective tissue remodeling and in the accelerated matrix breakdown and release of chemokines, cytokines, and other proteins associated with tumor cell invasion and metastasis (Egeblad and Werb, 2002). They are produced by stromal cells and macrophages near the invading tumor cells, and their activities are increased in malignant tumors (Duffy et al., 2008). High expression levels of a variety of MMPs, including MMP-9, have been reported to be associated with progression and poor prognosis of breast cancer (Egeblad and Werb, 2002; Duffy et al., 2008).
Although the aforementioned results are very supportive for the use of DNAzymes for cancer therapy, all of these preclinical studies of tumor growth were conducted via intratumoral injections. While intratumoral injections can illustrate the promise of DNAzymes relative to target selectivity and therapeutic potential, it is impracticable for clinical trials in humans. Thus, successful systemic application of DNAzymes in pathophysiological and therapeutic studies requires prior demonstration of significant uptake of DNAzyme by the target tissues.
In this study, we determined systemic distribution and cytotoxicity of AM9D in healthy mice and in the MMTV-PyMT transgenic mouse model of breast cancer to establish the efficacy of DNAzymes as a preventive and/or treatment regimen for many types of cancer. We have found that naked AM9D is safe when administered systemically and distributes to all major organs of healthy and MMTV-PyMT transgenic mice.
Materials and Methods
Synthesis of 35S-radiolabeled oligonucleotides
The catalytic DNAzymes used in these experiments were synthesised and purified by Integrated DNA Technology. The sequence of the DNAzyme targeting MMP-9 mRNA (AM9D) is, 5′-GTGGTGCCAGGCTAGCTACAACGATTGAGGTCG-3′. In the control DNAzyme (CD), 5′-CTAGTCAGCGGCTAGCTACAACGATAAGCTGCT-3′, the catalytic sequence of DNAzyme is flanked by 9 bases randomly chosen and not specific for any MMP coding sequence. AM9D was labeled at the 5′ end using ATPγ- 35S (Perkin Elmer) and T4 polynucleotide kinase (Promega). The 35S labeled AM9D ([35S]-AM9D) was then purified using a centri-10 column (Applied Biosystems). [35S]-AM9D was dissolved in sterile phosphate buffered saline (PBS). The concentration of the solution was adjusted so that 100 μL administered to 20g mice resulted in a dose of 10 mg/kg of body weight (specific activity, 0.275 mCi/kg). [35S]-AM9D was mixed with unlabeled molecules to obtain the desired concentration.
Animal experiments
All animal studies were conducted in accordance with guidelines established by the Animal Care and Use Committee at The University of Tennessee Health Science Center. Twenty-three 8-week-old female FVB mice weighing 20–25 g and twenty-two MMTV-PyMT transgenic mice (FVB strain) bearing mammary tumors of various sizes and weighing 17–22 g were used in this study. MMTV-PyMT transgenic mice develop tumors in 6–8 weeks of age. Mice bearing a range of early (<500 mm3) and late stage (>750 mm3) tumors were used.
Systemic distribution
Normal mice
[35S]-AM9D (100μL; 10 mg/kg; 0.275 mCi/kg) in PBS was administered to 23 healthy momentarily anesthetized mice via tail vein injection. Three animals receiving [35S]-AM9D were placed in metabolism cages, immediately following injection, for 7 days (168 hours). The urine and feces were collected daily for total radioactivity determination from each of the 3 animals. All samples were collected and stored at −80°C until analysis. The remaining 20 mice were divided into 5 groups of 4 animals each, and at 2, 6, 24, 48, and 72 hours following intravenous administration of [35S]-AM9D, one group of animals was euthanized and liver, heart, lung, kidney, spleen, intestines, brain, pancreas, and stomach were removed for determination of total radioactivity. Immediately prior to sacrifice, blood samples (0.5–1.0 mL) were collected from anesthetized mice via the retro-orbital vein.
MMTV-PyMT mice
DNAzyme was administered, as above, to 22 tumor bearing mice, and these were divided into 5 groups of 3–4 animals each and one group of 5 animals. At 2, 4, 8, 24, 48, and 72 hours following [35S]-AM9D administration, one group of animals was sacrificed. Tumor volumes were measured using calipers during the experiments, and after excision, wet weights were recorded. Tumors exceeding 1.37 g were highly heterogeneous upon gross visualization (i.e., necrotic areas and mixture of tumor consistency) and thus were not included in any statistical analysis of mammary tumors. Heart, lung, kidney, liver, spleen, and blood samples were removed for determination of total radioactivity.
Radioactive sample analysis
Organs were solubilized in at least 1 mL SOLVABLE™ according to manufacturer's instruction (Perkin Elmer) and incubated overnight at 50°C. Endogenous peroxidase activity was quenched with 10 mM EDTA and 6% hydrogen peroxide for 1 hour at 50°C. Blood samples were collected from each mouse and 200 μL of each sample was incubated with 1 mL SOLVABLE at 50°C for 1 hour. The endogenous peroxidase activity in blood was quenched with EDTA and hydrogen peroxide, as above. Approximately 10 mL of scintillation cocktail fluid was added to each sample and tissue radioactivity was measured using a Packard Tri-Carb 2900CA liquid scintillation analyzer. Counts per minute obtained from each sample were converted to microgram of AM9D equivalents and presented as the total micrograms of AM9D per either weight or volume of tissue, blood, urine, or fecal output. Initial and final rates of clearance were calculated based on the slope of the line obtained from plotting μg AM9D per gram of tissue or per mL of blood as a function of time (initial: 2–6 hours for normal mice and 2–8 hours for MMTV-PyMT mice, final: 24–72 hours). All data were expressed as mean±standard deviation of mean. The quenching effect of tissue on radioactive count was tested by adding appropriate amounts of [35S]-AM9D to scintillation vials containing solubilized normal organs. No tissue quenching effect was observed.
