
Abstract
Objective:
Characterization of the multidrug resistance (MDR) region in P. aeruginosa strain PA59 revealed the presence of antibiotic resistance genes, including blaIMP-45 and blaVIM-2, within a complex genetic landscape of mobile genetic elements.
Methods:
Carbapenem-resistant Pseudomonas aeruginosa (CRPA) strains were isolated from Shanghai Changhai Hospital. Polymerase chain reaction (PCR) was used to detect the β-lactamase genes in the isolated strains. Strains carrying two or more genes were subjected to whole-genome sequencing (WGS) and in-depth bioinformatics analysis.
Results:
A total of 94 CRPA strains were isolated, among which PA59 was determined to carry blaIMP-45 and blaVIM-2 genes. Compared with single-gene positive or other blaIMP and blaVIM dual-gene positive strains reported, PA59 exhibited a broader range of drug resistance. We discovered a multidrug resistant (MDR)–related region composed of various mobile elements in the PA59 chromosome. This region carried many resistance genes, including the target genes blaIMP-45 and blaVIM-2. By further comparing the mobile elements GI13 and Ph08, we speculated that this integron structure carrying blaIMP-45 and blaVIM-2 was initially integrated into the genomic island or prophage, forming a more complex genetic structure, and then further integrated into the PA59 chromosome through plasmids. Phylogenetic tree analysis showed limited sequence similarity between PA59 and other CRPA strains.
Conclusions:
This study identified PA59 as the first reported P. aeruginosa strain carrying both blaIMP-45 and blaVIM-2 on the chromosome. The assembly and annotation of the PA59 genome provide valuable insights into the genomic diversity and gene content of this clinically important pathogen, aiding the development of effective strategies against antibiotic resistance.
Introduction
P. aeruginosa is a prevalent opportunistic pathogen that can cause severe infections in immunocompromised individuals. One of the major concerns regarding P. aeruginosa infections is the emergence of multidrug-resistant strains, which are often associated with high morbidity and mortality rates. 1 Among the multidrug-resistant P. aeruginosa strains, the metallo-β-lactamase (MBL)–producing strains 2 are of particular concern due to their broad-spectrum resistance to β-lactam antibiotics, which are commonly used in clinical practice. MBLs, characterized by their active site containing a metal zinc, have the ability to hydrolyze almost all β-lactam antibiotics except monobactams, rendering them ineffective against bacterial infections. 3 In recent years, the detection of MBLs has increased significantly, with a wide distribution across various bacterial species and geographical regions, making them a focal point of clinical attention.4,5 The genotypes of acquired MBLs include imipenemase (IMP), Verona integron-encoded metallo-beta-lactamase (VIM), São Paulo metallo-beta-lactamase (SPM), Seoul imipenemase (SIM), German imipenemase (GIM), New Delhi metallo-beta-lactamase (NDM), and Alcaligenes faecalis metallo-beta-lactamase (AFM),6,7 among which IMP and VIM are the most predominant. Each genotype can be further classified into various variants based on the gene and translated amino acid sequences. MBL genes can be located on integrons, transposons, plasmids, chromosomes, or other genetic elements. Owing to their ability to be plasmid-mediated, MBLs are highly transferable, making them a significant contributing factor to bacterial resistance.
Francisco Toval et al. conducted at a Costa Rican hospital found a strain of P. aeruginosa carrying both the blaIMP and blaVIM genes, along with multiple integrons, potentially aiding in gene spread. 8 Another study 9 identified a Klebsiella pneumoniae strain with 25 resistance genes, including blaNDM-5 and blaKPC-2, resistant to most antibiotics except polymyxin B and tigecycline. Whole-genome sequencing (WGS) revealed that the strain also harbors numerous virulence-related genes and mobile genetic elements like genomic islands and phages. Therefore, when multiple carbapenem resistance genes coexist in a bacterial strain, their ability to carry various resistance and virulence-related mobile genetic elements broadens the spectrum of resistance and virulence in these bacteria.8–11 In this study, we investigated the resistance genes of 94 clinical isolates of Carbapenem-resistant Pseudomonas aeruginosa (CRPA) and found a strain (PA59) representing the first documented case of P. aeruginosa that harbors both blaIMP-45 and blaVIM-2 resistance genes on its chromosome. We further studied its genetic context aim to better understand its resistance mechanism and provide a theoretical basis for the development of effective prevention and control strategies against this multidrug-resistant pathogen.
