
Abstract
In Gram-negative bacteria, the bacterial cell wall biosynthetic mechanism requires the coordinated action of enzymes and structural proteins located in the cytoplasm, within the membrane, and in the periplasm of the cell. Its main component, peptidoglycan (PG), is essential for cell division and wall elongation. Penicillin-binding proteins (PBPs) catalyze the last steps of PG biosynthesis, namely the polymerization of glycan chains and the cross-linking of stem peptides, and can be either monofunctional or bifunctional. Their action is coordinated with that of other enzymes essential for cell-wall biosynthesis, such as lytic transglycosylases (LT). Here, we have studied SltB1, an LT from Pseudomonas aeruginosa, and identified that it forms a complex with PBP2, a monofunctional enzyme, which requires the presence of Ca2+. In addition, we have solved the structure of SltB1 to a high resolution, and identified that it harbors an EF-hand like motif containing a Ca2+ ion displaying bipyramidal coordination. These studies provide initial structural details that shed light on the interactions between the PG biosynthesis enzymes in P. aeruginosa.
Introduction
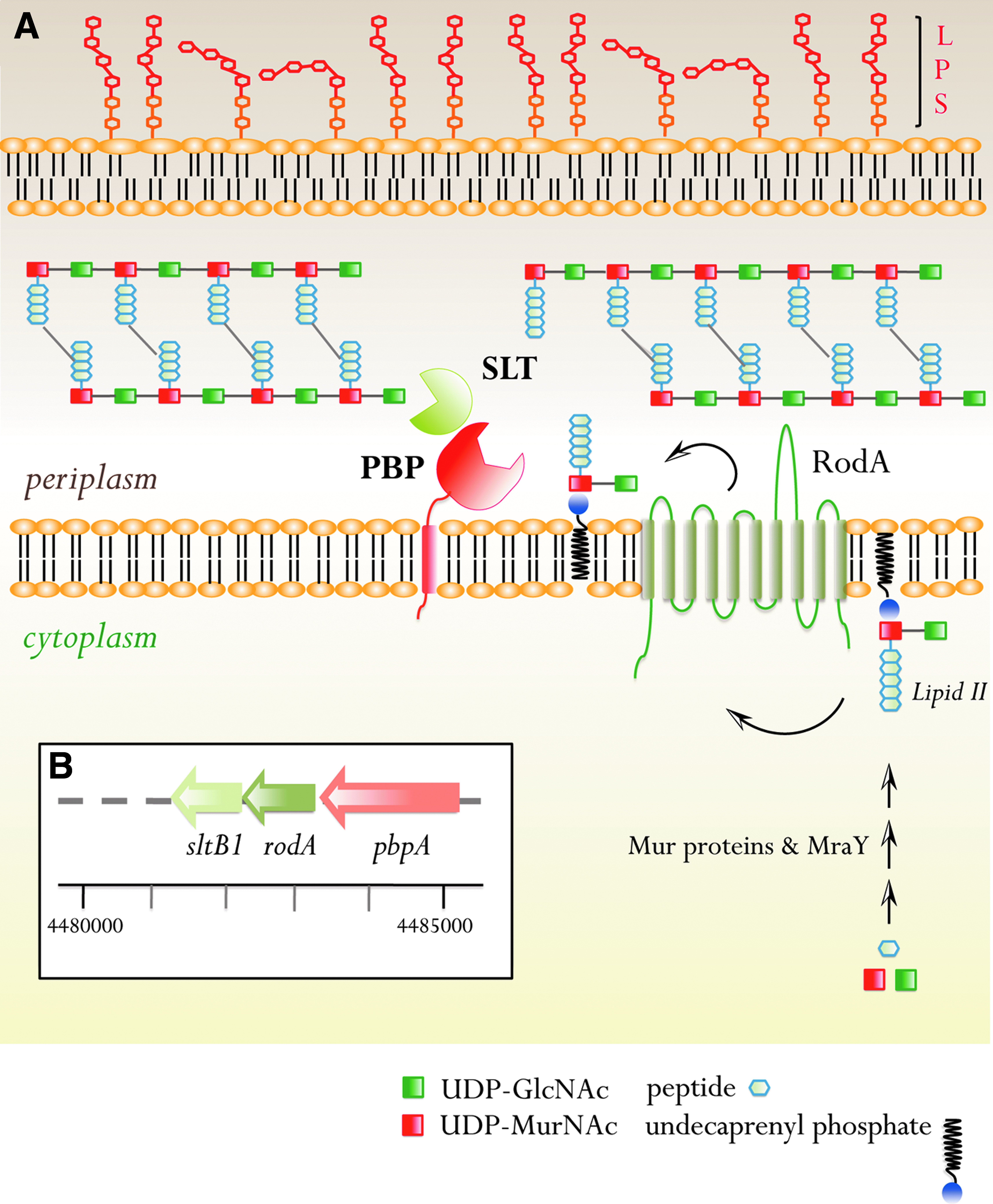
Peptidoglycan (PG) synthesis.