Cytotoxicity
Seven groups of 5 healthy mice were intravenously injected with PBS or various concentrations (10 mg/kg, 32 mg/kg, and 75 mg/kg of body weight) of either AM9D or CD and were sacrificed 7 days later. Following dosing, animals were observed daily for signs of toxicity including trouble grooming, lack of food consumption, and any other signs of lethargy; none were observed. At necropsy, liver, heart, lung, kidney, and spleen were collected and fixed in 4% paraformaldehyde, impregnated with 25% sucrose for cryoprotection, and were processed for histopathological evaluation (standard light microscopic examination of hematoxylin and eosin-stained tissue slides) and immunofluorescence studies. To further determine chronic toxicity of AM9D, mice were injected with 75 mg/kg of body weight (1500 μg/injection) of either AM9D or CD once a week for a total of 10 weeks. Mice were closely observed, sacrificed 7 days after the last injection, and organs collected as described above.
Immunogenesity and the effect of AM9D on innate immune system was determined by measuring the serum levels of anti-DNA antibody [immunoglobin G (IgG) and A (IgA)] and the inflammatory cytokines [interleukin (IL)-5, tumor necrosis factor (TNF)-α, monocyte chemotactic protein (MCP)-1, keratinocyte-derived chemokine (KC), interferon-inducible protein (IP)-10, IL-15, IL-6, RANTES (regulated on activation, normal T cell expressed and secreted), and interferon (IFN)γ] 4 hours after treating the animals with PBS or AM9D, respectively using MILLIPLEX MAP Mouse Cytokine/Chemokine immunoassay kit, as described by the manufacturer (Millipore).
In vitro and in vivo stability of AM9D
The in vitro stability of AM9D was determined by incubating 1 μg of [35S]-AM9D in freshly prepared mouse serum at 37°C. An equal amount was removed at various time points (1, 2, 4, 6, and 23 hours), loaded on 8% urea acrylamide gel, and visualized by autoradiograph of the gel. The stability of AM9D molecule in vivo was determined by intravenously administering [35S]-AM9D into MMTV-PyMT tumor bearing mice as stated above. The animals were euthanized 2 or 4 hours post [35S]-AM9D administration and mammary tumors were excised and stored at −80°C. Tumor tissues (100 mg) were homogenized and [35S]-AM9D was then isolated from the tissue homogenate and blood serum of the treated animals by Master Pure™ (Epicenter) following the manufacture protocol. The isolated DNA was subjected to 8% urea-polyacrylamide gel electrophoresis and visualized by ethidium bromide staining and the associated radioactivity was detected by exposing the gel to x-ray film.
In situ hybridization
The intact AM9D molecules in mammary tumor slices was detected by in situ hybridization using two independent oligomeric probes, 5′-AGCTAGCCTGGCACCACCAC-3′ and 5′-CGACCTCAATCGTTGT-3′, complementary to 5′ and 3′ ends of the AM9D respectively. The tumor slices were incubated with 3 μg of each 5′ end fluorescently labeled probes in hybridization buffer consisting of 25% formamide, 2×saline-sodium citrate (SSC), 0.5 mg/mL salmon sperm DNA, 1×Denhart's solution, 100 mM DTT, and 10% dextran sulfate, overnight at 37°C. The slides were then washed twice with 2×SSC, 0.1% sodium dodecyl sulfate for 5 minutes at room temperature followed by two washes with 2×SSC, 25% formamide for 20 minutes at 45°C, dried at room temperature, cover slipped, and visualized by fluorescence microscopy.
Results
Pharmacokinetics of DNAzyme in normal mice
The systemic distribution of DNAzyme in mice and its rate of clearance were determined by injecting naked [35S]-AM9D (10 mg/kg) in PBS via the tail vein into healthy FVB mice, and the amount of radioactivity present in blood and different organs was measured as a function of time. AM9D injected in this manner distributed to all major organs; liver, heart, spleen, kidney, and lungs (Fig. 1A). Two hours after AM9D injection, approximately 10% of the total DNAzyme was found in the major organs (Table 1). The order of percentage accumulation was: blood>liver>kidney>lung>spleen>heart. Blood contained a majority of AM9D, with 6.3% of the total DNAzyme in the first 2 hours; this declined to 2.1% after 6 hours (Table 1). Studies revealed 1.8%, 1.1%, and 0.4% of total AM9D injected was delivered to liver, kidney, and lung, respectively. Finally, spleen and heart each contained 0.2% of total AM9D in 2 hours post injection. The majority of the accumulated AM9D was cleared from these organs within the first 24 hours. Thirty-one percent of the delivered dosage of [35S]-AM9D was cleared from the system via feces and urine over a period of 72 hours.