Material and Methods
Bacterial isolates, identification, and antimicrobial susceptibility testing
Continuous and nonrepetitive CRPA strains were isolated from clinical specimens of patients in Shanghai Changhai Hospital between January 1, 2019, and December 31, 2019. The strains were identified using the Microflex matrix-assisted laser desorption/ionization time-of-flight mass spectrometer (Bruker, Germany). The antimicrobial susceptibility testing of the strains was performed using the VITEK 2 compact automated microbiology analyzer (bioMérieux, France). For drugs not included or with limitations in the analyzer, the Kirby-Bauer disk diffusion method (Oxoid, UK) was used as a supplementary test. The interpretation of drug susceptibility testing results followed the guidelines of the Clinical and Laboratory Standards Institute (CLSI) M100-S28 standard. 12 CRPA was defined as P. aeruginosa that were resistant to either meropenem (MIC ≥8 μg/mL) or imipenem (MIC ≥8 μg/mL). The quality control strain used for drug susceptibility testing was P. aeruginosa (ATCC 27853), provided by the Shanghai Clinical Laboratory Center. WHONET 5.6 software was used for strain distribution and drug resistance data analysis.
DNA extraction and detection of carbapenemase and porin genes by PCR
The detection of β-lactamase genes blaKPC, blaBIC, blaOXA-48, blaNDM, blaIMP, blaVIM, blaSPM, and outer membrane protein (OMP) OprD2 was performed using the polymerase chain reaction (PCR) method. The primer sequences used in this study have been previously reported (Table 1). The primers were synthesized by Shanghai Bioengineering Company. Positive amplification products were subjected to second-generation sequencing by Shanghai LingEn Biotechnology Co., Ltd. The sequencing results were aligned with the GenBank database to confirm the amplified product as the target gene fragment.
Primers for the Detection of Carbapenem-Resistant–Related Genes
Whole-genome sequencing using short and long reads
The strain carrying two or more carbapenem-resistant–related genes identified by PCR was cultured on agar plates in ambient air at 37°C or in fluid medium at 37°C with agitation. Genomic DNA sample was isolated from the cell pellets with a Bacteria DNA Kit (OMEGA, USA) according to the manufacturer’s instructions, and quality control was subsequently performed on the purified DNA samples. Genomic DNA was quantified by using TBS-380 fluorometer (Turner BioSystems Inc., Sunnyvale, CA). High-quality DNA sample (OD260/280 = 1.8∼2.0, >6ug) is used to construct fragment library.
This strain was sequenced by Shanghai Winnerbio Technology Co., Ltd. (Shanghai, China) using Illumina NovaSeq 6000 and PacBio Sequel II platforms. For Illumina sequencing, at least 1 μg genomic DNA was used for each strain in sequencing library construction. DNA samples were sheared into 400–500 bp fragments using a Covaris M220 Focused Acoustic Shearer following manufacture’s protocol. Illumina sequencing libraries were prepared from the sheared fragments. The prepared libraries then were used for paired-end Illumina sequencing (2 × 150 bp) on an Illumina NovaSeq 6000 machine. For PacBio sequencing, SMRTbell library inserts (20 kb) were sequenced, and subreads shorter than 500 bp were removed. The PacBio sequences were error corrected, binned, and then assembled using the Canu assembler (version 2.2, https://github.com/marbl/canu). Software Pilon (version 1.24, https://github.com/broadinstitute/pilon) was used for assemblies polishing with Illumina short reads to improve genome quality. To determine the presence of any plasmids, the filtered Illumina reads were mapped using SOAP (https://soap.genomics.org.cn/) to the bacterial plasmid database made by Shanghai Winnerbio Technology Co., Ltd. based on GenBank.