High-molecular-mass (HMM) PBPs can either carry both GT and TP domains (class A), or catalyze only the TP reaction (class B). Low-molecular-mass (LMM) PBPs, on the other hand, can be either carboxypeptidases or endopeptidases, thus playing a role in the regulation of the level of PG cross-linking by cleaving peptide bonds within the stem peptide.13,14,17,22,27 Notably, PG precursors are added by PBPs to the pre-existing PG in gaps in the cell wall generated by lytic transglycosylases (LTs) and endopeptidases, enhancing the turnover of old material, which suggests that such enzymes could be potentially involved in a macromolecular complex whose role is to coordinate the action of these antagonistic activities during the cell cycle.23,27
LTs have been grouped into four families according to amino-acid sequences and the conservation of motifs, and are either periplasmic or attached to the inner leaflet of the outer membrane through a lipoyl moiety. In Escherichia coli, at least seven different LTs have been identified and studied: Slt70, which is soluble, and six membrane-bound enzymes (MltA, MltB, MltC, MltD, MltE, and EmtA). 27 In support of the hypothesis that PBPs interact with LTs in a macromolecular complex, PBP2, a HMM class B enzyme, was shown to interact with MltA in Neisseria meningitidis 8 ; in E. coli, MltA immobilized on an affinity column was shown to recognize PBP1c, PBP2, PBP3, and PBP1b, the latter through an interaction with the structural protein MipA 28 ; and Slt70 was shown to bind to PBP3, PBP7, and PBP8. 21 LTs have, thus, been shown to interact with HMM as well as LMM PBPs.
Pseudomonas aeruginosa is a major human pathogen, the causative agent of nosocomial infections, and a particular threat to immunocompromised and cystic fibrosis patients. 12 P. aeruginosa expresses a class B PBP (PBP2) within a gene cluster that also harbors the RodA flippase and a periplasmic transglycosylase (LT), SltB1.1,10,20 In order to further conduct the studies of Legaree and Clarke, who provided the first evidence regarding the interaction between PBP2 and SltB1, 9 we purified both proteins independently and used surface plasmon resonance (SPR) spectroscopy to show that the interaction between PBP2 and SltB1 requires the presence of Ca2+. In addition, we solved the crystal structure of P. aeruginosa SltB1 to a high resolution, which revealed that it carries an EF-hand-type Ca2+-binding loop that could potentially play a role in the recognition of partner molecules such as PBP2. These results provide an initial framework for understanding the structural requirements for interactions between PG biosynthesis enzymes in P. aeruginosa.
Experimental Procedures
Cloning
Regions from genes pbpA and sltB1, corresponding to PBP2 (residue 39–646) and SltB1 (residue 40–341), respectively, were initially amplified from a clinical P. aeruginosa strain PAO1. SltB1 was cloned into the first site of a pETDuet-1 vector (EcoRI/HindIII), thus downstream from a hexahistidine tag, and a thrombin cleavage site was added to the vector by polymerase chain reaction. PBP2 was cloned into pET30b (NdeI/HindIII), resulting in a noncleavable C-terminal fusion to a hexahistidine tag.
Protein expression and purification
PBP2
E. coli BL21(DE3) STAR (Invitrogen) carrying the expression vector just described were grown at 37°C in Luria-Bertani medium (LB), supplemented with 15 mg/L kanamycin. Expression was induced by the addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 0.4 mM at an optical density (OD) at 600 nm of 0.3 A.U., and the cells were harvested by centrifugation after overnight growth at 16°C. The pellet was resuspended in lysis buffer (50 mM NaH2PO4 pH 8.0, 300 mM NaCl, 10% glycerol, 20 mM imidazole, 1% 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate [CHAPS]) and sonicated. Cell debris were removed by centrifugation, and the cleared lysate was incubated with Ni-chelating resin (GE Healthcare) pre-equilibrated in lysis buffer. Hexahistidine-tagged protein was retrieved by an imidazole gradient (20–500 mM). The fractions containing PBP2 were pooled and further purified by size-exclusion chromatography (Superdex 200 HR 10/30) equilibrated in 25 mM HEPES pH 7.5, 150 mM NaCl. The protein was concentrated in a 50 kDa cutoff concentrator (Vivaspin).
SltB1
E. coli BL21(DE3) C43 cells carrying the expression vector just described and the chaperone-expressing vector pG-KJE6 were grown at 37°C in LB, supplemented with 100 mg/L ampicillin and 34 mg/L chloramphenicol. The expression of the chaperones was induced by the addition of 0.5 mg/ml L (+) arabinose, 10 ng/ml tetracycline, and 20 mM benzyl alcohol at an OD600nm of 0.4 A.U.; and cells were further grown at 16°C until an OD600nm of 0.6 A.U. was reached. Expression of SltB1 was induced by the addition of IPTG to a final concentration of 0.2 mM, and bacteria were harvested by centrifugation after overnight growth at 16°C. The pellet was resuspended in lysis buffer (25 mM Tris-HCl pH 8.0, 250 mM NaCl, 10% glycerol, 20 mM imidazole, and 1% CHAPS), and the cells were disrupted by sonication. The lysate was clarified by centrifugation and incubated with Ni-chelating resin (GE Healthcare) pre-equilibrated in lysis buffer. Hexahistidine-tagged protein was retrieved by an imidazole gradient to 500 mM, which was followed by size-exclusion chromatography (Superdex 200 HR 10/30 in 25 mM HEPES pH 7.5, 150 mM NaCl) and ion exchange (Mono Q in 25 mM HEPES pH 7.5, 20 mM–1M NaCl gradient). For crystallization trials, the protein was further concentrated to 22 mg/ml in a 10 kDa cutoff concentrator (Vivaspin).