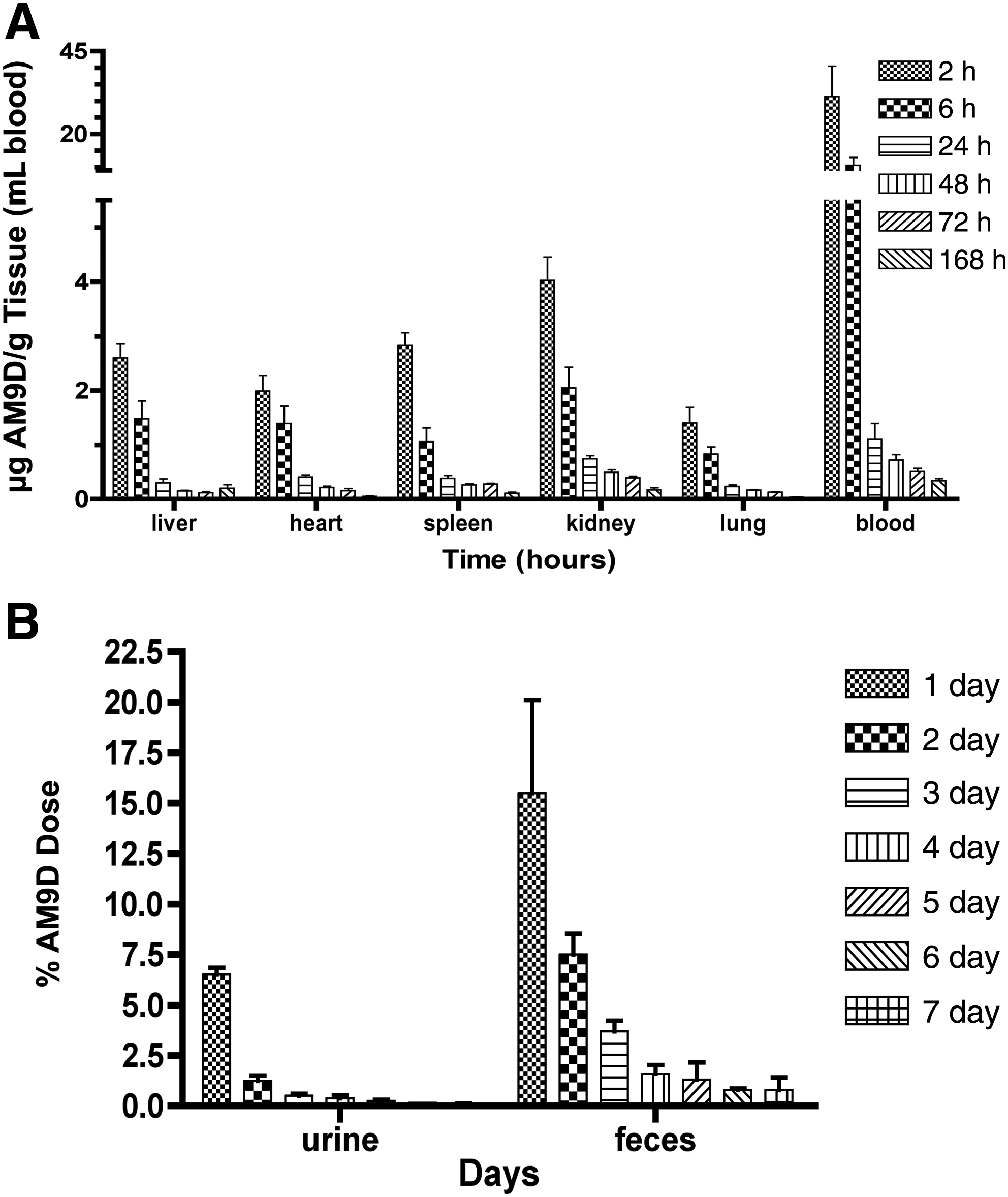
Numbers in parentheses give the range of data.
AM9D, anti-MMP (matrix metalloproteinase)-9 DNAzyme.
The rate of initial clearance (from 2 to 6 hours) and final clearance (from 24 to 72 hours) of AM9D from different organs were calculated based on the amount of AM9D remaining in tissues as a function of time (Table 2). The rate of initial clearance of AM9D from blood was faster than kidney, followed by spleen>liver>heart≥lung (Table 2). The rates of initial and final clearance of AM9D from kidney were 0.495 μg AM9D/g of tissue/hour and 0.0074 μg AM9D/g of tissue/hour, respectively. The rate of initial clearance of AM9D from heart and lung are equivalent; however, AM9D was retained in lung longer as its rate of final clearance in lung is approximately two-fold slower than that of the heart (Table 2).
Initial clearance is between 2 and 6 hours.
Final clearance is between 24 and 72 hours.
Fecal excretion represented the major pathway of AM9D elimination (Fig. 1B). The amount of AM9D excreted in feces (32%) was 3-fold greater than the amount released from urine (11%) over the 7-day period. As expected, AM9D excretion was time-dependent with the majority being eliminated in the first 72 hours.
Pharmacokinetics of AM9D in mammary tumor bearing mice
Female MMTV-PyMT transgenic mice bearing early to late stage mammary tumors were used to test the distribution of AM9D to mammary tumors. Naked [35S]-AM9D in PBS was administered to tumor bearing animals via tail vein injection. The animals were then euthanized at different times and the amount of [35S]-AM9D present in different organs, blood, and mammary tumors was determined as a function of time (Fig. 2A, B). The order of percentages of [35S]-AM9D accumulation in different organs of MMTV-PyMT mice was mammary tumor>liver≈blood>kidney>spleen>lung>heart. [35S]-AM9D concentration in the mammary glands was markedly higher than in other tissues. In these mice, 4.1% of the total injected AM9D was accumulated in the mammary tumors in 2 hours (Table 3). Mammary tumors of transgenic mice were of varying sizes, and the amount of AM9D accumulated in mammary tumors was proportional to the weight of the tumor burden (Fig. 3). Interestingly, the accumulated AM9D in the mammary tumor is initially cleared at a slower rate compared with the other organs, with a rate of initial clearance (2–8 hours post AM9D injection) of 0.068 μg AM9D per gram of tissue per hour (Table 4). The mammary tumors characterized as small tumors (<0.7 g) continue accumulating AM9D 2–4 hours post injection (Fig. 2B). This corresponds with the initial clearance of AM9D from blood 2–4 hours post DNAzyme injection (Fig. 2A). Similar to AM9D distribution in normal mice, the percentages of the total DNAzyme accumulated in blood, liver, kidney, lung, spleen, and heart, 2 hours post AM9D injection, was 3.5%, 3.9%, 1.3%, 0.4%, 0.5%, and 0.3%, respectively (Table 3). However, the rate of initial clearance of AM9D from spleen was slower than blood but faster than any other organs tested followed by liver≈kidney>heart>lung>mammary tumors (Table 4). The majority of AM9D was eliminated from the organs and blood within the first 24 hours (Fig. 2B). The rate of initial clearance from liver and kidney was found to be the same (4.3μg AM9D/g tissue/h) during the first 8 hours of administration and slightly increased in kidney between 24 and 72 hours. In heart, the rate of clearance in 2–8 hours (initial) and 24–72 hours (final) post administration was 2 times and 3.4 times faster, respectively, than that in lung (Table 4).