Annotation and bioinformatic methods
The rRNA genes were found by using the Barrnap (version 0.9, https://www.cbs.dtu.dk/services/RNAmmer/) and tRNA genes by using tRNAscan-SE (version 2.0.8, http://trna.ucsc.edu/software/) with default settings. Prodigal (version 2.6.3) was used to predict the open-reading frame 15 with default parameters. The predicted gene sequences were translated and searched against the National Center for Biotechnology Information (NCBI) nonredundant database, the UniProt/Swiss-Prot database, the protein families (Pfam) database, the Clusters of Orthologous Group (COG) database, and the Kyoto Encyclopedia of Genes and Genomes (KEGG) database for annotation. Additional annotation was performed using the following databases: Pathogen Host Interactions, Virulence Factors of Pathogenic Bacteria, Antibiotic Resistance Genes Database, Carbohydrate Active enZYmes, and antiSMASH platform (version 6.0.0, https://antismash.secondarymetabolites.org/). 16 Prophages were identified using the PHAST database (http://phast.wishartlab.com), while clustered regularly interspaced short palindromic repeats (CRISPR) elements were identified by CRISPRCasFinde with default parameters (https://crisprcas.i2bc.paris-saclay.fr/CrisprCasFinder/Index). 17 Genomic islands were determined with the web tool IslandViewer (version 4) using two the independent methods Islander and IslandPath-DIMOB with default parameters. 18
Results
Strain source, drug resistance genes, and phenotypic analysis
A total of 108 nonduplicate CRPA strains were isolated from the Microbiology Department of Shanghai Changhai Hospital from January 1 to December 31, 2019. Among them, 94 CRPA strains were successfully recovered and subjected to PCR gel electrophoresis. Among these, 47 strains showed the absence of the OprD2 gene, 11 strains were positive for the blaKPC gene, 3 strains were positive for the blaVIM gene, and 1 strain was positive for the blaIMP gene. In addition, there was one strain carried both the blaIMP and blaVIM genes. Through second-generation sequencing, the three blaVIM-positive strains were identified as blaVIM-24 (PA60) and blaVIM-2 (PA61 and PA62), respectively. The blaIMP-positive strain was identified as blaIMP-45 (PA53). The strain carrying both the blaIMP and blaVIM genes was named PA59 (blaIMP-45 and blaVIM-2).
The characteristics of PA59 chromosome
This whole-genome shotgun project has been deposited at GenBank under the accession CP123953; preliminary alignment results between PA59 and other genomes at the whole-genome level were showed as a comparative genomic circle plot generated using the BLAST Ring Image Generator (BRIG) software. (Fig. 1). PA59 chromosome has a total length of 7,299,968 bp and a GC content of 65.41%. It contains large numbers of antibiotic resistance genes (ARGs), virulence genes, integrons, prophages, genomic islands, transposons, CRISPR, and other mobile genetic elements (MGEs). The alignment results show that the main differences between PA59 chromosome and other P. aeruginosa sequences lie in the insertion of a long sequence in the region from 5,074,026 to 5,713,717 bp. This sequence consists of various MGEs and carries numerous antibiotic resistance genes. We refer to this region as the PA59 MDR-related region. Importantly, we have identified that the target genes blaIMP-45 and blaVIM-2 are both located on the PA59 MDR-related region. On either side of this region, there are two prophages, containing 12 genomic islands and 4 integrons. These elements include genes mediating resistance to fosfomycin (fosA), aminoglycosides (aac(6’)-Ib3, aac(3)-IId, aadA1, rmtB), macrolides (mph(A), ere(A)), quinolones (qnrVC6), folate pathway antagonists (sul1, dfrB1), beta-lactams (blaOXA-1, blaIMP-45, blaTEM-1B, blaVIM-2), quaternary ammonium compounds (qacE), and amphenicols (catB3).