SPR experiments
Real-time monitoring of the interaction between PBP2 and SltB1 was performed through the immobilization of PBP2 onto a CM5 sensorchip (GE Healthcare) using a BIAcore 3000 instrument (GE Healthcare). Immobilization was performed at a flow rate of 10 μl/min in HBS-P buffer (10 mM HEPES pH 7.5, 150 mM NaCl containing 0.005% (v/v) surfactant P20). The flow cells of a CM5 chip were first activated with 70 μl of 0.2 M N-ethyl-N′- (diethylaminopropyl)-carbodiimide and 0.05 M N-hydroxysuccimide. Afterward, soluble PBP (24 μg/ml in 10 mM acetate buffer pH 5.0) was injected over one of the activated flow cells until a level of 1,900 response units was reached. The un-reacted groups were then blocked by an injection of 70 μl of 1 M ethanolamine pH 8.5 over the two flow cells. One hundred microliters of the analyte (SltB1) was injected in HBS-P over the different flow cells at a flow rate of 30 μl/min, followed by 180 sec of dissociation. The background signal recorded on the control surface without immobilized protein served as a blank sensorgram for subtraction of the bulk refractive index background. Data were analyzed by the global fitting of both the association and dissociation phases for six SltB1 concentrations (ranging from 0.105 to 1.26 μM) simultaneously, using the BIAevaluation software (GE Healthcare).
Crystallization
Initial crystallization conditions were found using the high-throughput crystallization facility at the HTX lab (Partnership for Structural Biology, Grenoble). Crystals were obtained at 20°C in 1.9 M malonate pH 6.5. Subsequently, we manually produced diffraction quality crystals by mixing 1 μl SltB1 (20 mg/ml) and 1 μl precipitant (1.5 M malonate pH 6.5, 3 mM CaCl2) as hanging drops in 24-well VDX™ plates (Hampton Research). The crystals were cryoprotected using mother liquor supplemented with 30% (vol/vol) glycerol.
Data collection and structure determination
Diffraction images of SltB1 crystals were collected at the European Synchrotron Research Facility (ESRF) beamline ID23-EH2. A native data set was collected to 1.8 Å. Images were processed and scaled with XDS, 9 revealing a P3221 space group with one monomer per asymmetric unit. In order to obtain the phases, we performed a molecular replacement experiment by employing the coordinates of Slt35 from E. coli (lacking the first 18 amino acids 25 ; PDB code 1QUS) as a search model, using PHASER. 23 Automated protein model building and structure refinement were performed using ARP/wARP, 3 leading to an R-factor of 0.178 (Rfree 0.211). The structure was improved by several rounds of manual refinement using REFMAC 5.5 18 and model building using Coot. Data collection, structure solution, and refinement statistics can be found in Table 1.
rmsd, root mean square deviation.
Results
The interaction between PBP2 and SltB1 is Ca2+ dependent
The production of recombinant SltB1 using a previously described method 10 only yielded low amounts of soluble protein. In order to circumvent this problem, we coexpressed SltB1 in the presence of molecular chaperones (DnaK, GroEL, DnaJ, GroES, and GrpE) and also performed chemical treatment of the growth media with the heat-shock inducer benzyl alcohol.4,19 These two modifications to the original expression protocol allowed us to obtain large amounts of highly soluble, pure SltB1 (Fig. 2) that was employed for both SPR experiments and crystallization assays. PBP2 was expressed in E. coli and purified by using classical nickel affinity and gel filtration chromatographies.
