
Abstract
Streptococcus pneumoniae protects itself from components of the human immune defense system by a thick polysaccharide capsule, which in most serotypes is covalently attached to the cell wall peptidoglycan. Members of the LytR-Cps2A-Psr (LCP) protein family have recently been implicated in the attachment of anionic polymers to peptidoglycan in Gram-positive bacteria, based on genetic evidence from Bacillus subtilis mutant strains and on the crystal structure of S. pneumoniae Cps2A containing a tightly bound polyprenol (pyro)phosphate lipid. Here, we provide evidence that Cps2A and its two pneumococcal homologs, LytR and Psr, contribute to the maintenance of normal capsule levels and to the retention of the capsular polysaccharide at the cell wall in the capsular type 2 S. pneumoniae strain D39. GFP fusions of all three LCP proteins showed enhanced localization at mid-cell, indicating a role in cell wall growth. Single cps2A or psr mutants produced a reduced amount of capsule. A cps2A lytR double mutant showed greatly impaired growth and cell morphology and lost approximately half of the total capsule material into the culture supernatant. We also present the crystal structure of the B. subtilis LCP protein YwtF and provide crystallographic evidence for the phosphotransferase activity of Cps2A, supporting an enzymatic function in the attachment of capsular polysaccharides to cell wall peptidoglycan.
Introduction
In the majority of serotypes, the capsular polysaccharide genes are located between dexB and aliA. 5 Here, we focus on the serotype 2 strain, D39, the capsular region of which begins with the cps2A–D genes (Fig. 1A). All 17 capsular genes in this region are under control of the promoter upstream of cps2A. 17 The first gene in the region, cps2A, encodes for a member of the LCP protein family (LytR, Cps2A, and Psr homologs), which is widespread in Gram-positive bacteria and does not occur in Gram negatives. The pleiotropic phenotypes of LCP mutant strains of various species have led to the suggestion that this protein family is a transcriptional regulator of cell wall processes. 27 Cps2A (also called Wzg), Cps2B (Wzh), Cps2C (Wcd), and Cps2D (Wze) have all been implicated in the regulation of capsule synthesis in S. pneumoniae. Indeed, Cps2D is an autophosphorylating tyrosine kinase that appears to be regulated by Cps2C4,30 and both might be spatial regulators that connect capsule synthesis to cell division. 21 Cps2A is required for the full expression of capsule; cps2A mutant strains contain significantly less capsule material attached to the cell wall. 31 However, like other LCP family members, Cps2A is an integral membrane protein, the bulk of which is located on the outside face of the cytoplasmic membrane (Fig. 1B) 19 ; it is not immediately apparent that how to reconcile this observation with any role in DNA binding. A recent report by Kawai et al. provides strong evidence that LCP proteins are the long searched for enzymes catalyze the covalent attachment of anionic cell wall polymers, like teichoic acids and capsular polysaccharides, to peptidoglycan. 24 This publication detailed genetic and biochemical evidence from the characterization of mutant strains of Bacillus subtilis and the crystal structure of the soluble part of Cps2A. Interestingly, the conserved, LCP domain of Cps2A contained a (pyro)phosphorylated polyprenol lipid, the precise chemical identity of which was dependent upon the Escherichia coli expression strain used for purification of the protein. Preliminary assays showed that Cps2A had weak, metal-ion-dependent pyrophosphatase activity, supporting an enzymatic function. Most likely, Cps2A binds the undecaprenol-pyrophosphoryl-linked capsule precursor to transfer the phosphorylated capsule chain to C6-OH of MurNAc residues in the peptidoglycan precursor or polymer.
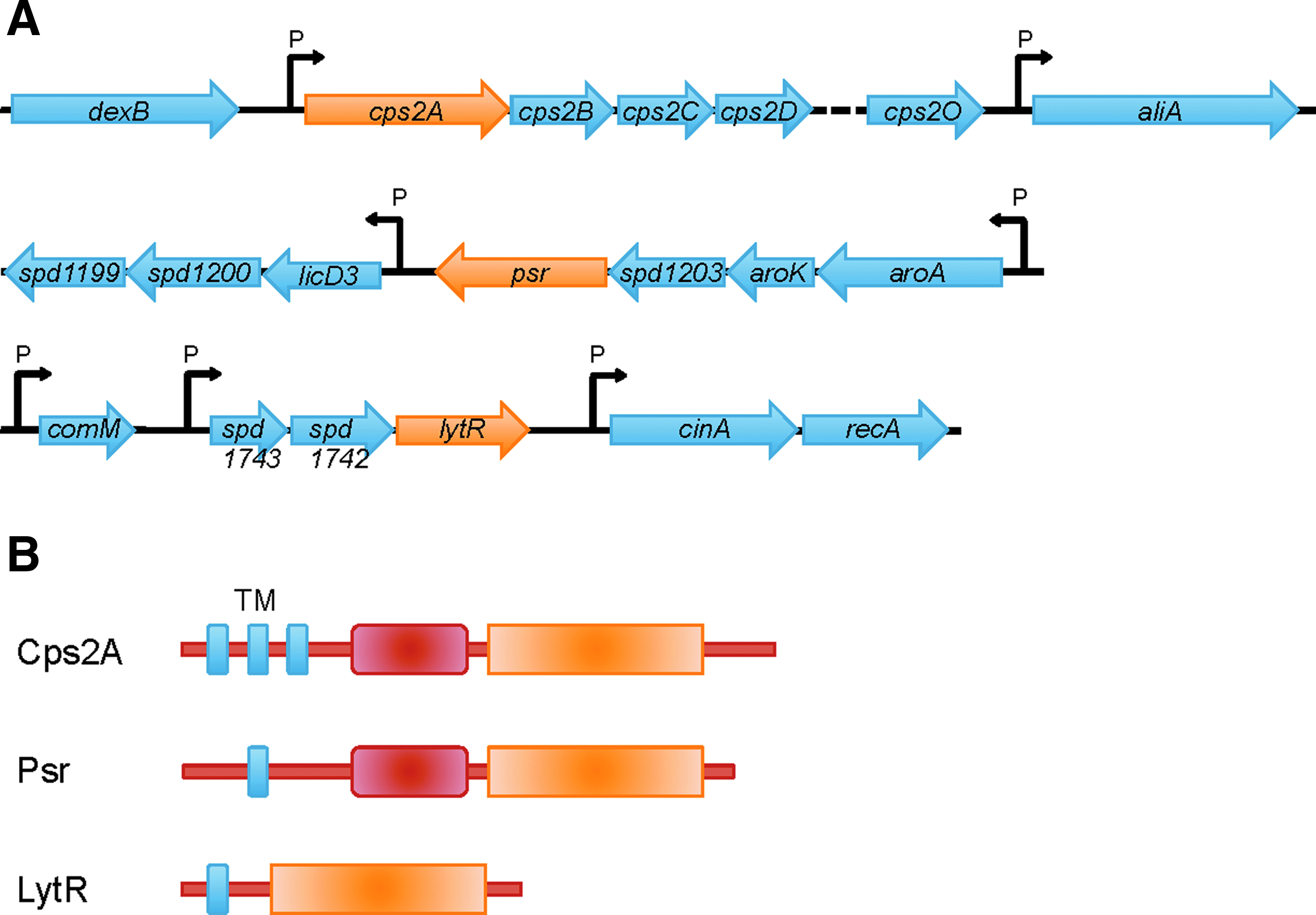
The LCP genes and proteins in Streptococcus pneumoniae.
In this study we have investigated the role of Cps2A, LytR, and Psr in the attachment of capsular polysaccharides to the cell wall of capsule type 2 strain D39. We show that all LCP proteins localize to the cell membrane and are enriched at mid-cell. Cps2A and psr mutants have a reduced amount of capsule. Inactivation of lytR proved difficult in the background of the encapsulated D39 strain, leading to mutants that were not viable in liquid culture. A suppressor mutant of a D39 cps2A-lytR double mutant grew with strongly distorted morphology in liquid culture, where it lost a large portion of its capsule material into the supernatant. LytR, together with Cps2A, thus plays a role in retaining the capsule at the cell wall. The crystal structure of the LytR homolog from B. subtilis, YwtF, and the pyrophosphorylysis observed by Cps2A support an enzymatic function of these proteins. These data and our previous observations 24 are consistent with a model whereby Cps2A is responsible for the covalent attachment of capsular polysaccharide to the pneumococcal cell wall, and that LytR can take over this function in the absence of Cps2A.
Materials and Methods
Bacterial strains, growth conditions, and genetic transformation
Table 1 contains the strains used in this study. S. pneumoniae R626,36 and D392 were grown at 30°C or 37°C in C+Y (pneumococal C medium with yeast extract) medium containing 1 mg/ml yeast extract. 41 To induce the expression of gfp+ fusions under PczcD control on plasmid pJWV25 and its derivatives, 0.15 mM ZnCl2 was added to the C+Y medium. Blood agar plates were made of tryptic soy broth (TSB) medium containing 1.5% agar and 3% defibrinated sheep blood. For growth curves, S. pneumoniae was grown overnight at 37°C on TSB blood agar plates with the appropriate antibiotics. C+Y medium without antibiotics was inoculated from colonies grown on plates to an OD620 of 0.05–0.07, incubated in a water bath at 37°C, and the OD620 was measured at the indicated time intervals.