Blastn comparisons of the PA59 chromosome and selected sequences with high similarity (T2101, AG1, PABL012, N17-1, AA2) for comparison. The labels for antibiotic resistance genes (in red) are based on the prediction results from resfinder, while the virulence factors (in army green) are represented using results from VFDB with an identity% ≥90%, considering the limitations of the plot size, aesthetic considerations, and result reliability. VFDB, virulence factors of pathogenic bacteria.
Genetic contexts of MDR-related region in PA59
We categorized the MDR-related region on the PA59 chromosome into distinct structural components based on the MGEs it contained, as illustrated in Figure 2. It was evident that there were overlaps between certain MGEs within this part of the sequence. For instance, the prophage Ph07 encompassed the entire genomic islands GI11 and a partial sequence of GI12. Integron int1 was situated within GI13, while int2 and int3 resided within GI19, and int4 was located within Ph08. Notably, among these MGEs, GI13, GI19, Ph08, and their respective four integrons, all harbored ARGs. In addition, only GI17 was identified to carry mph(A) and aac(3)-IId compared with the other MGEs. Importantly, the carbapenem-resistant–related genes blaIMP-45 and blaVIM-2 were positioned within int1 and int4, respectively.

The distribution of mobile elements and genetic contexts of the multidrug resistance-related region of P. aeruginosa PA59 chromosome. The purple rectangles indicate prophages. The brown rectangles indicate genomic islands. The dark blue rectangles indicate integrons. The sequence of MDR-related region in the figure below corresponds to the figure above and shows the similarity with pPA30_1 and pWTJH6.
We also conducted a separate Blastn alignment for the PA59 MDR-related sequence to analyze its origin. The results revealed that the sequences with high similarity to it mainly belonged to plasmids of the Pseudomonas genus. Therefore, we suspect that this segment of the sequence may have been integrated into the PA59 chromosome from an MDR plasmid. In addition, there is no clear synteny between the prophages and genomic islands of the PA59 MDR-related region and the similar sequences (Fig. 2), indicating that they do not exhibit high conservation. As shown in Figure 2, the alignment results between the PA59 MDR-related region and the two most homologous complete plasmid sequences suggest a higher similarity to the complete pPA30-1 plasmid, suggesting the possibility of overall plasmid integration, although we did not find any T4SS-related genes on the PA59 chromosome. Interestingly, all four integrons (integron 1 CP045003.1, integron 2 MF144194.2, integron 3 MN961670.1, genomic islands GI13 CP104871.1) can be found in the NCBI database with highly similar sequences (100% query coverage and ≥99.98% identity), although these sequences are rare and mostly found on plasmids or chromosomes of the Pseudomonas genus. Integron1 carrying blaIMP-45 shows high homology with plasmids carried by P. aeruginosa or P. putida, rather than the chromosome sequence, indicating that integron1 likely originated from a plasmid. Therefore, this may be the first discovery of the complete integration of the PA59-integron1 sequence into the P. aeruginosa chromosome. On the other hand, integron4 carrying blaVIM-2 shows high homology with both the chromosome and plasmids, and it cannot be determined from the alignment whether integron4 originated from a plasmid or a chromosome sequence, suggesting that the acquisition of integron4 is likely associated with transposons.
Genetic contexts of blaIMP-45 and blaVIM-2 alleles in PA59
To further elucidate the genetic structures carrying blaIMP-45 and blaVIM-2, we compared the differences between the mobile elements GI13 and Ph08, where blaIMP-45 and blaVIM-2 are located, and their homologous sequences (Figs. 3 and 4). It can be observed that the homologous region of pPA30_1 to our PA59-GI13 can be divided into two parts, separated by 45,199 bp (the left and right sequences of pPA30_1 with homology to PA59-GI13 are plotted separately in Fig. 3). The left sequence of pPA30_1 (418,527–445,620 bp) carries mercury resistance-related genes (merA, merC, merP, merT, and merR), multiple recombinase/integrase genes, and numerous hypothetical proteins, with IS6100 at the end. The right sequence (37,570–46,449 bp) contains the ISCR1-sul1-qacEΔ1 structure and integron1, with the difference being the insertion of ISPa7 in the intI1 flank of PA59-GI13. Integron1 has a complete integron structure, carrying four resistance genes catB3, blaOXA-1, blaIMP-45, and aac(6’)-Ib3.