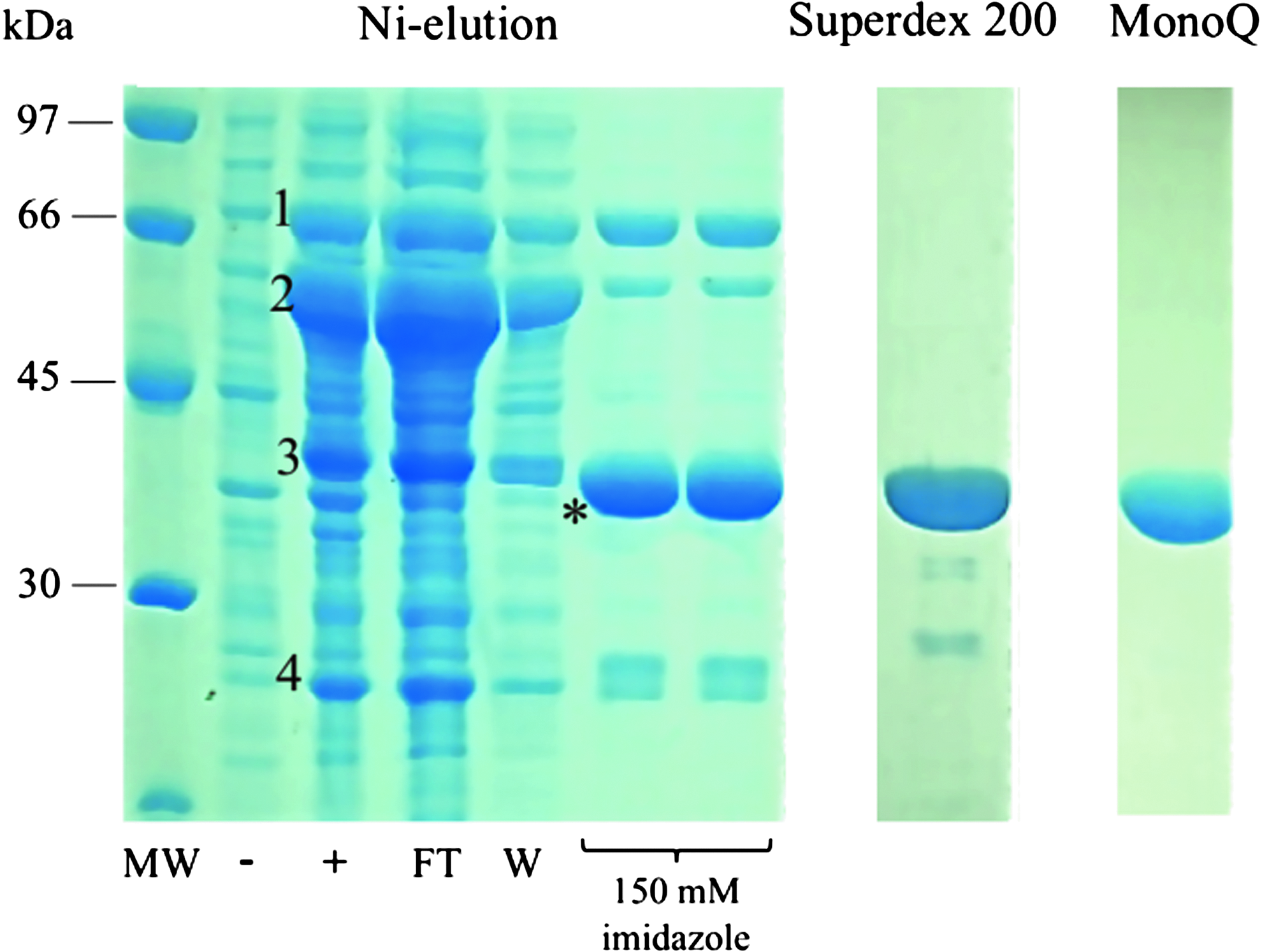
SDS-PAGE analysis of coexpressed SltB1 with molecular chaperones and benzyl alcohol. Recombinant Escherichia coli BL21(DE3) C43 cells were grown at 37°C and induced with isopropyl β-D-1-thiogalactopyranoside, tetracycline, L-arabinose, and benzyl alcohol. The majority of coexpressed chaperones could be removed during immobilized metal ion affinity chromatography purification (FT). 1, DnaK; 2, GroEL; 3, DnaJ; 4, GrpE; *, SltB1; −, uninduced cells; +, induced cells; FT, flowthrough; W, wash fraction; MW, molecular weight marker.
We then investigated the interaction between PBP2 and SltB1 by SPR analysis. Increasing concentrations of recombinant SltB1, ranging from 0.105 to 1.26 μM, were injected over a CM5 sensor chip on which PBP2 was the immobilized ligand (Fig. 3). Analysis of the SPR-binding data using the BIAevaluation software showed that the best fit for the interaction between immobilized PBP2 and soluble SltB1 was provided by the Langmuir 1:1 reaction model, yielding values for the association rate constant (ka) and dissociation rate constant (kd) of 2.52×104 M−1×s−1 and 5.98×10−3 M−1×s−1, respectively. The resulting apparent equilibrium dissociation constant (KD) of 230 nM (X 2 =1.07) was calculated from the kd/ka ratio and is comparable to that measured by Legaree and Clarke 10 of 130 nM, in which a similar experiment was performed, but with SltB1 immobilized on the sensor chip and PBP2 tested as the soluble analyte. Interestingly, in our hands, this interaction was completely abolished when SltB1 was purified in the presence of 1 mM EDTA. Even at the highest concentration of tested analyte (6.34 μM), no interaction could be detected. The interaction between immobilized PBP2 and soluble SltB1 could only be recovered after the dialysis of SltB1 into a buffer containing 2.5 mM CaCl2. This observation suggested that the interaction between SltB1 and PBP2 requires the presence of Ca2+.