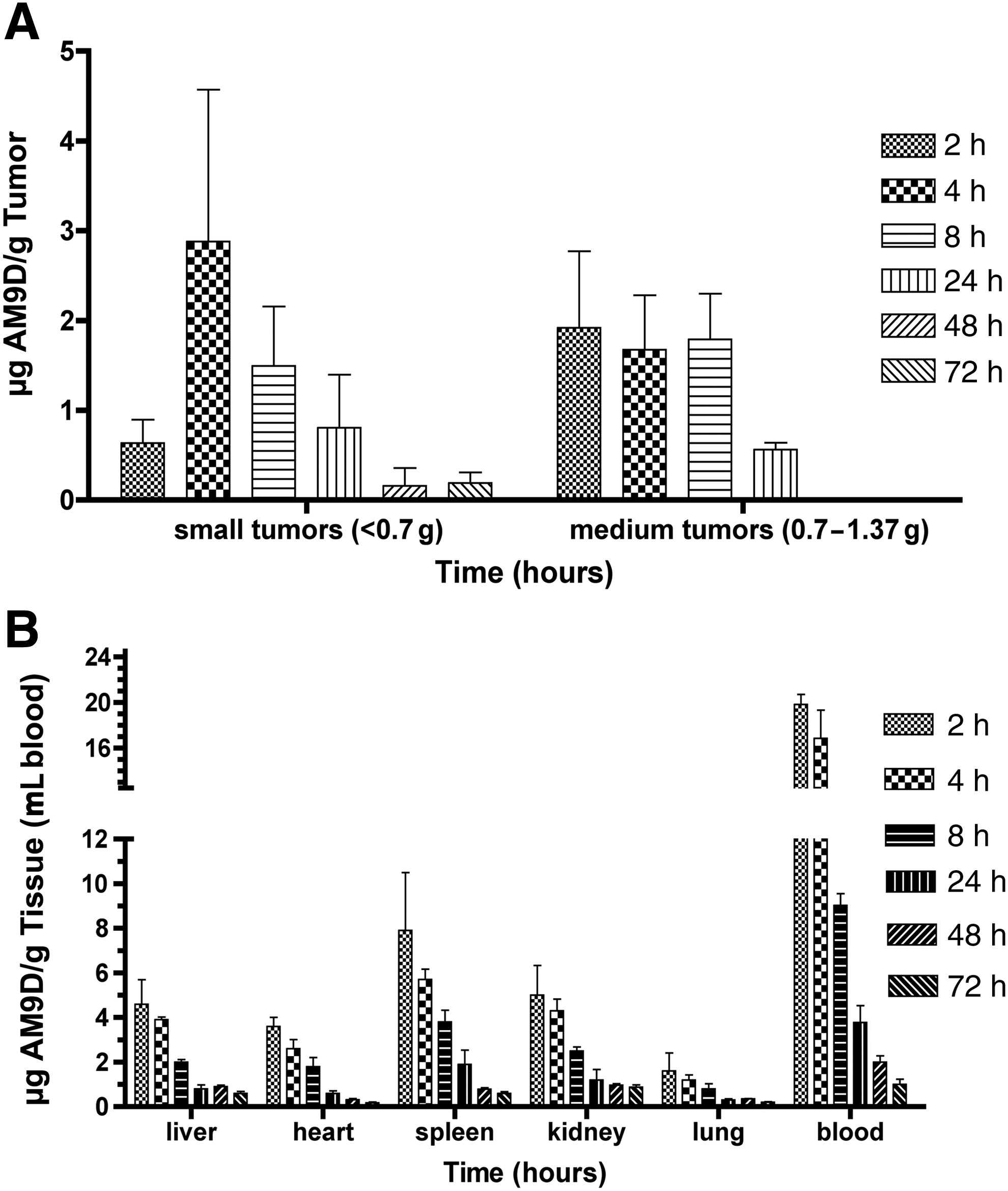
Distribution profile for [35S]-AM9D in MMTV-PyMT mice.