The genetic contexts of blaIMP-45 carrying genomic island 13 and integron 1 in P. aeruginosa PA59. Open arrows represent coding sequences (red arrows, resistance genes; blue arrows, integrases; green arrows, insertion sequences; orange arrows, other proteins; gray arrows, hypothetical and unclassified proteins; white arrows, integration sites or promoter regions) and indicate the direction of transcription.

The genetic contexts of blaVIM-2 carrying prophage 8 and integron 4 in P. aeruginosa PA59. Open arrows represent coding sequences (red arrows, resistance genes; blue arrows, integrases; green arrows, insertion sequences; orange arrows, other proteins; gray arrows, hypothetical and unclassified proteins; white arrows, integration sites or promoter regions) and indicate the direction of transcription.
Regarding PA59-Ph08, where the blaVIM-2 gene is located, no homologous sequences with obvious similarity to its overall structure were found in the database. After comparison, we believe that the left sequence (5,690,189–5,700,112 bp) originated from a transposon structure similar to the DZ-B1 strain, which includes integron4 carrying the resistance genes blaVIM-2, aac(6’)-Ib3, and dfrB1. The reference sequence pNY5709-IMP also contains integron4, and it was found that multiple copies can exist. The right sequence (5,700,113–5,713,717 bp) shows high similarity to a partial sequence (4,004–17,577 bp) of pOZ176, including phosphatase genes and multiple recombinase/integrase genes (Fig. 4). We speculate that this integrated structure carrying blaIMP-45 and blaVIM-2 was initially integrated into a genomic island or prophage to form a more complex genetic structure and then further integrated into the PA59 chromosome through plasmids.
SNP analysis and phylogenetic tree construction
Using P. aeruginosa PAO 1 (NC_002516) as the reference genome, we performed a comparative analysis between 16 isolates of carbapenem-resistant copper–green P. aeruginosa strains obtained from Shanghai Changhai Hospital and the reference genome. In addition, we selected samples with high similarity sequences from the NCBI database (Table 2) and aligned them with PA59 also using P. aeruginosa PAO1 (NC_002516) as the reference genome. The gene alignment results between the two groups of samples were obtained, and based on this, a SNP matrix was extracted from the aligned sequences and converted into a fasta file. Using this file, a maximum likelihood method was used to construct the phylogenetic tree. Compared with other strains in the NCBI database, although the MDR-related region of PA59 showed high similarity to the complete pPA30-1 plasmid, the phylogenetic tree revealed that RJ19-28 was the closest to PA59 at the whole-genome level (Fig. 5A). Compared with strain RJ19-28, PA59 was also isolated in Shanghai, China, and belonged to ST708 and serotype 07. However, RJ19-28 carried blaIMP-45 and blaAFM-1 on the plasmid, 19 while PA59 carried blaIMP-45 and blaVIM-2 on the chromosome. It was observed that the similarity between the PA59 strain and other carbapenemase-producing strains screened in our hospital in 2019 (Fig. 5B) was limited, indicating low homology. The ST type of the PA59 strain was identified as ST 708, and the serotype was O7, which was inconsistent with all other strains.