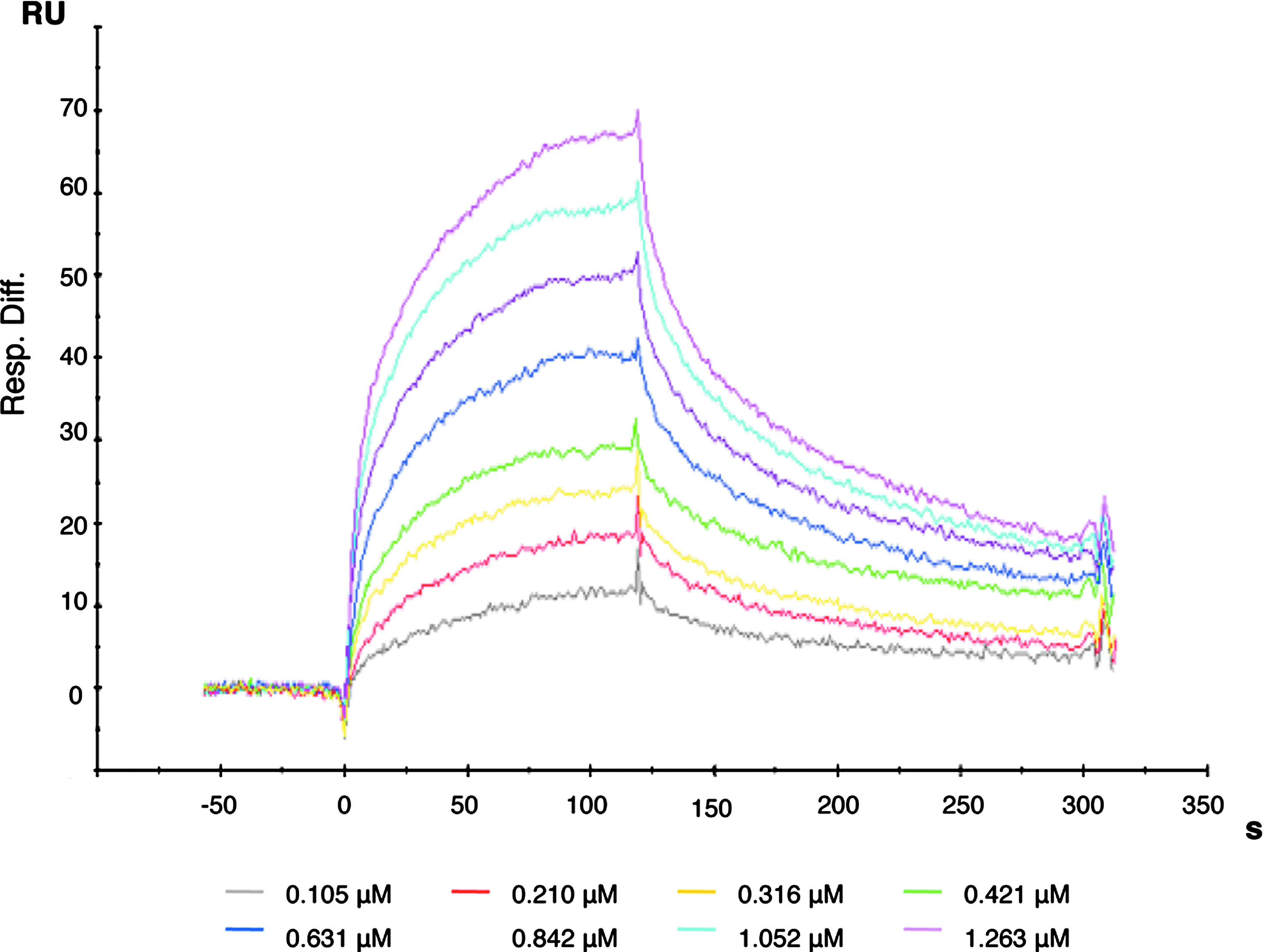
Interaction of PBP2 with SltB1 demonstrated by SPR analysis. PBP2 was immobilized by coupling amino groups to a carboxylated dextran surface. SltB1 was injected for 180 s at concentrations ranging from 0.105 to 1.26 μM.
Crystal structure of SltB1
The structure of SltB1 was solved to 1.8 Å by performing a molecular replacement experiment using the structure of Slt35 from E. coli 26 as a search model; a sequence comparison between SltB1 and Slt35 reveals that the two enzymes share 46% sequence identity (Fig. 4). All residues included in the SltB1-expressing clone (40–341) were visible in the electron density map, with the exception of a loop formed by residues 281–283. The fold of the SltB1 monomer is very similar to that of Slt35 (a soluble, active form of the 40 kDa membrane-bound MltB) 26 ; a comparison of the two structures reveals a root mean square deviation of 0.969 Å for 243 superimposed Cα atoms.