E. coli DH5α (Invitrogen) was grown in lysogeny broth (LB) medium at 37°C with aeration. Where necessary, erythromycin was added at a concentration of 1 mg/ml for E. coli and 1 μg/ml for S. pneumoniae, tetracycline was added at 12.5 μg/ml for E. coli and 2.5 μg/ml for S. pneumoniae, and kanamycin was added at 50 μg/ml for E. coli and 400 μg/ml for S. pneumoniae.
Transformation of S. pneumoniae
DNA was introduced to the pneumococcal strains by means of natural transformation using 100 ng/ml of CSP-1 to induce competence in the recipient strain. 32 Cultures of S. pneumoniae were grown at 37°C until they reached an OD620 of 0.15–0.2, at which point CSP-1 and the transforming DNA were added, and the cultures were further incubated for 90–120 min at 37°C before plating on selective medium to identify transformants. S. pneumoniae transformants were selected by plating on TSB blood agar plates supplemented with the appropriate antibiotic.
Polymerase chain reaction amplification, cloning, and sequencing
Chromosomal DNA from S. pneumoniae was isolated using the Promega Wizard® genomic DNA purification system. For this, pneumococcal cells from liquid culture were harvested by centrifugation and resuspended in lysis buffer (50 mM Tris/HCl [pH 8.5], 5 mM MgCl2, 25 μg/ml RNase, and 0.4% DOC). To obtain chromosomal DNA, single colonies were propagated on TSB blood plates after transformation and cell material from the plate was resuspended in lysis buffer, and then incubated at 37°C for 10 min. Plasmids were purified with the Qiagen plasmid midi kit. Restriction enzymes and T4 ligase were purchased from Fermentas. DNA fragments were purified by using the QIAquick® PCR purification kit. DNA was amplified with Phusion® high-fidelity DNA polymerase from Finnzymes. Oligonucleotides listed in Table 2 were supplied by Sigma. Sequencing was performed at GATC, Germany.
Cloning of gfp+ fusions
For production of GFP+-tagged proteins in S. pneumoniae, the zinc-inducible promoter of plasmid pJWV25 was used. 13 To generate gfp+ fusions of cps2A, lytR, and psr in the background of R6 and D39, cps2A was amplified using the oligonucleotides cps2A_F and cps2A-R, lytR was amplified using the oligonucleotides lytR_F and lytR_R, and psr was amplified using the oligonucleotides psr_F and psr_R. The polymerase chain reaction (PCR) products were digested with the appropriate restriction enzyme (Table 2), ligated with digested pJWV25, and transformed into E. coli DH5α selecting on tetracycline. Transformants were screened by colony PCR using oligonucleotides pJWV25_scr_F and pJWV25_scr_R. The integrity of the insert in pJWV25 was confirmed by sequencing using oligonucleotides pJWV25_scr_F and pJWV25_scr_R. Correct plasmids were transformed into R6 and D39 while selecting for tetracycline resistance, resulting in strains R6 P czcD -gfp+-cps2A, D39 P czcD -gfp+-cps2A, R6 P czcD -gfp+-lytR, D39 P czcD -gfp+-lytR, R6 P czcD -gfp+-psr, and D39 P czcD -gfp+-psr. The correct integration of the pJWV25 derivatives into the bgaA region was confirmed by PCR amplification with the oligonucleotides bgaA_check_F and bgaA_check_R.
Gene deletions
D39 cps2A::ermB was created by replacing the cps2A gene by ermB followed by the promoter of cps2A to ensure native transcription levels of the downstream genes. Therefore, a construct consisting of four PCR fragments was consecutively cloned into pGEM-3Z: the 1 kb upstream flanking region of cps2A (oligonucleotides cps2A_up_F and cps2A_up_R), the ermB gene (oligonucleotides ermB_F and ermB_R), the promoter region of cps2A (oligonucleotides Pcps2A_F and Pcps2A_R), and the 1 kb downstream flanking region of cps2A (oligonucleotides cps2A_down_F and cps2A_down_R). Transformants of DH5α were screened by colony PCR using the oligonucleotides pGEM3Z_scr_F and pGEM3Z_scr_R. The final knockout construct was sequence verified and transformed into D39, selecting for erythromycin resistance. To confirm correct integration of the construct into the capsular region, the locus was PCR amplified with the oligonucleotides Δcps2A_seq_F and Δcps2A_seq_R and sequenced. D39 cps2A::ermB was transformed with pJWV25-cps2A, resulting in D39 cps2A::ermB P czcD -gfp+-cps2A, which was complemented with gfp+-cps2A upon zinc induction.
D39psr contains an insertion-inactivation of psr. This mutant was generated using the pJDC9-based insertion-deletion mutagenesis method. 10 The first 500 bp of psr (omitting the start codon ATG) was cloned in pJDC9 using the oligonucleotides psr_F and psr_R. The insert was verified by sequencing and the plasmid was transformed into D39. Correct integration of the plasmid into the psr gene was verified by PCR amplification. With oligonucleotides intF_psr located in the chromosome and intR_pJDC9 on the plasmid, the upstream region of psr was amplified. The downstream region was checked by PCR using oligonucleotides intF_pJDC9 and intR_psr.
Construction of D39 cps2A::ermB lytR::kan PczcD-lytR and D39 cps2A::ermB psr::kan
To construct the D39 cps2A::ermB lytR::kan mutant, a kanamycin resistance cassette flanked by DNA corresponding to the upstream and downstream regions of the psr locus was designed. A DNA fragment corresponding to the upstream region of psr was amplified by PCR from strain D39 and subsequently fused to the 5′-end of the kanamycin cassette in a second PCR. Next, a PCR fragment corresponding to the downstream region of psr was amplified and fused to the 3′-end of the resistance cassette. The final fragment was integrated into the psr locus of D39 cps2A::ermB by natural transformation.
To construct the D39 cps2A::ermB lytR::kan Pczcd-lytR strain, pJVW25 was inserted into the genome of the D39 cps2A::ermB strain as described previously. 13 The resulting strain was subsequently made streptomycin resistant by transformation with genomic DNA from strain CP1415. 33 Next, a kan::rpsL Janus cassette 39 was integrated downstream of the zinc-inducible PczcD promoter of the chromosomal pJVW25. The Janus cassette was subsequently replaced by a PCR fragment consisting of the coding sequence of D39 lytR. Finally, the wt+ copy of lytR was removed by natural transformation with a PCR cassette as described previously, 22 and colonies were selected on agar containing 0.15 mM ZnCl2.
D39 cps2A::ermB lytR::kan was created by transforming D39 cps2A::ermB with chromosomal DNA isolated from D39 cps2A::ermB lytR::kan P czcD -lytR and selecting colonies for kanamycin resistance and, therefore, the replacement of lytR with the kanamycin resistance cassette. The resultant colonies were screened by PCR for the absence of lytR at its locus using the oligonucleotides lytR_ext_F and lytR_ext_R. The general absence of lytR was verified using lytR_int_F and lytR_int_R and the absence of insertions at bgaA locus was checked using bgaA_int_F and bgaA_int_R. To exclude the possibility that any mutation occurred in the psr gene, psr was amplified and sequenced using intF_psr, seq_psr_middle_F, and psr_flank_R.
Expression of GFP+ fusion proteins in S. pneumonia and fluorescence microscopy
Strains expressing GFP+ fusion proteins were grown and samples were prepared as published previously. 13 The GFP+ signal was recorded using a DeltaVision experimental setup and the application of an exposure time of 2 sec using an excitation filter (Chroma) and 100% transmission (excitation at 490 nm and emission at 535 nm). For phase-contrast microscopy, the specimens were exposed for 100 ms with 100% transmission. Images taken were deconvoluted using the DeltaVision deconvolution software (ratio conservative, number of cycles 15, wavelength 528 nm, medium noise filtering).
Western blot and dot blot analyses
The expression and the correct size of the GFP+ fusion proteins were verified by western blotting. Whole cell lysates of the strains expressing GFP+ fusions were run in equal amounts on sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). The proteins were blotted onto nitrocellulose and the GFP+ fusions were detected with anti-GFP antibody (rabbit polyclonal; Invitrogen). A secondary horseradish peroxidase (HRP)-labeled anti-rabbit antibody (goat polyclonal; Invitrogen) was used in combination with an ECL kit (Amersham) for visualization of the antibody complexes.
Quantification of capsule per colony
D39 cps2A::ermB, D39, and R6 were transformed with genomic DNA from D39 cps2A::ermB lytR::kan P czcD -lytR and selected on TSB blood plates containing kanamycin. Agar pieces containing a single colony were excised from the plates and stored at −20°C. Thawed samples were mixed with 60 μl of lysis buffer and the agar was melted for 10 min at 100°C. In total, 7 serial 1:3 dilution steps in H2O were made and 2 μl of each dilution was applied to a nitrocellulose membrane. The membrane was air-dried for 2 h at room temperature and 15 min at 60°C, blocked in TBS + 0.5% casein for 1 h at room temperature, incubated with anti-serotype 2 serum in 1:10,000 (from rabbit; Statens Serum Institute) in TBS + 0.5% casein overnight at 4°C, washed 3 times for 5 min in TBS + 0.2% Tween20, incubated with a secondary HRP-labeled anti-rabbit antibody (goat polyclonal; Invitrogen), washed 3 times for 5 min in TBS + 0.2% Tween20, and detected using an ECL kit (NBS Biologicals) on photographic films (GE Healthcare).