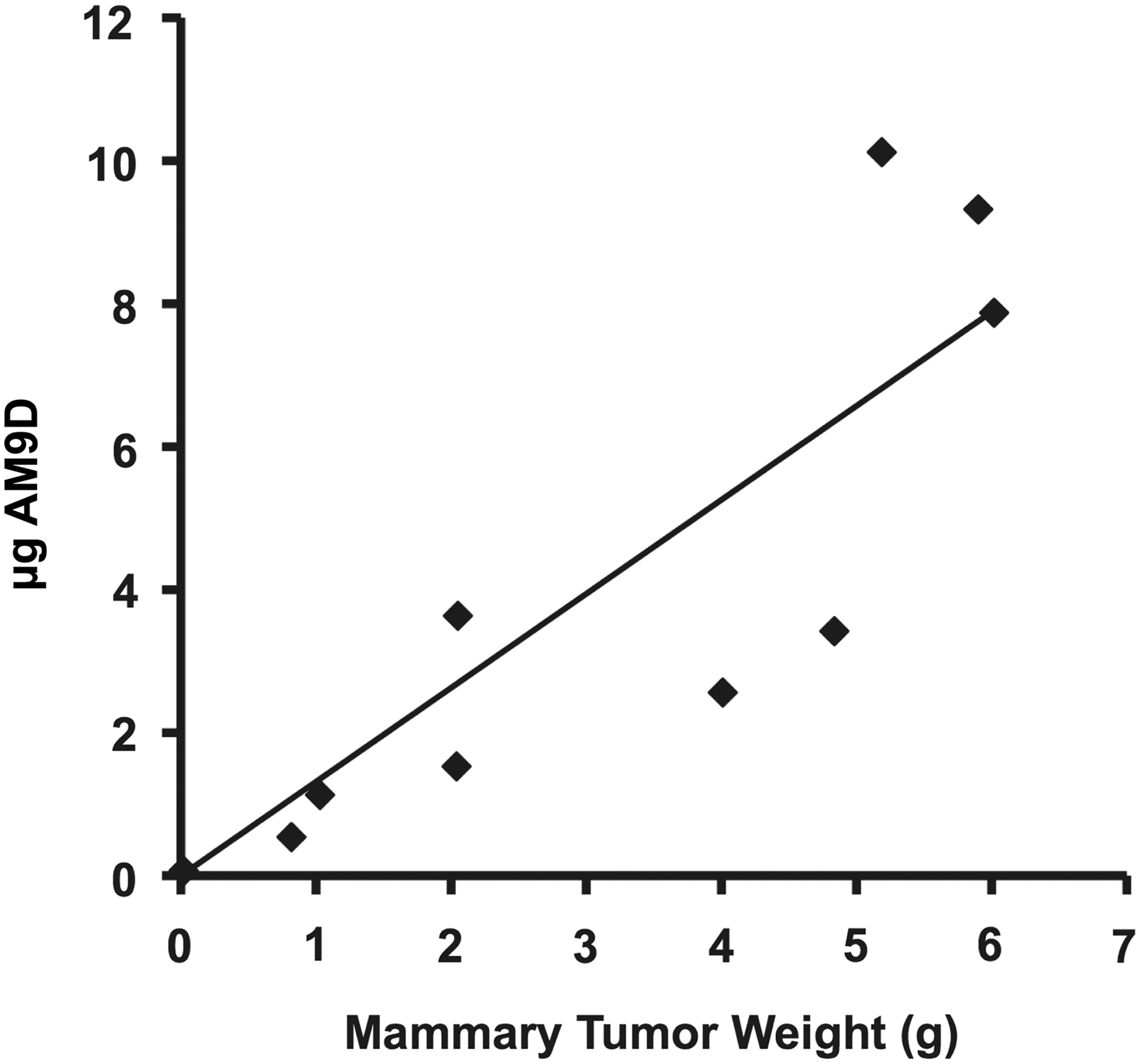
Distribution of [35S]-AM9D in MMTV-PyMT mammary tumors. Total tumor burden was measured in grams and the total μg AM9D found in tumors were calculated. AM9D injected into mice bearing tumors of varying sizes show a proportional correlation between tumor size (all tumors combined per mouse) and AM9D distribution (Y=1.4045x – 0.4159, R2=0.7764).
Numbers in parentheses give the range of data.
Tumors larger than 1.37 g were excluded from calculations.
Initial clearance is between 2 and 8 hours.
Final clearance is between 24 and 72 hours.
Tumors larger than 1.37 g were excluded from calculations.
These results indicated that DNAzyme oligonucleotides could be delivered intravenously to all organs without the use of lipids or other carrier molecules, as naked AM9D was injected, and that the [35S]-AM9D was present in various organs including mammary tumor tissues and organs of high incidence for metastasis (i.e., lung and liver). In addition, mammary tumors appear to take up AM9D faster and retain it longer than any other organs.
Stability of AM9D in serum and tumor tissues
The stability of [35S]-AM9D molecules in serum and in mammary tumor tissue was demonstrated by (1) in vitro incubation of [35S]-AM9D at 37°C in the presence of serum for 23 hours, (2) isolation of [35S]-AM9D DNA from serum and mammary tumors of MMTV-PyMT mice intravenously treated with [35S]-AM9D, and (3) in situ hybridization of tumor tissue. An autoradiogram of [35S]-AM9D incubated with mouse serum showed no detachment of 35S from the DNAzyme over time and that about 70% of AM9D molecule was intact after 23 hours at 37°C (Fig. 4A). This was confirmed by demonstrating that [35S]-AM9D isolated from intravenously injected MMTV-PyMT mouse serum, and resected tumors were intact as shown by autoradiograph (Fig. 4B). In situ hybridization further demonstrated that the majority of the naked AM9D present in the tumor tissues are intact molecules as the independent 5′ and 3′ probes complementary to AM9D are colocalized within mammary tumor tissue (Fig. 4C). These data clearly show that AM9D molecule is stable in vivo post intravenous injection for at least 23 hours and verify that the detected radioactivity (35S) in tissues indeed corresponds to intact [35S]-AM9D molecule. This is consistent with our previous observation showing that AM9D is stable for at least 72 hours in MDA-MB-231 cells and for 10 days post single intratumoral injection in mammary tumors of MMTV-PyMT transgenic mice (Hallett et al., 2013). These results are also consistent with Cai et al.'s published data demonstrating that DNAzyme targeting c-jun (33P-DZ13) is stable for up to 120 hours in serum at 37°C, and 33P labeled Dz13 was retained in serum for at least 24 hours after a single intravenous administration in Sprague-Dawley rats (Cai et al., 2012). Cai and coworkers were also able to isolate intact 33P-Dz13 from tumors 1 hour after intratumoral 33P-Dz13 delivery in T79 SCC tumor-bearing SCID mice (Cai et al., 2012). In our hand, modification of AM9D with an inverted T at the 3′ end (Santoro and Joyce, 1998) did not result in improved inhibition of MMP-9 or stability of AM9D in vitro or in vivo when compared with the unmodified version (data not shown). Together, these data suggest that naked AM9D can be useful as a therapeutic agent to treat human patients with breast cancer or other solid tumors in which targeting MMP-9 has been shown to be antitumorigenic.