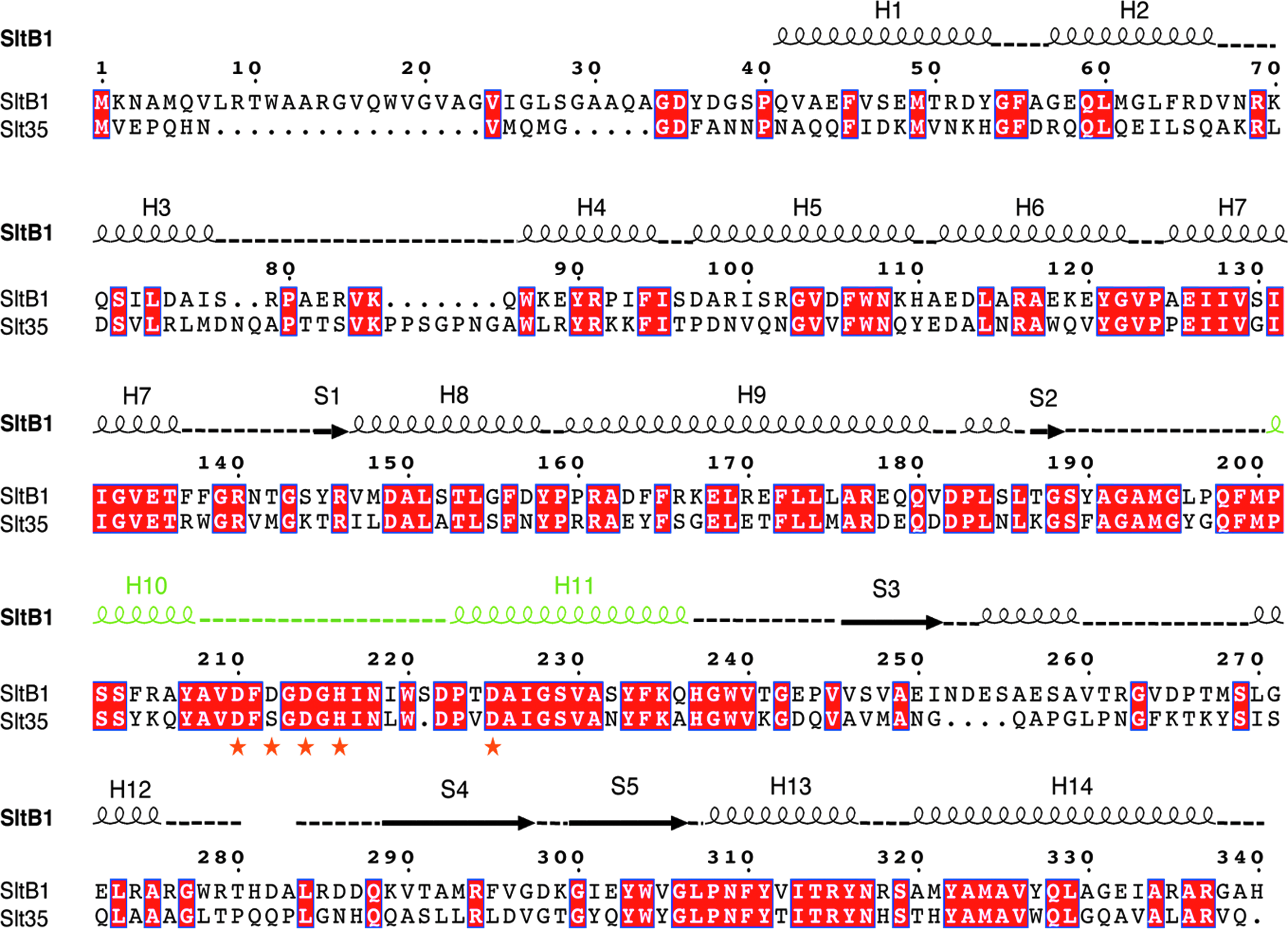
Structural alignment of Pseudomonas aeruginosa SltB1 and E. coli Slt35. Identical residues are shown with a red background. The EF-hand-like motif is highlighted in green, whereas the interacting ligands are presented as orange stars. Secondary structure elements visualized in the SltB1 structure are indicated above the sequences. H, helix; S, sheet.
SltB1 harbors three major domains: an N-terminal α domain, a core domain that resembles the fold of lysozyme, and a C-terminal β-sheet domain (shown in blue, red, and green, respectively, in Fig. 5A). The α domain is formed by five helices, encompassing residues 40–78 (H1–H3) and 143–188 (H8 and H9). The β domain (residues 242–307) comprises one α helix (H12) and a small twisted β-sheet (S3–S5) packed against three helices (H4, H13, and H14) of the core domain. In this β domain, four residues (281–283) are missing from our model, due to the absence of electron density in the map, suggesting a high flexibility for this region.