Quantification of capsule in culture lysate and supernatant
D39 cps2A::ermB, D39psr, D39 cps2A::ermB lytR::kan, D39 cps2A::ermB psr::kan, D39, and R6 were grown on plates containing the appropriate antibiotic. C+Y medium was inoculated from plates to OD620 of 0.08 and grown at 37°C. To quantify total capsular material, an aliquot of culture was lysed at OD620 0.2 with 0.4% DOC, DNase, and RNase (10 μg/ml each) at 37°C for 10 min and centrifuged at 14,000 rpm for 10 min at room temperature. The supernatant was stored at −20°C. A second aliquot of the culture was centrifuged at 13,000 rpm for 10 min at 4°C, and the supernatant was mixed with 0.4% sodium deoxycholate before storage at −20°C. Samples were thawed at 50°C and diluted 1:1.5 in 12 steps with H2O. About 2 μl of each dilution was applied onto nitrocellulose membrane, air-dried, and processed as described for colony blots, but here the samples were washed with TBS without Tween20. The electrochemiluminescence signal was detected using Image Quant LAS 4,000 mini (GE Healthcare).
Visualization of proteins in culture lysate and supernatant
Whole culture, or culture supernatant (after centrifuging at 13,000 rpm for 15 min at 4°C) collected at an OD620 of 0.2 was mixed with sample buffer, and boiled 4 times for 10 min at 100°C and vortexed for 1 min. The samples were stored at −20°C before separation by 12% SDS-PAGE and visualization by silver staining (Proteo Silver Staining Kit; Sigma).
Capsule negative staining with Congo Red
The capsule was visualized by mixing 1 μl Congo Red solution (2% Congo Red in H2O) with 1 μl of liquid culture (or a colony picked from plate) on a glass slide. The mixture was spread on the slide and let to air-dry. Cells were visualized with a Nikon Eclipse Ti microscope with phase contrast using a 100×oil-immersion objective. The capsule was visible as a clear area surrounding darker cell bodies on red background.
Electron microscopy
Cells were grown in C+Y at 37°C. At an OD620 of 0.23 (strain D39 cps2A::ermB lytR::kan) or 0.4 (D39), samples were fixed in 2% glutaraldehyde in sodium cacodylate buffer (pH 7.4) (TAAB Laboratory Equipment) overnight. Samples were processed by the Electron Microscopy Research Service of Newcastle University. Briefly, samples were incubated in 1% osmium tetroxide (Agar Scientific) for 1 h, and then dehydrated in an acetone-graded series before being impregnated with a graded series of epoxy resin (TAAB Laboratory Equipment) in acetone and finally embedded in 100% resin and set at 60°C for 24 hr. Cells were sectioned and counterstained with 2% uranyl acetate and lead citrate (Leica) before being imaged on a Philips CM100 Compustage Transmission Electron Microscope (FEI) with an AMT CCD camera (Deben).
Cloning of B. subtilis ywtF and purification and crystallization of YwtF
The genetic region encoding the soluble domain of YwtF (amino acids 44–322, Δtm-YwtF) was amplified from B. subtilis 168 genomic DNA by PCR, using the primers YwtF FW and YwtF RVII for the first reaction and this PCR product, with the primers YwtF FW and YwtF RV, for the second round of PCR. The PCR products were digested with the appropriate restriction enzymes (Table 2), ligated with similarly restricted pET28a, and transformed into E. coli XL-1 Blue, selecting for kanamycin resistance. The product of this reaction, pET28a-Δtm-YwtF, placed the open-reading frame for Δtm-YwtF under a T7, IPTG-inducible promoter and added a C-terminal hexahistidine tag to the expressed protein. Sequencing confirmed the correct DNA sequence of pET28a-Δtm-YwtF.
Expression and purification of Δtm-YwtF
pET28a-Δtm-YwtF was transformed into E. coli BL21(DE3) selecting for kanamycin resistance and grown at 37°C with shaking in LB media containing kanamycin to an OD595 of 0.6 before expression of Δtm-YwtF was induced by the addition of 1 mM IPTG. The temperature of the culture was reduced to 18°C and the growth was continued for a further 16 h before the cells were pelleted by centrifugation at 4,000 g at 4°C (Beckman J-26-XP, JLA 8.1000 rotor).
Cell pellets were resuspended in 20 ml of Ni-NTA buffer A (50 mM Tris-HCl [pH 8.0], 300 mM NaCl, and 10 mM imidazole) before the addition of 1 ml of 25×EDTA-free complete protease inhibitor (Roche Diagnostics) and 20 μl of DNAse (Sigma; final concentration of 5 μg/ml). The cell suspension was sonicated (Bandelin sonopuls HD2070) on ice for 3 min at 80% power and 70% cycle before being clarified by centrifugation at 20,000 g, 4°C for 25 min (Beckman J-26-XP with JA 25.50 rotor).
Soluble cell lysate was loaded manually using an ÄKTA prime (GE Healthcare) at 1 ml/min onto a 5 ml Ni2+-NTA (Ni-NTA) cartridge (Qiagen), pre-equilibrated in Ni-NTA buffer A. The column was washed with 40 ml of Ni-NTA buffer A to remove any nonspecifically bound proteins. The tightly bound proteins were eluted with Ni-NTA buffer B (50 mM Tris-HCl [pH 8.0], 300 mM NaCl, and 250 mM imidazole). The presence of protein was monitored by absorption at 280 nm and confirmed by SDS-PAGE of the collected fractions.
Fractions containing the protein of interest were pooled and concentrated (Amicon Ultra-15 10K; Millipore) to 1 ml before size-exclusion chromatography on a Superdex S75 HiLoad 16/60 (GE healthcare) column pre-equilibrated in gel filtration buffer (10 mM Tris-HCl [pH 8.0] and 250 mM NaCl) using an ÄKTA purifier (GE Healthcare). Eluted proteins were fractionated by volume and monitored by absorption at 280 nm. SDS-PAGE confirmed the presence and purity of Δtm-YwtF.
Crystallization
Crystals of Δtm-YwtF were obtained in hanging drops by mixing equal volumes of protein at 25 mg/ml in 10 mM Tris (pH 7.5), 100 mM NaCl with a well solution of 0.1 M HEPES (pH 7.5), 20% PEG-3350, 0.2 M MgCl2 at 22°C. Crystals were cryoprotected by first transferring them into a drop of the well solution, followed by a drop of well solution supplemented with 10% PEG 400, before flash freezing in liquid nitrogen.
Data collection and processing
Diffraction data were collected at the Diamond Light Source synchrotron on beamline I02. A total of 500 images of 0.4° oscillations were collected at a wavelength of 0.9795 Å for Δtm-YwtF. Diffraction data were integrated using MOSFLM 28 and scaled and reduced with SCALA. 15 The crystal structure was solved by molecular replacement using MolRep 42 from the CCP4 suite (Collaborative Computational Project, Number 4, 1994) and with the previously solved, lower-resolution crystal structure of Δtm-YwtF (unpublished; PDBid 3MEJ) as the search model. The correctly positioned protein model was subjected to one round of rigid body refinement in REFMAC5, 34 to optimize its location in the crystallographic asymmetric unit. The resultant protein model was subjected to numerous cycles of restrained refinement in PHENIX REFINE 1 interspersed with manual model building in COOT 14 until the refinement converged (Table 3).
Mass spectrometry of Δtm-YwtF
The purified protein was dialyzed extensively against 10 mM ammonium acetate (pH 8.0) and then concentrated to 1 mg/ml. Samples were analyzed at the Mass Spectrometry facility at the Astbury Centre for Structural Molecular Biology at the University of Leeds, United Kingdom.
Purification and crystallization of Cps2A(R267A)
Cps2A(R267A) was purified by Ni-NTA affinity and size-exclusion chromatography as described previously. 24 Crystals were grown in hanging drops with well solutions of either 0.1 M sodium acetate (pH 4.6), 8% PEG-4K or 0.2 M di-ammonium citrate (pH 5.0), 15% PEG-3350. The former crystals were cryoprotected by direct transfer to 0.1 M sodium acetate (pH 4.6), 6% PEG-4K, 30% ethylene glycol and then flash frozen in liquid nitrogen, whereas the latter were transferred to 0.2 M di-ammonium citrate (pH 5.0), 15% PEG-3350, 20% PEG 300 before flash freezing in liquid nitrogen.
Diffraction data were integrated using MOSFLM 28 for the crystals grown in acetate and with XDS 23 for the crystals grown in citrate; both datasets were scaled with SCALA. 15 An initial model was obtained by fitting the structure of the wild-type protein (PDB entry 2XXP 24 ) into the data by molecular replacement with PHASER. 29 The structures were subsequently refined by iterative rounds of refinement (with REFMAC 34 for acetate crystals and PHENIX 1 for citrate crystals) and rebuilding in COOT. 14
Phosphate release assay
Fifty-microliter samples of Cps2A and Cps2A(R267A) (8 mg/ml) were incubated for 5 days at 22°C in 20 mM di-ammonium citrate (pH 5.0). About 6 μl of a stock solution of 1 M Tris-HCl (pH 8.0) was added and the release of inorganic phosphate was measured using the colorimetric PiPer assay (Invitrogen).