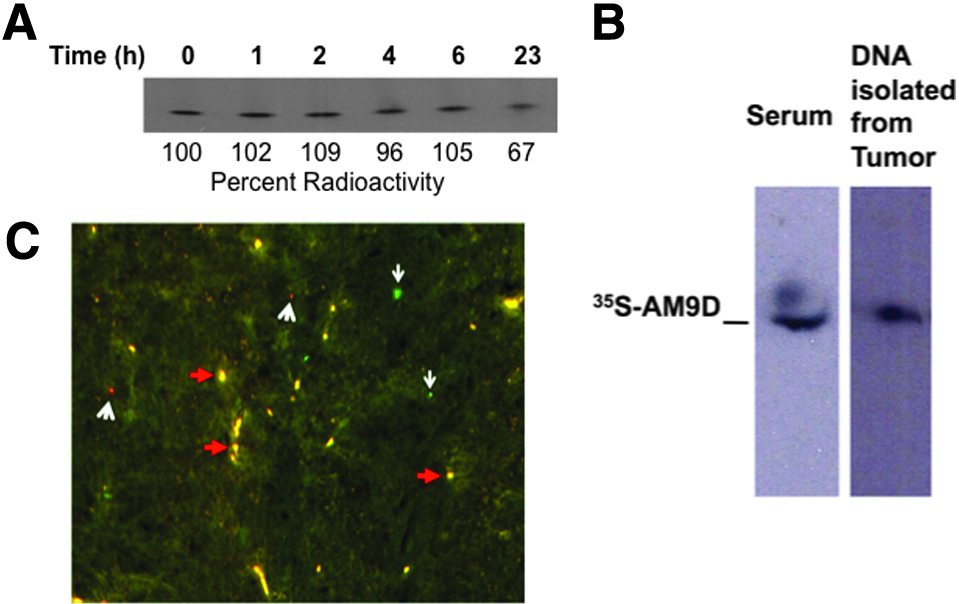
Stability of AM9D in serum and in mammary tumors.
Acute and chronic tissue cytotoxicity of intravenous, bolus administration of AM9D in healthy mice
The possibility of acute tissue toxicity arising from intravenous AM9D delivery was examined in healthy FVB mice following a single injection of escalating concentrations of AM9D or CD (10 mg/kg, 32 mg/kg, and 75 mg/kg of body weight) in PBS. PBS was used as a control. Seven days post AM9D injection, mice were sacrificed, and liver, spleen, lung, heart, and kidney were isolated and analyzed for cytotoxicity using hematoxylin and eosin stain (H&E) (Fig. 5A). All tissues were evaluated by a pathologist, blindly, for evidence of capillary leakage, acute cellular damage including necrosis or apoptosis, acute inflammation and mitotic activity. No evidence of acute cytotoxicity, mitogenesis, or cell death, local or widespread, was observed in the organs examined.
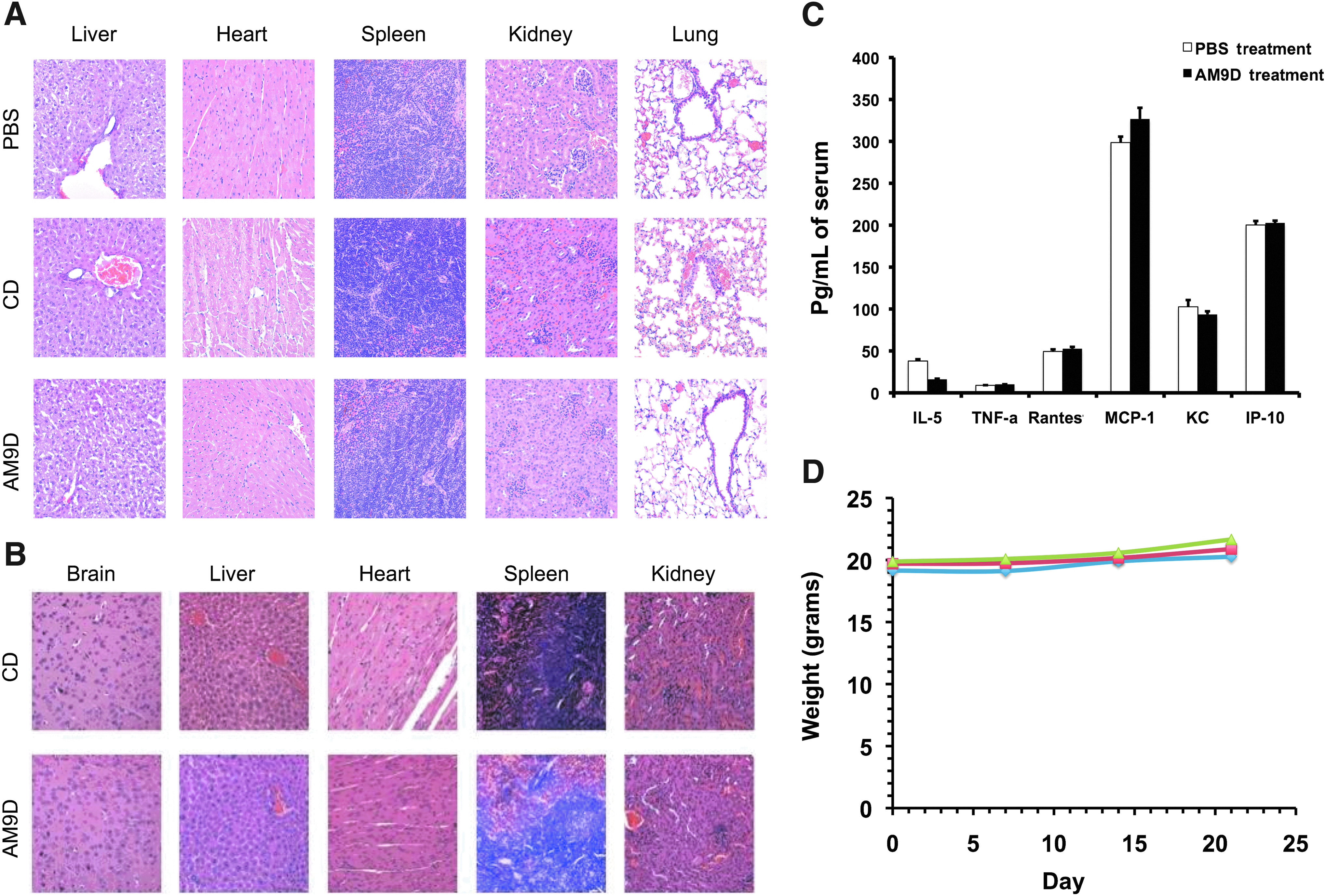
Acute and chronic toxicity associated with AM9D treatment.