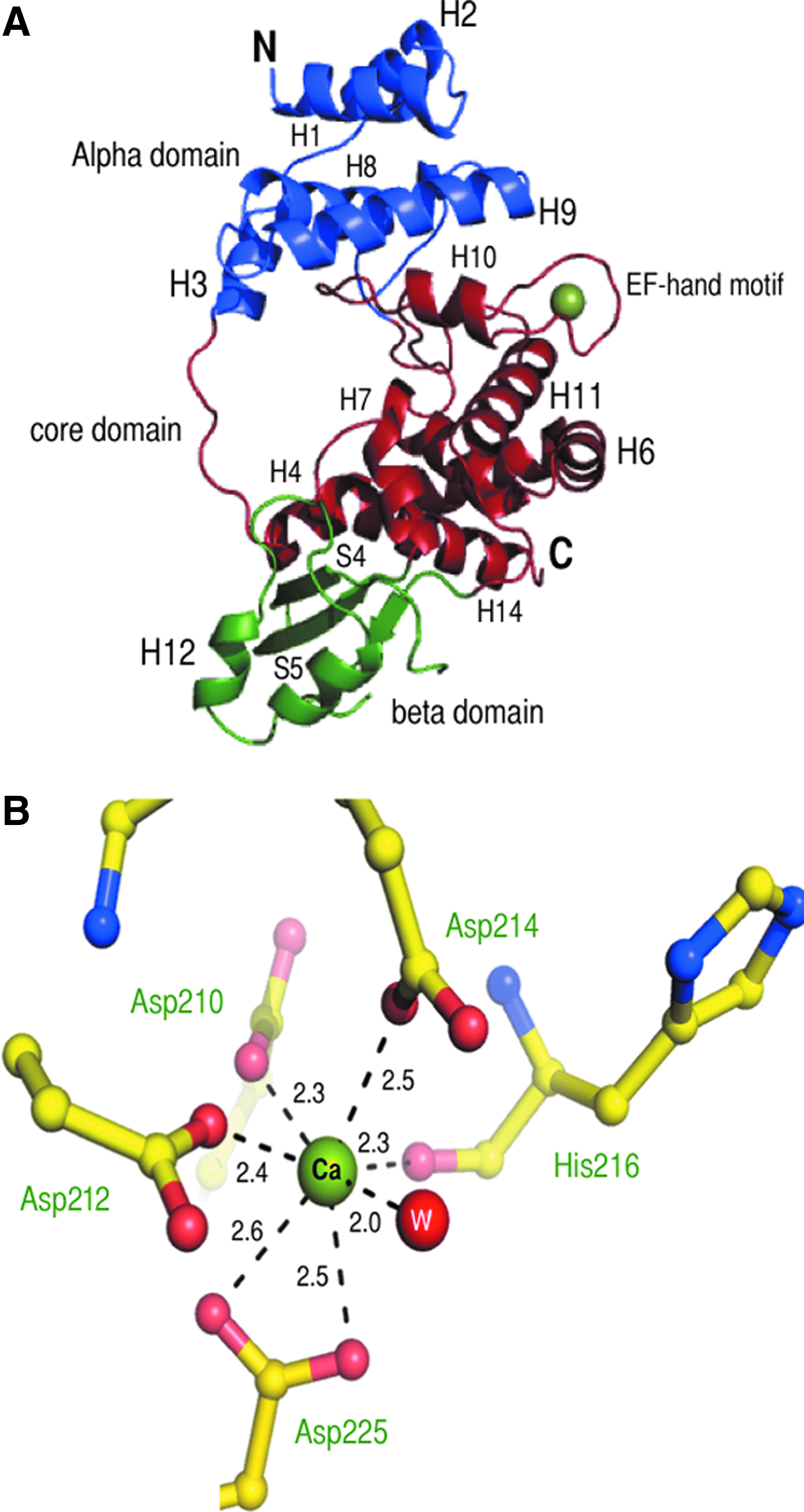
Three-dimensional structure of SltB1.
The core domain consists of three segments comprising eight helices (H4-H7 residues 79–142; H10 and H11, residues 189–243; H13 and H14, residues 308–339). This domain is sandwiched between the α and β domains and is of particular interest, as it bears the putative catalytic site and an EF-hand like motif (see next).
Substrate and metal-binding regions
Superposition of the structure of E. coli Slt35 with our SltB1 structure shows that the catalytic acid/base in Slt35, Glu162, is located at the same position as Glu135 in SltB1, suggesting that the latter could play the role of catalytic acid/base in the P. aeruginosa enzyme. Furthermore, Glu135 is located in a groove that is present within the core domain, which, by analogy with the structure of other enzymes in the family, harbors the substrate-binding region. 26 In SltB1, the groove is mainly lined by aromatic residues (Tyr90, Phe94, Phe164, Phe165, Tyr190, Phe199, Tyr207, Tyr233, Tyr316, and Tyr322). Notably, the aromatic residues present in the groove of E. coli Slt35 were shown to be involved in PG recognition, and, thus, the involvement of the groove region for interaction with the substrate by SltB1 could also be possible.