Results
Based on the role of LCP proteins in the cell wall assembly process of B. subtilis, and the crystal structure of the soluble part of Cps2A, we inferred that the three homologs in S. pneumoniae (Cps2A, LytR, and Psr; Fig. 1) might have roles in the attachment of capsule and teichoic acid to peptidoglycan, which are the last steps in connecting the different cell wall polymers to build the final cell wall. To obtain more insights into their cellular function, we first localized fluorescent fusions of the three LCP proteins in S. pneumoniae using a recently published plasmid tool. 13
Cellular localization of GFP-Cps2A, GFP-LytR, and GFP-Psr
To localize the three members of the LCP family in S. pneumoniae, the cps2A, lytR, or psr genes were cloned into pJWV25 in such a way that their 5′-ends were fused to gfp+. 13 The resulting plasmids were transformed into the type 2 capsule strain D39 or the nonencapsulated strain R6. The expression of GFP+-LCP fusion proteins was induced by the addition of Zn2+ to the growth medium. SDS-PAGE/western blot analyses of cell lysates confirmed that GFP+ fusion proteins of Cps2A, LytR, or Psr with the correct size were present in the lysate of the corresponding expression strains, detected with an anti-GFP antiserum, whereas no GFP band was visible in negative control lysates (Fig. 2A). Importantly, GFP bands with lower molecular weights were either not detectable, or had a low intensity, indicating that the expressed GFP-fusion proteins were not significantly degraded in the cell.
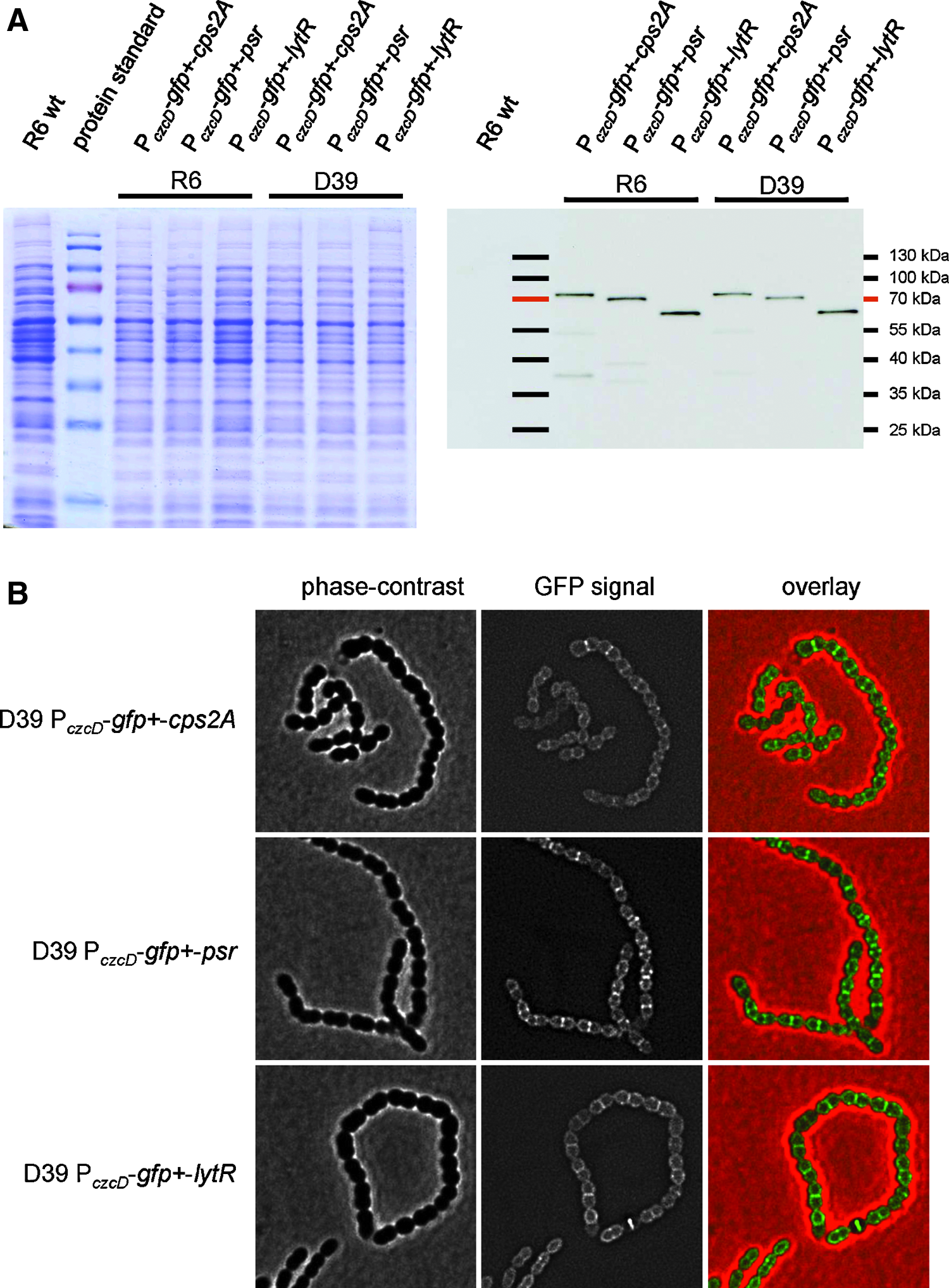
Cellular localization of GFP+-Cps2A, GFP+-Psr, or Gfp+-LytR.
Next, we localized the GFP+ fusion proteins in D39 P czcD -gfp+-cps2A, D39 P czcD -gfp+-psr, and D39 P czcD -gfp+-lytR strains by fluorescence microscopy. 13 All three GFP-fusion proteins localized at the cell membrane (Fig. 2B), consistent with the predicted membrane anchor present in Cps2A, Psr, and LytR (Fig. 1B). Interestingly, all three proteins were enriched at the septal, mid-cell region of the cells, and this effect was most apparent for GFP+-Psr (Fig. 2B). A similar localization pattern was observed in the nonencapsulated strain R6 (data not shown), indicating that the membrane localization of the three proteins, and their enrichment at the septum, does not depend on the presence of the capsule.
Capsule attachment in lcp mutant strains grown in liquid medium
In B. subtilis, the three LCP proteins have recently been implicated in the attachment of anionic polymers to the cell wall peptidoglycan. Because the B. subtilis LCP proteins appear to have redundant roles in vivo, we aimed to test the contribution of each of the pneumococcal LCP proteins in the capsule attachment to the cell wall. For this, we have produced strains D39 cps2A::ermB, in which cps2A is replaced by an erythromycin cassette allowing the expression of the downstream capsule genes under their native promoter, and D39psr, in which the psr gene is interrupted by the integrative plasmid pJDC9. Despite several attempts, we were unable to stably grow transformants of D39 lytR::kan in liquid C+Y growth medium (see next section). When growing in liquid medium, strain D39 cps2A::ermB contained a reduced amount (29%) of total capsule material compared with the parental D39 strain (Fig. 3), confirming previous studies. 31 D39psr contained slightly reduced amount of capsule (71%). Like the parental D39 strain, the cps2A and psr mutant strains did not release significant amounts of capsule into the supernatant, indicating that the capsule, although present in reduced amount, remains attached to the cell surface (Fig. 3). We next aimed to investigate the role of lytR in the attachment of capsule to the cell wall.
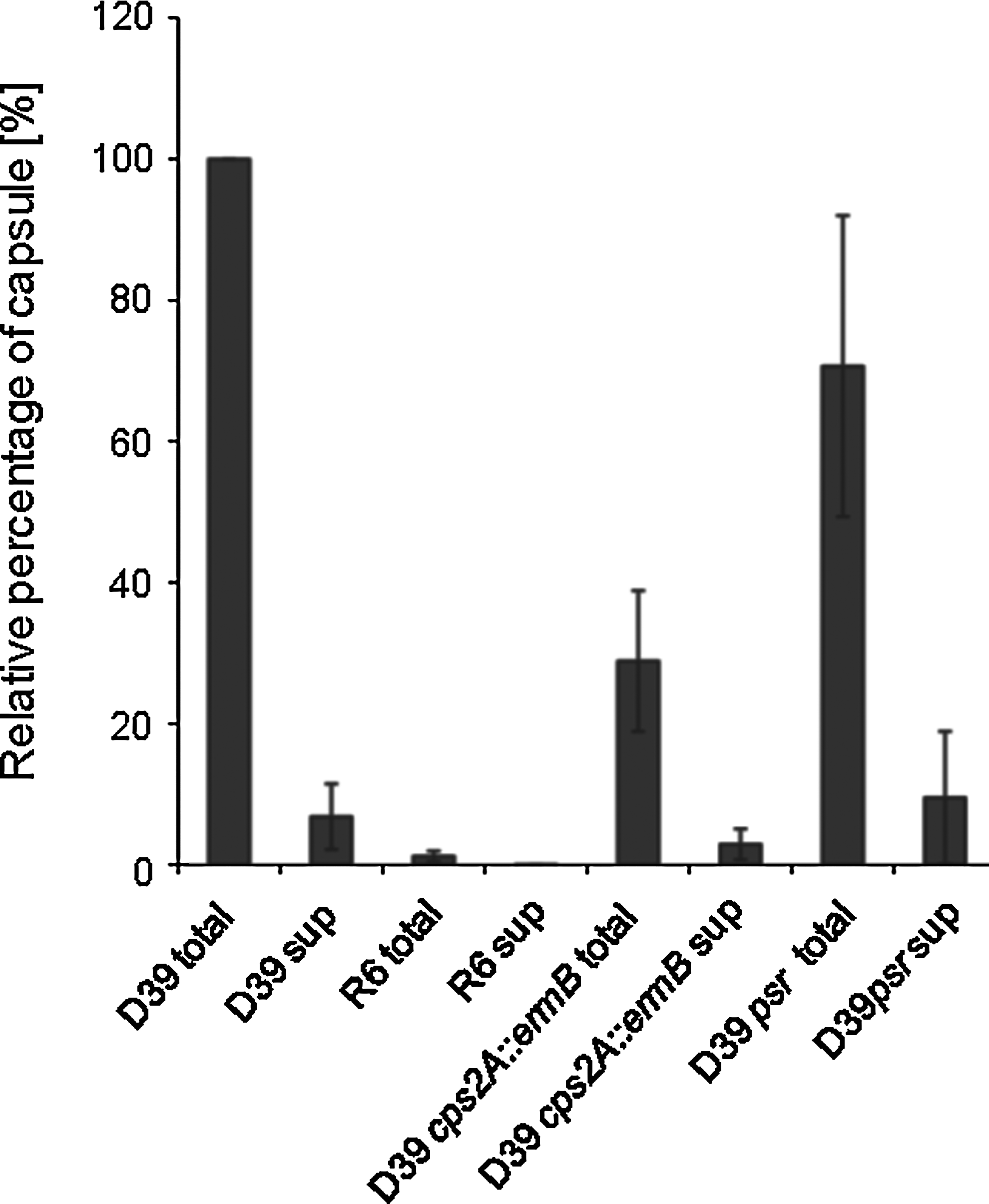
The relative percentages of capsule in total cell lysates and in culture supernatants of various strains. Capsules were quantified by dot blot analysis using an anti-type 2 capsule antiserum. The values are the mean±SD of three independent samples.