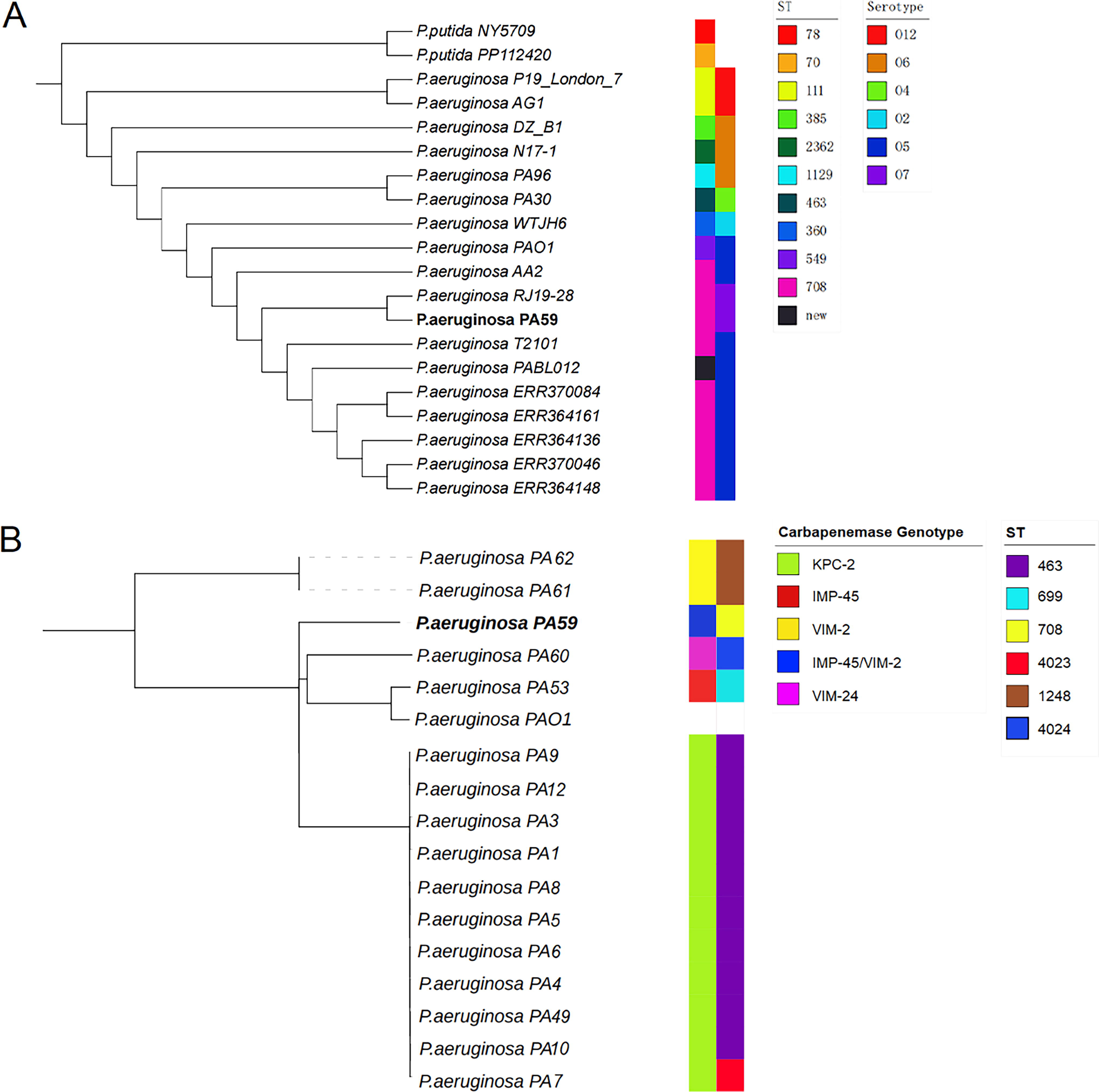
Phylogenetic tree (ML tree based on core SNPs).