Chronic cytotoxicity of AM9D was determined in healthy FVB mice that received 10 weekly intravenous injections of 75 mg/kg AM9D or CD; PBS was used as a control. Seven days following the final DNAzyme injection (11 weeks total exposure), mice were sacrificed and all major organs were isolated. Pathological evaluation of H&E stain of tissue slices revealed that DNAzyme is safe, and no evidence of capillary leakage, cellular damage including necrosis or apoptosis, inflammation, or mitotic activity was observed (Fig. 5B). Oligonucleotide-based molecules have been shown to have off-target effects and induce inflammation (KRIEG, 2004). To determine if AM9D is immunogenic and would induce inflammatory responses, the level of anti-DNA antibody (IgG and IgA) and the inflammatory cytokines and chemokines (IL-5, TNF-α, MCP-1, RANTES, KC, IP-10, IL-15, IL-6, and IFNα) in the serum of PBS and AM9D treated animals were measured. We did not detect by ELISA anti-DNA antibody, IL-6, IL-15, or IFNα, which would be an indication of a Toll-like receptor 9 (TLR9) response (Greten et al., 2004; Rakoff-Nahoum et al., 2004; Hagemann et al., 2007; Sasai et al., 2010), in serum of MMTV-PyMT mice intravenously treated with AM9D or PBS. Also, no significant difference was observed between the serum levels of inflammatory cytokines or chemokines in PBS and AM9D treated animals (Fig. 5C). Further, careful pathological evaluation of organs of animals treated with AM9D or CD revealed no macrophages and dendritic cells upon chronic exposure to DNAzyme (Fig. 5A, B). These findings therefore suggest that naked AM9D is not immunogenic and is safe for intravenous administration.
Discussion
The present study shows, for the first time, that anti-MMP-9 DNAzyme (AM9D) administered intravenously without the use of lipids or other carrier molecules to healthy or tumor-bearing MMTV-PyMT transgenic mice is safe and distributes to all tissues, including brain and mammary glands and is cleared from the system in a time dependent manner.
Recently, RNA cleaving DNA enzyme (DNAzymes) have emerged as novel, highly selective inhibitors of gene expression and reports of studies demonstrating the efficacy of DNAzyme based cancer therapies are beginning to emerge (Dass et al., 2008; Yang et al., 2010; Cai et al., 2012; Cho et al., 2013). Cho and colleagues (Cho et al., 2013) have recently, in a phase 1 clinical trial, demonstrated that DNAzyme targeting c-jun (DZ13) was safe and well tolerated after single intratumoral injections at all doses. DZ13 had previously shown to suppress basal cell and squamous cell carcinomas growth in both immunodeficient and immunocompetent syngeneic mice and reduce lung nodule formation in a model of metastasis (Cai et al., 2012). Toxicological studies in cynomolgus monkeys, minipigs, and rodents demonstrated that DZ13 is safe and well tolerated. Although no study results have been posted, Lun-Quan Sun and coworkers have completed a phase 1/2 clinical study where EBV-LMP1 targeted DNAzyme in combination with radiation therapy has been used to treat nasopharyngeal carcinoma (clinicaltrials.gov identifier: NCT01449942). However, thus far, all of the preclinical and clinical studies testing the effect of DNAzyme on tumor growth were via intratumoral or subcutaneous injections. In spite of the fact that tumor growth is an important factor in cancer progression, metastasis is ultimately the determining factor for cancer survival for many types of cancers (Vasselli et al., 2003; Berger et al., 2005; Phadke et al., 2008). Thus, while intratumoral injections can show promise as to the DNAzymes specificity to its target and future therapeutic capabilities, it is not widely applicable in humans. To advance the therapeutic potential of DNAzyme toward clinical trial, it is necessary to design an anti-metastatic DNAzyme and demonstrate its anti-metastatic potential based on its systemic distribution, safety, and efficacy.
Expression of MMP-9 has been shown in breast tumors isolated from patients (Jones et al., 1999) and is one of the most important factors for metastatic behavior of tumor cells (Gavrilovic et al., 1990; Duffy et al., 2000; Noel et al., 2008). MMP-9 degrades type 4 collagen, one of the most abundant collagens in the extracellular matrix(Birkedal-Hansen et al., 1993; Pourmotabbed et al., 1994), and is involved in cell proliferation, angiogenesis, and apoptosis (Bergers et al., 2000; Egeblad and Werb, 2002; RAO, 2003). Increasing evidence suggests that MMPs contribute to the formation of a microenvironment that promotes tumor growth during early stages of tumorigenesis. Our previous observations that AM9D treatment decreased MMP-9 production and reduced invasive behavior of human MDA-MB-231 (Hallett et al., 2013) and glioma cells (Batson et al., unpublished data) suggest that AM9D has a great potential as an antitumorigenic/antimetastatic agent. Thus, to utilize AM9D as a therapeutic agent in breast cancer preclinical and clinical studies, healthy and MMTV-PyMT transgenic mice bearing early to late stage tumors were used to test the distribution of AM9D to different organs and mammary tumors. The order of percentage accumulation of AM9D in healthy mice in different organs was blood>liver>kidney>lung>spleen>heart. In addition to these organs, [35S]-AM9D was also distributed to brain, intestines, stomach, pancreas, and skeletal muscle of MMTV-PyMT mice (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/nat), suggesting its potential as a therapeutic adjuvant for treatment of metastatic tumors.