Capsule attachment in cps2A lytR cells
Inactivation of lytR in the nonencapsulated strain R6 leads to a severe phenotype where cells are mis-shapened and enlarged and often display misplaced septa. 22 Transformation of the encapsulated strain D39 with DNA from R6 lytR::kan yielded small colonies but we were unable to propagate these cells in liquid culture. Therefore, we first aimed to use strains with a Zn2+-controlled lytR gene, allowing its repression in the absence of Zn2+ in the growth medium. 13 We constructed D39 cps2A::ermB lytR::kan P czcD -lytR, which contains the lytR::kan and cps2A::ermB mutations and, in addition, an ectopic copy of lytR at the bgaA site under the control of the Zn2+-inducible promoter P czcD . However, this strain readily lost the repression of the ectopic lytR gene in the absence of Zn2+, resulting in constitutive expression of lytR from the bgaA site. Presumably, there is a strong selective pressure to generate suppressor mutations in the Zn2+ induction system leading to the expression of lytR without Zn2+.
Because none of the encapsulated lytR mutants grew stably in liquid culture, we next quantified the amount of capsule in whole colonies obtained after transforming strains D39 or D39 cps2A::ermB with DNA from D39 cps2A::ermB lytR::kan P czcD -lytR with selection for kanamycin resistance (Fig. 4). The state of the capsule in these transformants was also visualized by microscopy after staining with Congo Red. Interestingly, while the D39 transformants were homogenous, the kanamycin-resistant D39 cps2A::ermB transformants produced two visibly different types of colonies that differed in the amount of capsule present (Fig. 4). The majority of transformants, herein referred to as D39 cps2A::ermB lytR::kan, were small and contained a low amount of, or perhaps no, capsule, comparable to that of R6 lytR::kan transformants. We were not able to grow cells from this colony type in liquid medium. At low frequency, we also obtained larger colonies containing capsule, although at a reduced level to the D39 transformants. We conclude that these transformants have acquired a secondary, as yet unknown, mutation that allows the retention of the capsule in the cps2A lytR background. These transformants could be grown in liquid culture (see next section), and we refer to this suppressor strain as D39 cps2A::ermB lytR::kan (S). DNA from both types of kanamycin-resistant D39 cps2A::ermB transformants was extracted, and we confirmed by PCR analysis that the cps2A gene was inactivated by the erythromycin cassette. We also confirmed that the bgaA region was unchanged and the lytR gene could not be amplified. In addition, in both transformants, the psr gene sequence was identical to that in D39. Hence, both types of transformants were devoid of cps2A and lytR and had an intact psr gene, indicating that the differences in the amounts of capsules were not due to a difference in their LCP genes.
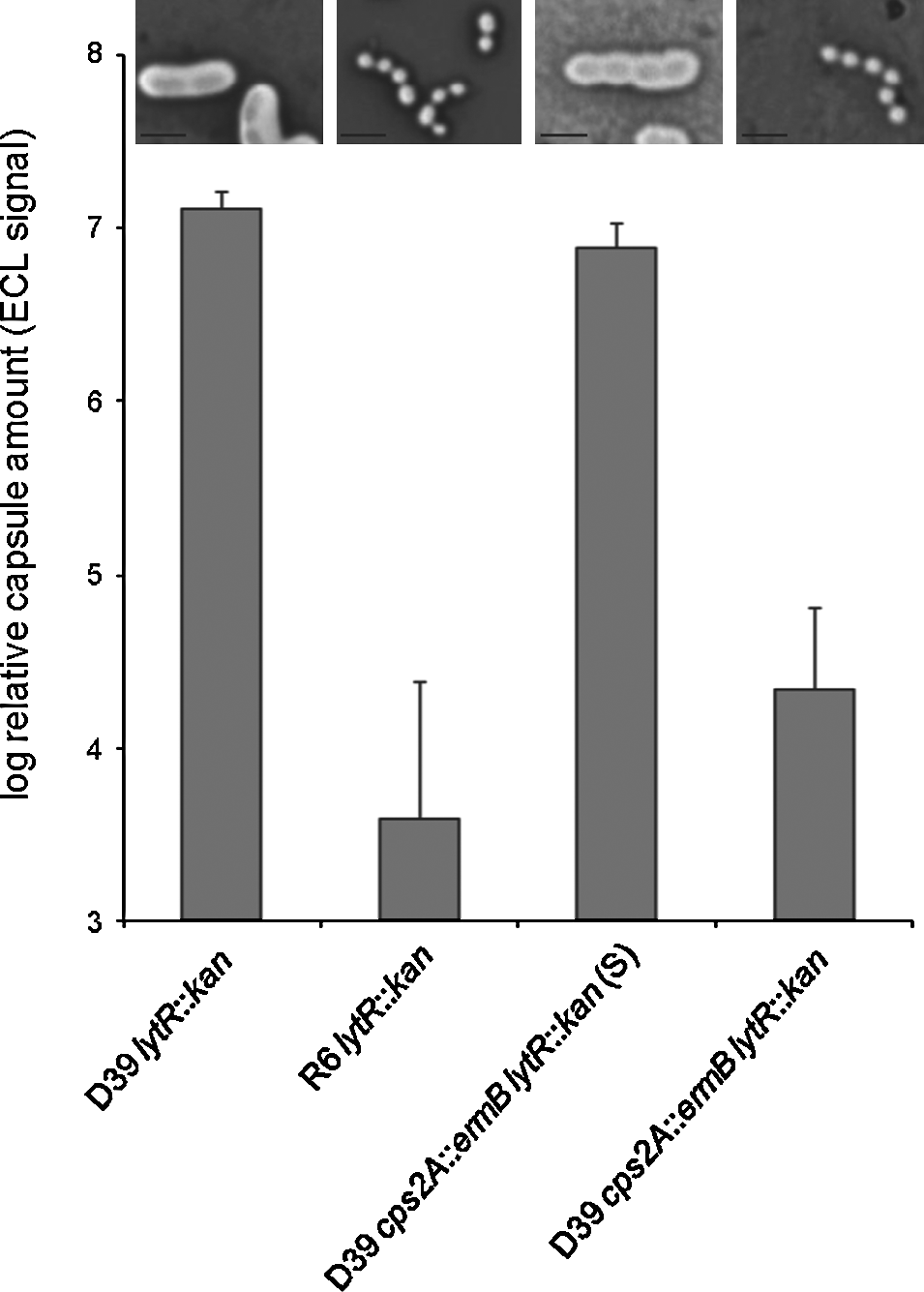
Relative capsule amount in whole colonies of transformants. Strains were transformed to introduce the lytR::kan mutation. Whole colonies of transformants were taken and resuspended, and the cells were lysed prior to quantification of the capsule by dot blot analysis. The values are the mean±SD of three independent samples. The suppressor mutant D39 cps2A::ermB lytR::kan (S) contained far less capsule than D39 lytR::kan, whereas D39 cps2A::ermB lytR::kan and R6 lytR::kan have virtually no capsule. Micrograph insets above the histograms show capsule-stained cells of the strains.
The cps2A lytR (S) suppressor mutant loses capsule material into the supernatant
The suppressor strain D39 cps2A::ermB lytR::kan (S) was able to grow in liquid C+Y growth medium, although growth was strongly impaired as indicated by the slower generation time compared with single cps2A, lytR, or psr mutant; the double cps2A psr mutant; and the parental strain (Fig. 5A). The cps2A lytR (S) suppressor mutant grew only to an optical density of 0.2–0.3 before lysing when inoculated from a fresh agar plate, and the strain did not grow at all when it was inoculated from liquid culture.