Reference Genome Highly Similar to the PA59 Genome
Discussion
In this study, we identified a strain of P. aeruginosa-PA59 that carries both blaIMP-45 and blaVIM-2 metallo-β-lactamase genes. The phenomenon of P. aeruginosa strains carrying two or more metallo-β-lactamase genes has increased in recent years. A previous study 8 reported the combination of blaIMP-18 and blaVIM-2 genes in P. aeruginosa in Costa Rica. In addition, Turton et al 20 collected a P. aeruginosa isolate in the United Kingdom that had both blaVIM-2 and blaIMP-18 genes. Castillo-Vera et al described P. aeruginosa isolates carrying both blaVIM-2 and blaVIM-11 in Mexico. 11 Others10,21,22 also reported isolates that coproduce blaIMP-1 and blaVIM genes. In China, it appears that there is a relatively high occurrence of P. aeruginosa strains carrying two or more metallo-β-lactamase genes.23,24 However, only Fang YF et al 25 and Zhang X et al 19 reported a carbapenem-resistant P. aeruginosa isolate coproducing blaIMP-45 and blaAFM-1. No specific subtyping analysis has been conducted for the others. The PA59 strain we discovered in our study is the first reported MBL-producing P. aeruginosa that carries both blaIMP-45 and blaVIM-2 resistant genes. In addition, a unique aspect of our study is that the blaIMP-45 and blaVIM-2 genes in the isolated P. aeruginosa (PA59) are located on the chromosome, similar to the finding of blaIMP-18 and blaVIM-2 in P. aeruginosa. 8 Other reported cases of two or more metallo-β-lactamase genes coexisting in P. aeruginosa are located on plasmids.
blaIMP-45 is a variant of blaIMP-9 with a single amino acid substitution (G214S), first reported in a canine-origin P. aeruginosa strain from Beijing, China in 2014. 26 Another study reported an outbreak of CRPA carrying blaIMP-45 in a tertiary hospital in Shanghai and investigated the role of IncP-2 plasmids in the dissemination of blaIMP-45 among P. aeruginosa. 20 However, this is the first time that the blaIMP-45 gene has been found on the chromosome of P. aeruginosa.
P. aeruginosa carrying two or more resistant genes is a result of the acquisition of resistance genes through horizontal gene transfer. 27 This process involves the transfer of genetic material between bacteria through mechanisms such as conjugation, transduction, and transformation. P. aeruginosa has a highly adaptable genome, allowing it to rapidly develop resistance to antibiotics by acquiring resistance genes. This is facilitated by mobile genetic elements such as plasmids, integrons, and transposons, which can carry multiple resistance genes and transfer them between bacteria.28,29 Our study suggests that the integration of the blaIMP-45 and blaVIM-2 genes into the P. aeruginosa chromosome initially occurred in a genomic island or prophage, forming a more complex genetic structure, and then further integrated into the PA59 chromosome through plasmids. The reference sequence AG1 chromosome carries both blaIMP-18 and blaVIM-2 genes, but their insertion positions are significantly different from our PA59 chromosome, and the MDR-related region found in PA59 is not present. 8 The MDR-related region containing blaIMP-45 and blaVIM-2 may represent a novel pattern related to the insertion and integration of resistance genes.
In conclusion, our study has identified a rare strain of P. aeruginosa carrying both blaIMP-45 and blaVIM-2 on its chromosome, shedding light on its resistance mechanism. Our findings highlight the importance of continuous surveillance of multidrug-resistant plasmids and their integration into bacterial chromosomes. The assembly and annotation of the PA59 genome provide new insights into the genomic diversity and gene content of this important human pathogen and lay the foundation for the development of effective strategies to combat the spread of antibiotic resistance.
Footnotes
Acknowledgments
We would like to thank Shanghai Winnerbio Technology Co., Ltd. (Shanghai, China) for their assistance with bioinformatics support, which was invaluable to the success of this study.
Authors’ Contributions
W.M. and J.G.: Conceptualization, Methodology, Writing—Original Draft Preparation. C.D.: Data Curation, Formal Analysis. X.H. and L.X.: Investigation, Resources. W.M. and Y.S.: Software, Validation. Q.Q. and W.M.: Funding Acquisition, Project Administration. Q.Q.: Conceptualization, Methodology, Supervision, Writing—Review and Editing.
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by Biosafety Research Special Project (