Approximately, 43% of the delivered dosage of [35S]-AM9D was cleared from the system via feces and urine over a period of 72 hours (Fig. 1B). This data is consistent with Agrawal et al.'s observation that 40% of intravenously administered antisense phosphorothioate oligonucleotides complementary to HIV RNA in mice, is excreted in urine and 16% in feces within the first 48 hours (Agrawal et al., 1991). Contrary to these data, however, the unmodified AM9D oligonucleotides were observed to have a higher excretion of DNAzyme in feces than in urine (Fig. 1B).
Interestingly, when [35S]-AM9D was administered to MMTV-PyMT transgenic mice, the amount of DNAzyme accumulated in mammary tumors 2 hours post injection was 0.6% and 0.2% higher than in blood or liver, respectively, and its rate of initial clearance from mammary tissue was slower than from the other organs (Tables 3 and 4). The amount of AM9D accumulated in mammary tumors was proportional to the tumor burden (Fig. 3), suggesting a favorable therapeutic outcome. This is expected considering that mammary tumors can be highly vascularized (Lau et al., 1999), and tumors have been reported to increase nucleic acid and metabolites uptake. According to the results obtained from the intratumoral treatment (Hallett et al., 2013), 1 mg/kg of AM9D intratumorally is sufficient to significantly inhibit the expression of MMP-9, decrease the rate of tumor growth, and reduce the size of the mammary tumors spontaneously formed in MMTV-PyMT transgenic mice by 50% (Hallett et al., 2013). Therefore, considering the stability and biodistribution profile of intravenously administered AM9D (Fig. 1), 25 mg/kg intravenous delivery of AM9D should be sufficient to significantly reduce the tumor burden in an animal model of breast cancer.
Systemic administration of the naked AM9D is safe and at 75 mg/kg of body weight caused no observable mortality or organ toxicity in mice (Fig. 5). The safety and tolerability of AM9D was tested by several criteria. AM9D did not adversely affect body weight (Fig. 5 D), it did not stimulate immune system response and/or induce inflammation (Fig. 5C), and finally, blinded pathological studies did not reveal any evidence of acute or chronic toxicity, mitogenesis, or cell death, local or widespread, in any organs examined (Fig. 5A, B), contrary to those described for other oligonucleotides (Dass and Choong, 2010). Several investigators have reported hepatocyte toxicity and necrosis (Agrawal et al., 1991; Dass and Choong, 2010) and renal tubular structural perturbations (Agrawal et al., 1991) with oligonucleotide administration as low as 20 mg/kg (Dass and Choong, 2010) and as high as 30 mg/kg/day over 14 days (total 420 mg/kg) (Agrawal et al., 1991). This. however, was not observed in this study possibly due to the high specificity of AM9D toward MMP-9 mRNA (Hallett et al., 2013) and lack of functional CpG motifs in AM9D. CpG motifs have been shown to activate TLR9 and lead to stimulation of the immune system (Chuang et al., 2002; KRIEG, 2006) and upregulation of TNF-α, and IL-6 (Greten et al., 2004; Rakoff-Nahoum et al., 2004; Hagemann et al., 2007). Although AM9D contains two CpG regions, one in the catalytic domain and one in the binding arm, it did not stimulate TLR-9 as no TNF-α or IL-6 was detected in serum of AM9D treated animals by ELISA. Our data is consistent with recent studies reported by Dicke and colleagues demonstrating that the DNAzyme targeting GATA-3, a transcription factor crucial for the development of allergic immune reactions, containing two CpG motifs did not activate TLR9 or neutrophils in primary innate immune cells and did not stimulate cytokine production (IL-6, IL-12p40, and TNF-α) by macrophages (Dicke et al., 2012). Similar to these findings, careful observation of major organs of animals exposed to AM9D or CD for 10 weeks revealed no macrophages or dendritic cells (Fig. 5A, B). These studies suggest the suitability of AM9D as an adjuvant drug candidate for treatment of solid metastatic tumors.
In conclusion, systemic distribution and safety/toxicity studies revealed that naked AM9D is taken up by the cells in vivo without a carrier, is nontoxic when systemically administered and has favorable pharmacological properties with respect to intravenous administration and tissue distribution. Most importantly, mammary tumors can take up the naked AM9D. The amount of AM9D accumulated in the mammary tumor is higher and its rate of clearance is slower than any other organs tested. Although cells equally take up both AM9D and CD, only AM9D has anti-tumor capabilities (Hallett et al., 2013). These factors may permit the development of AM9D as a cancer therapeutic agent with much improved toxicity profiles as compared with conventional drugs.
Footnotes
Acknowledgments
The authors wish to thank Drs. Tiffany Seagroves, Edward Rosloniec, and Yasser Abdelrahman from University of Tennessee Health Science Center for generously providing the MMTV-PyMT mice to begin our colony, for performing cytokine assay, and for providing technical assistance, respectively. We also thank Drs. Yi Lu, R. K. Rao, and Tiffany Seagroves for their constructive criticism and input. Dr. Stanley Kosanke, Associate Professor of Veterinary Pathology at the University of Oklahoma Health Science Center, provided tissue toxicity analysis and pictures of organs. Sincere thanks to Dr. Barbara Fingleton for comments and suggestions. The technical assistance of Mrs. Jyothi Parvathareddy is appreciated.
The work was supported by the National Institutes of Health [CA107183 to T.P., F31CA144572 to M.H.] and UTRF Maturation Grant to T.P.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.