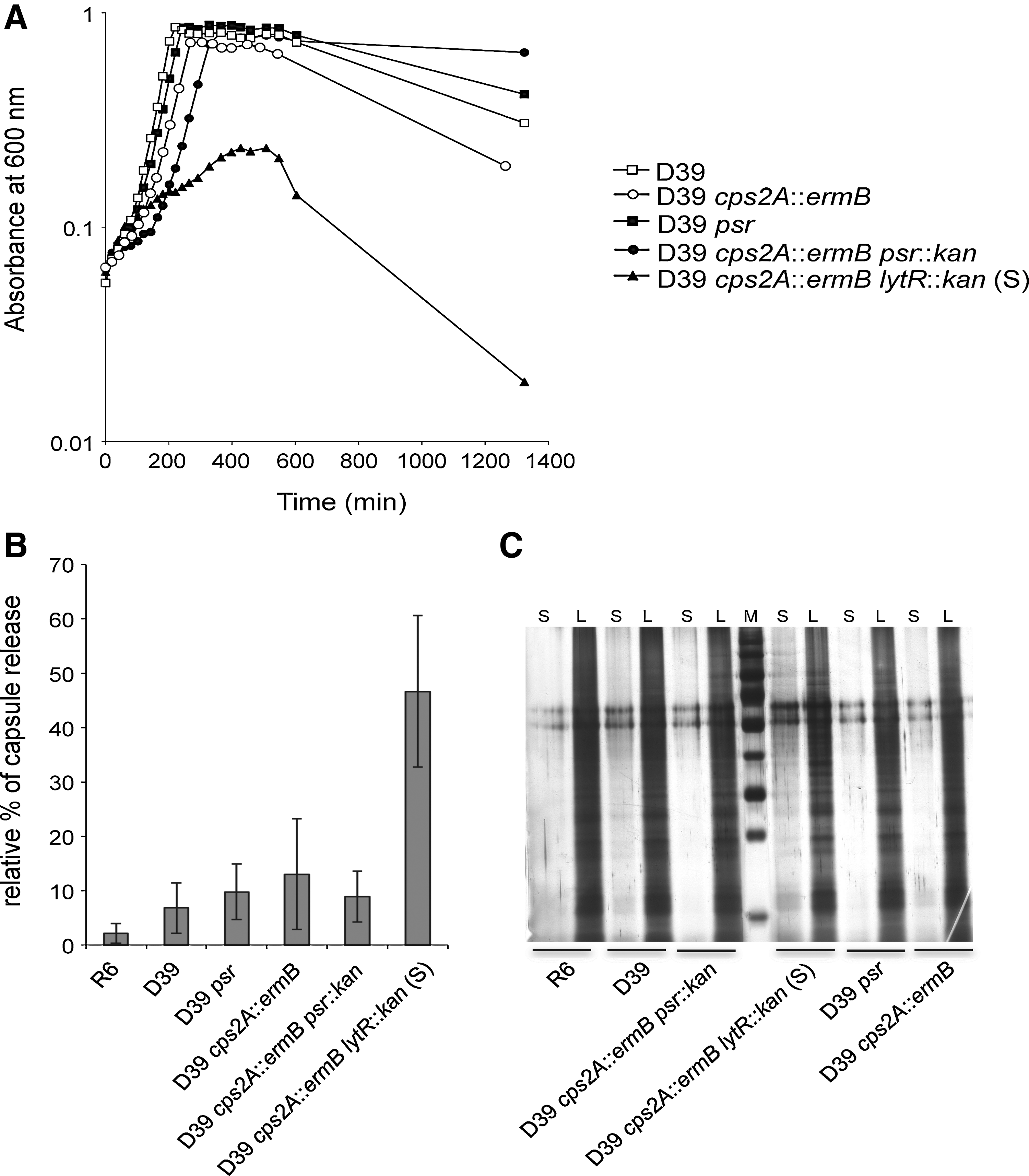
Characterization of the suppressor mutant D39 cps2A::ermB lytR::kan (S).
We next quantified the capsule material in the supernatant of these cultures relative to the total capsule present in lysate (Fig. 5B). Interestingly, the D39 cps2A::ermB lytR::kan (S) suppressor strain released as much as 47% of its total capsule material into the supernatant, which was significantly more than the parental strain, the cps2A and psr single mutants, and the cps2A psr double mutant. The higher amount of capsule in the supernatant of D39 cps2A::ermB lytR::kan (S) was not due to lysis of cells, because the supernatant of this strain contained similar levels of proteins to those of the other strains (Fig. 5C).
When grown in liquid medium, D39 cps2A::ermB lytR::kan (S) produced 31% of the total capsule amount of D39 (data not shown), which is a similar reduction to that seen when the strain is grown on agar plates (Fig. 4). Indeed, electron micrographs of thin sections of cells from liquid culture show a strong reduction in surface-attached capsule compared with strain D39 (Fig. 6). Further, cells of D39 cps2A::ermB lytR::kan (S) were inhomogeneous in size and had misplaced septa, similar to R6 lytR mutant cells. Taken together, these data show that LytR is required for normal cell morphology and that it contributes with Cps2A to retaining the capsule at the cell surface.
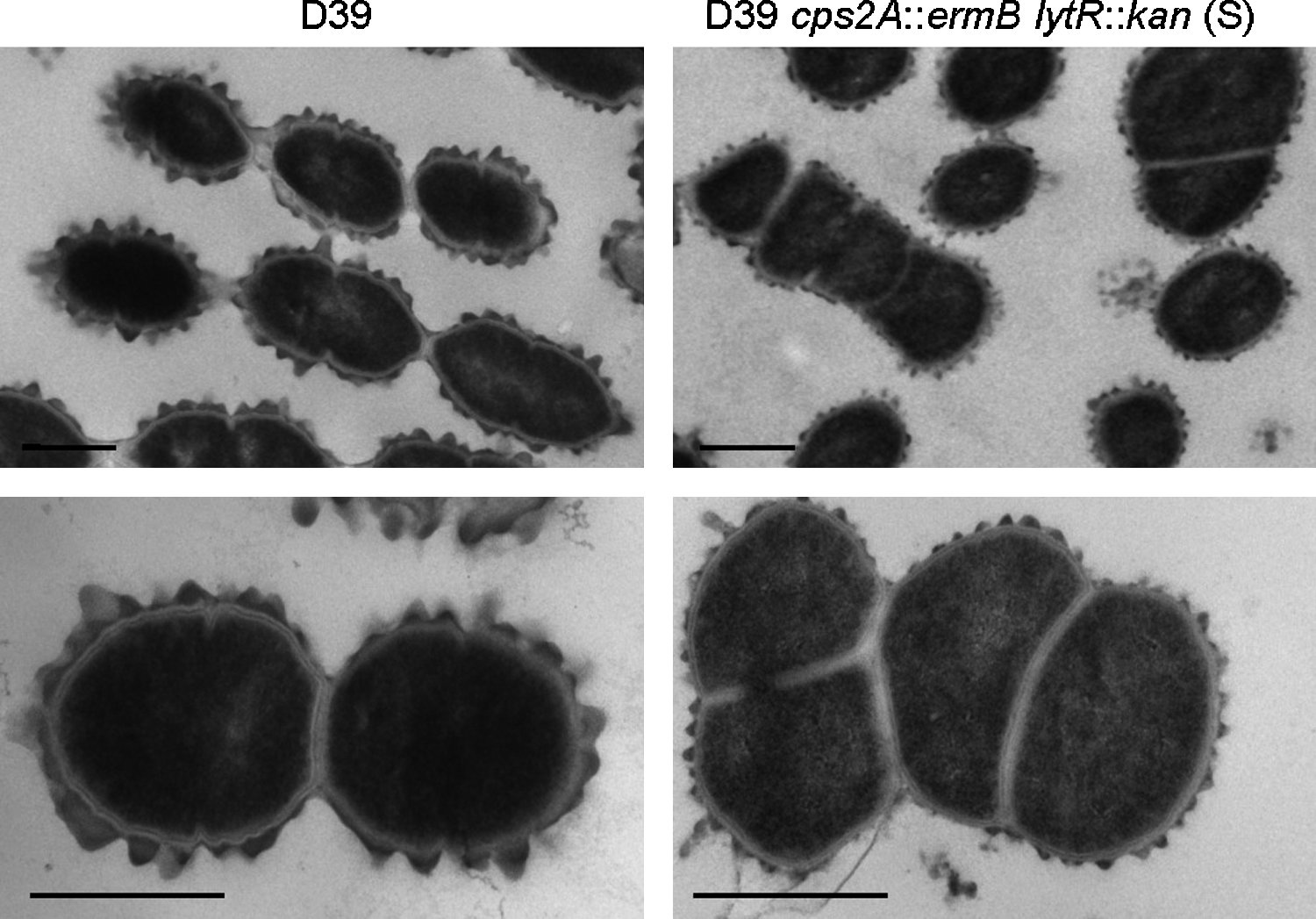
Visualization of surface-attached capsule in D39 and D39 cps2A::ermB lytR::kan (S). Electron micrographs show reduced amount of cell-surface-associated capsule and aberrant septum placement and cell size. Bar, 0.5 μm.
Crystallographic evidence for polyprenol pyrophosphatase activity of Cps2A
In a previous report we showed that Cps2A and the B. subtilis homolog YwtF have a pyrophosphatase activity toward polyprenoid pyrophosphate lipid substrates, a reaction resembling the transfer of teichoic acid or capsular polysaccharides onto peptidoglycan. 24
We have now obtained crystallographic evidence that corroborates the ability of Cps2A to hydrolyze the phosphate-phosphate bond in polyprenol pyrophosphate lipids. When the extracellular domain of Cps2A is overexpressed in E. coli, it co-purifies with endogenous polyisoprenoid phosphate lipids. Such Cps2A preparations have been crystallized as complexes with endogenous octaprenyl-pyrophosphate and decaprenyl-phosphate lipids. 24 We have subsequently found that the crystallization conditions affect the nature of the lipid bound to the protein. Identical batches of the Cps2A(R267A) mutant were crystallized under similar, but slightly different, conditions (0.1 M sodium acetate [pH 4.6] 8% PEG-4K vs. 0.2 M di-ammonium citrate [pH 5] 17% PEG-3350) and both crystal forms had the same space group with near-identical unit cell dimensions. In the structure of the wild-type Cps2A, Arg267 interacts with the lipid β-phosphate. The Cps2A(R267A) mutant protein proved to be, serendipitously, particularly amenable to crystallography. The major lipid in this preparation of Cps2A(R267A) was shown by mass spectroscopy to be octaprenyl-pyrophosphate. 24 However, in the refined crystal structures (refinement statistics in Table 3), the lipid composition of the crystallized protein differed. The crystals grown in acetate buffer clearly show a pyrophosphate lipid head group, even in electron density maps with phases calculated from only the protein atoms (Fig. 7A). In the crystals grown from citrate buffer, only a monophosphate lipid is visible. Further, an arginine residue, Arg242, has a significantly different position in the two crystal forms, which can be attributed to a salt-bridging interaction between Arg242 and the lipid β-phosphate.