The second segment of the core domain contains an EF-hand-like motif (green region in Fig. 4, residue 201–236), one of the most widespread calcium-binding folds, which is found in a number of metal-binding proteins. The EF-hand of SltB1 differs slightly from canonical EF-hand patterns. First, the EF-hand loop of SltB1 contains 15 residues instead of the typical 12-residue loop observed in canonical EF-hand proteins. 29 Second, the bidentate ligand at position 12 is, in fact, located at position 16 (Asp225). SltB1 can be, thus, classified as a class 4 EF-hand-like protein based on the classification of Zhou et al. 29
E. coli Slt35 carries an EF-hand-type calcium-binding loop, and Ca2+ was shown to be important for stability of the molecule. 24 In the SltB1 structure, presented here, the metal ion bound in the EF-hand loop could be successfully modeled as a Ca2+, which was also present in the crystallization solution. The Ca2+ ion is coordinated by six protein oxygen atoms (Oδ1 of Asp210, Oδ1 of Asp212, Oδ1 of Asp214, O of His216, Oδ1 and Oδ2 of Asp225) and one water molecule with an average distance of 2.3–2.6 and 2.0 Å, respectively (Fig. 5B). The water molecule is located between the Ca2+ ion and two other residues in the EF-hand loop, namely Asp225 and Asp214. The amino acids that participate in metal coordination in the EF-hand motif are highly conserved (orange stars in Fig. 4), with the exception of the residue in an analogous position to that of His216, which can be Arg, Glu, or Lys, as the interaction is made with the backbone carbonyl. 25 The coordination of the metal ion, therefore, represents the typical bipyramidal configuration for EF-hand-like motifs. Notably, in SltB1, the EF-hand loop is one of the regions that has the highest B-factors, revealing flexibility and a potential requirement for interaction with other protein partners for stabilization.
Discussion
Proteins that are involved in the biosynthesis of PG have been postulated as being a part of a macromolecular complex that includes cytoplasmic, membrane-inserted, and periplasmic members. PBPs and LTs exert antagonistic functions within the PG biosynthesis process, namely Lipid II polymerization/cross-linking and glycan strand cleavage, and the regulation of their activities has been proposed to be facilitated by their proximal localization/direct interaction.7,14,27
In P. aeruginosa, PBP2 has been shown to play a key role in the rod-shaped morphology of the cell; deletion of the pbpA gene, which codes for PBP2, generates strains that display a spherical shape. In addition, such strains also display increased susceptibility to β-lactams, indicating that PBP2 plays a role in the development of resistance to such antibiotics. 11 In this work, we have furthered the studies of Legaree and Clarke 10 regarding the interaction between PBP2 and a molecular partner in the PG elongation process, SltB1, a LT.
In order to perform these studies, it was necessary to develop a protocol to obtain large amounts of soluble SltB1. This was made possible by coexpressing SltB1 in the presence of chaperones and of benzyl alcohol, but not by using the additives separately. It is conceivable that the addition of benzyl alcohol induced the expression of other endogenous chaperones (such as IbpA/B or ClpB), allowing them to work in concert with the plasmid-encoded chaperones GroEL-GroES and DnaK-DnaJ-GrpE in order to prevent aggregation and to mediate the correct refolding of SltB1. 4 Subsequently, we employed SPR spectroscopy to study the interaction between PBP2 and SltB1 in the absence and presence of calcium ions. Notably, binding was only detected in the presence of Ca2+, suggesting that the metal could play a role in the stabilization of SltB1 and be potentially important for the interaction itself. Temperature-scanning circular dichroism and fluorescence spectroscopy experiments performed on purified E. coli Slt35 also revealed that Ca2+ played a role in the stabilization of the enzyme. 25 It is also of note that the interaction between SltB1 and PBP2 was identified by using the soluble form of the PBP (which does not carry the transmembrane region), attesting to the fact that it is the periplasmic region of PBP2 which is involved in recognition of SltB1 (in agreement with Legaree and Clarke 10 ).
Our high-resolution structure of SltB1 reveals a three-domain fold that is highly similar to that of other soluble LTs, and, especially, Slt35 from E. coli. 26 The middle, core domain (Fig. 5A) displays a classic EF-hand fold with a Ca2+ ion coordinated by seven oxygen atoms (Fig. 5B), which is often seen in other EF-hand-carrying proteins.15,29 Markedly, eukaryotic proteins that carry multiple EF-hand regions often undergo conformational modifications on binding Ca2+, an event that plays a role in signal transduction.6,29 It is conceivable that the requirement of Ca2+ for the interaction between SltB1 and PBP2 in P. aeruginosa reflects a conformational modification of the EF-hand of SltB1 on recognition of its partner; a detailed understanding of this process will require a solution to the high-resolution structure of the PBP2:SltB1 complex. However, our SPR studies indicated that SltB1 had to be in its metal-bound form in order to recognize PBP2, which suggests that this specific region could play a role in PBP recognition. Interestingly, Romeis and Holtje identified that E. coli PBP8, an endopeptidase, not only binds to Slt70 but also stimulates its activity and protects it from degradation, 21 pointing to the interdependence of these proteins for optimal functionality. Thus, the data presented here, combined with studies on LTs and PBPs from other bacterial species, define an initial framework for the understanding of the relationship between these antagonistic enzymes in the cell-wall biosynthesis process.
Footnotes
Acknowledgments
The authors would like to thank J. Marquez and the HTX Lab team (Partnership for Structural Biology, Grenoble, PSB) for access to and help with high throughput crystallization, I. Bally (IBS, PSB) for access to the SPR facility, and the European Synchrotron Radiation Facility (ESRF, PSB) for access to beamlines. This work was supported by a grant from the Netherlands Organization for Scientific Research and the Fondation pour la Recherche Médicale (FRM DEQ20090515390). I.N. was supported by a grant from the ZON-MW AMR program.
Disclosure Statement
No competing financial interests exist.