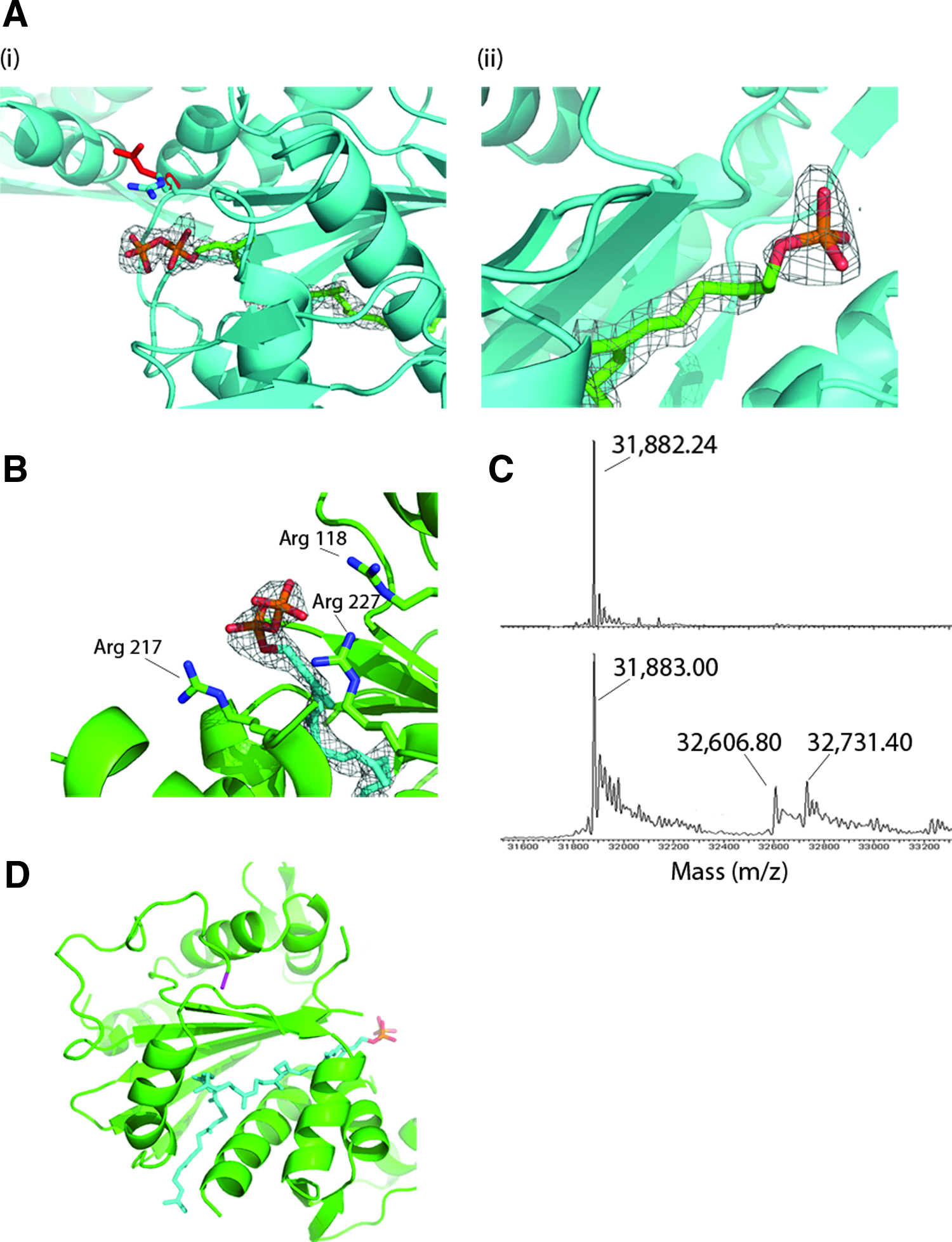
Crystallographic evidence for pyrophosphatase activity of LCP proteins.
As the crystals form over a much longer timescale in citrate buffer, not even appearing under a microscope for a month, the difference in lipid content is likely to be attributable to a change in the lipid content of the sample occurring during crystallization, presumably because of the pyrophosphatase activity described previously for Cps2A. 24 The Cps2A(R267A) mutant does indeed have pyrophosphatase activity in the di-ammonium citrate buffer used for crystallization. Using a colorimetric PiPer assay, a sample of 190 μM Cps2A(R267A) released 67±1.5 μM of inorganic phosphate after 5 days of incubation at 22°C in 20 mM di-ammonium citrate (pH 5.0). It might be argued that the Cps2A(R267A) sample contains a pre-existing mixture of lipids and the change in conditions favors crystallization of a particular protein-lipid complex from the mixture. However, it is hard to reconcile such a dramatic change in behavior with such a slight change in buffer pH and with the identical crystal packing that exists in the two crystal forms. Further, there are no obvious interactions of buffer ions with the lipid-binding site that could explain any buffer-specific stabilization of either protein-lipid complex. A change in lipid composition over the lengthy timescale of crystallization in citrate buffer, due to the innate pyrophosphatase activity of Cps2A, therefore, appears to be the most plausible explanation for this major change in crystallization behavior.
The B. subtilis LCP protein YwtF binds polyisoprenoid lipids
To simplify analysis of the proposed enzymatic function of LCP proteins, we sought to identify a homolog that could be isolated with an empty lipid-binding site. Structures of seven other LCP family proteins are currently in the PDB, none of which report a bound lipid molecule, though polyethylene glycol fragments have been built in the hydrophobic tunnel in PDB entry 3OWQ. The B. subtilis homolog of Cps2A, YwtF, was purified using a method similar to that of Cps2A and subjected to sparse matrix crystallization screening. Crystals were formed under identical conditions to those reported in the PDB (entry 3MEJ) and diffracted to a maximum resolution of 1.79 Å. 3MEJ was used as a starting model for refinement and was refined to an Rfree of 0.24. This structure determination revealed a ribbon of electron density that corresponds to the bound lipid molecule when the structure of the Cps2A-lipid complex is superimposed on YwtF.
Electrospray mass spectrometry was subsequently used to assess whether the sample contained bound polyisoprenoid lipids (Fig. 7C). Spectra were recorded under both native and denaturing conditions and were consistent with the mass of the intact protein, minus initiating methionine (31,883 Da). Additional peaks at 32,607 Da and 32,731 Da were uniquely present in the native spectra. The differences in mass relative to the peak from the intact protein, 724 and 849 Da, are consistent with octaprenyl-pyrophosphate and undecaprenyl-phosphate, respectively, lipids which have been identified to co-purify with Cps2A. 24 Octaprenyl-pyrophosphate provided a better fit to the electron density map in the hydrophobic tunnel than the undecaprenyl-phosphate; the crystal packing also did not allow sufficient space to accommodate the longer lipid. The lipid most convincingly filled the electron density in the all-cis configuration, rather than in the all-trans configuration (Fig. 7D), particularly for the isoprenoid group adjacent to the pyrophosphate. That the enzyme is stereospecific selective for lipid binding is underlined by fact that trans-polyprenoid lipids are more abundant in E. coli than cis-polyprenoid lipids, because of their roles as intermediates in quinone biosynthesis. 40 The electron density can accommodate di-trans, penta-cis octaprenyl-pyrophosphate, in which the two chiral double bonds furthest from the head group have the trans configuration. Di-trans, penta-cis octaprenyl-pyrophosphate is an intermediate in the synthesis of undecaprenyl-pyrophosphate 40 and may be sequestered preferentially by LCP proteins when expressed in the cytoplasm. Indeed, the final Rfree in refinement was in actual fact at its lowest (0.22) when di-trans, penta-cis octaprenyl-pyrophosphate was modeled into the active site.
Attempts to model the longer lipid undecaprenyl-phosphate into the electron density resulted in a steric clash between the hydrophobic tail of the lipid and an adjacent protein molecule in the crystal lattice. In full-length YwtF the hydrophobic tail may extend out of the protein tunnel to interact with the membrane. This potential “exit” path from the hydrophobic lipid tunnel is reasonably orientated with respect to the N-terminus of the protein, to which the transmembrane helix is attached, for this to occur (Fig. 7D). Overall, these structural results support further an enzymatic role of LCP proteins in the transfer of phosphorylated chains of anionic cell wall polymers from an undecaprenyl-pyrophosphoryl-linked precursor to peptidoglycan on cleavage of the pyrophosphoryl bond.
Discussion
LCP proteins are widespread in Gram-positive bacteria and are often present in multiple versions in one species. Lcp mutant strains of various species show pleiotropic phenotypes and, therefore, LCP proteins have been suggested to be involved in the regulation of some aspects of cell wall metabolism. However, a recent publication 24 provided strong evidence that LCP proteins are not regulatory factors, but are in fact phosphotransferase enzymes for one of the last steps in cell wall assembly, the attachment of anionic cell wall polymers like capsular polysaccharides and teichoic acids, to peptidoglycan. This hypothesis is strongly supported by crystal structures of the extracytoplasmic part of pneumococcal Cps2A, which, depending on the E. coli expression strain from which it was produced, contained either a phosphorylated or pyrophosphorylated polyprenol lipid; these are the substrate and product mimics of the phosphotransferase reaction. The reaction presumably leads to the teichoic acid or capsule chains becoming attached to C6-OH of MurNAc residues in peptidoglycan by the formation of a phosphodiester bond. It is this last reaction in cell wall synthesis, connecting the major cell wall polymers with each other, which produces the final and functional cell wall in Gram-positive bacteria. Perhaps, it is therefore not surprising that affecting this reaction often leads to pleiotropic phenotypes as observed in lcp mutants.
The aim of this work was to investigate the role of the three pneumococcal LCP proteins in the attachment of the capsule to the cell wall. One of them, CpsA, is encoded by the first gene of the large capsular gene cluster present in the different serotypes. A noticeable exception is serotype 3, which does not contain an lcp family gene in its capsule cluster and which does not covalently attach its capsule to peptidoglycan. Cps2A, the CpsA protein present in serotype 2 strains like D39, has been implicated previously in the attachment of capsule to the cell wall because a cps2A mutant strain has a reduced quantity (∼30%–40%) of cell-wall-attached capsular polysaccharide. Because the residual capsule material is attached to the cell wall, we hypothesized that other LCP proteins, LytR and Psr, might contribute to capsule attachment in the absence of Cps2A. We also considered the possibility that Cps2A, LytR, and Psr attach teichoic acid chains to peptidoglycan to form wall teichoic acid, or attach teichoic acid chains to the glycolipid anchor to form lipoteichoic acid. Either there is one enzyme for each of these three attachment reactions, or LCP proteins have (semi)redundant roles in cell wall assembly. The latter possibility is supported by the available crystal structures of Cps2A and its B. subtilis homologs, which bind the lipid and pyrophosphoryl part of the anionic polymer precursor strongly. These structures also display a surface groove for binding peptidoglycan chains, but they do not appear to recognize much or any of the repeating chain of the anionic polymer. Hence, it is possible that some of these enzymes are “nonspecific” in that they are able to attach different polyprenol pyrophosphoryl-linked polymers to peptidoglycan. This hypothesis is supported by the work presented in this article, which shows that LytR, at least, and perhaps Psr contribute to capsule–cell wall attachment in the absence of Cps2A. A role in cell wall assembly is also supported by the localization of GFP-fusions of all three LCP proteins at mid-cell (Fig. 2) where the pneumococcal cell wall grows. The fusion proteins also localized at mid-cell in the capsule-free strain R6 (not shown), indicating that capsule precursors are not required for their localization, supporting the hypothesis that LytR and Psr are active in cell wall assembly, presumably in the final assembly of teichoic acids. However, the precise role of each pneumococcal LCP protein in teichoic acid assembly remains to be determined.
D39 strains that lacked cps2A but expressed all other capsule genes had significantly reduced amounts of capsule material (Fig. 3), in confirmation of a previous report and indicating that LytR and Psr cannot complement fully the lack of Cps2A in the retention of the capsule. A psr mutant had slightly reduced amounts of capsule. Perhaps, Psr contributes to capsular assembly, or in this mutant Cps2A and LytR take over the Psr function to result indirectly in a reduced amount of capsule.
We were also unable to obtain a stable lytR mutant in the D39 background. LytR can be inactivated in the capsule-free R6 strain, leading to abnormal cell morphology and reduced growth. Presumably, the presence of a fully functional capsule assembly pathway adds to the growth problems of lytR mutants that prevent the isolation of viable mutants. Consistent with this premise, the inactivation of lytR in the D39 cps2A::ermB background, which contained less capsule material than D39 (Fig. 3), produced viable transformants. Interestingly, we have obtained two types of transformants, both of which were identical genetically, in that they both contained the cps2A and lytR deletions and retained an intact psr. The more abundant transformant type had little, if any, capsule and was unable to grow in liquid medium; we infer that this is the true phenotype of a D39 cps2A lytR double mutant. The second type of transformant, less abundant and most likely a D39 cps2A lytR double mutant with one or more suppressor mutation(s), contained capsule and was able to grow in liquid medium, although at a slow growth rate and only for one culture cycle. Interestingly, these cells lost massive amounts of capsule material into the growth medium without showing significant lysis, indicating that the capsule material was secreted into the growth medium.
The most likely explanation for our results is as follows. All three LCP homologs have semiredundant roles in the attachment of capsular polysaccharide and teichoic acid precursor chains to peptidoglycan, and perhaps of teichoic acid precursor chains to the glycolipid anchor. Deleting one or two of the corresponding genes results in an imbalance in the assembly of cell wall polymers causing growth defects, in particular when lytR is deleted. This indicates that LytR has specific function(s) that cannot readily be taken over by the two homologs. It could be expected that lcp mutants accumulate lipid-linked teichoic acid or capsule precursors in their cell membrane. However, in several lcp mutant strains investigated, the lipid-linked precursors do not accumulate but the final cell wall polymer is present at a reduced level. We hypothesize that the cytoplasmic synthesis and polymerization of teichoic acid and capsular polysaccharide chains, their transport across the membrane, and their attachment to the cell wall peptidoglycan are coupled, explaining the reduced amount of anionic polymers in lcp mutant strains (Figs. 2, 5, and 6). In the D39 cps2A lytR suppressor strain, this feedback regulation appears to be lost and thus the strain produces an excess of capsule material that cannot be retained in the cell wall and is lost into the culture supernatant. We are currently investigating how the capsule material is released from the cell membrane in this strain, and which genes are mutated to produce this phenotype.
At the molecular level, the new structures reported here bolster the evidence that LCP proteins are the phosphotransferases that catalyze the transfer of anionic cell wall polymers (teichoic acids and capsular polysaccharides) from lipid-linked intermediates onto peptidoglycan. First, the head group of the lipid changes when the timescale of crystallization is extended, consistent with pyrophosphorolysis of the lipid occurring within a crystallization drop. This pyrophosphorolysis is consistent with our previous observation that inorganic phosphate accumulates in purified preparations of the Cps2A-octaprenyl-pyrophosphate complex. 24 The pyrophosphorolysis reaction has chemistry resembling that of the phosphotransfer of teichoic acid or capsule onto peptidoglycan.
Second, we have also shown that the B. subtilis LCP protein YwtF binds lipids that resemble the substrate of the phosphotransfer reaction. The presence of exogenous E. coli octaprenyl-pyrophosphate lipid in the crystallized protein, in addition to undecaprenyl-phosphate, was confirmed by mass spectroscopy. On examination of the structure of YwtF bound to octaprenyl-pyrophosphate, many of the interactions between charged residues and the lipid head group resemble those seen in Cps2A. 24 The only exception is Asp82 (equivalent to in Asp234 in Cps2A). In Cps2A, Asp234 coordinates a magnesium ion that interacts with the pyrophosphate head group, whereas in YwtF, Asp82 is more distant from the phosphate and is at the start of a disordered loop region that could not be modeled. In Cps2A, this loop region is in close proximity to an additional protein domain that is unique to Cps2A. The proximity of this domain may stabilize the loop by restraining its conformational mobility, which may in turn enable Asp234 to coordinate the magnesium ion. The disorder in this loop in YwtF may explain why there is no magnesium bound to the pyrophosphate head group in the YwtF structure, despite the presence of 0.2 M MgCl2 in the crystallization conditions. Modulation of the mobility of this loop due to interactions with other components of the cell wall synthetic machinery could in fact provide a mechanism to regulate the activity of the enzyme.
The disorder in this loop may also explain the apparent weaker binding of the lipid to YwtF. In comparing the crystallographic B factors of the lipid to those of the surrounding protein, the octaprenyl-pyrophosphate appears to be more mobile in the YwtF structure (Table 3). Further, purified preparations of YwtF catalyze the pyrophosphorolysis of exogenously added geranyl pyrophosphate but Cps2A, purified in the same manner, is inactive in the same assay. 24 These observations suggest that the endogenous E. coli lipids are more loosely bound to YwtF than in Cps2A, and that the active site is, therefore, more accessible to exogenously added substrates.
In summary, our genetic data have provided insights into the complex mechanisms of capsule attachment in S. pneumoniae, pointing to a semiredundant role of all three LCP proteins in this process. Our structural data have reinforced the proposed role of LCP proteins as the enzymes that catalyze the transfer of teichoic acid and/or capsular polysaccharides onto peptidoglycan. Although the precise role each of these enzymes plays in cell wall assembly has yet to be defined, their inactivation would seem likely to impair pneumococcal growth and/or survival at an infection site. Therefore, we consider Cps2A and its homologs as valid targets in the search for antimicrobials that target pneumococci and, potentially, other Gram-positive pathogens.
Footnotes
Acknowledgments
This work was supported by a grant from the UK Biotechnology and Biological Sciences Research Council to W.V. and R.J.L. The authors thank the Electron Microscopy Research Service of Newcastle University for sample preparation, the Mass Spectroscopy Service of Leeds University's Astbury Centre for mass spectroscopy, and Dr. Arnaud Balsé of Newcastle University and the staff of beamlines at Diamond Light Source for help during diffraction data collection.