
Abstract
Podoplanin (PDPN), a 36-kDa type I transmembrane O-glycoprotein, is expressed in normal cells, including renal epithelial cells (podocytes), lymphatic endothelial cells, and pulmonary type I alveolar cells, and in cancer cells, including brain tumors and squamous cell lung carcinomas. PDPN activates platelet aggregation by binding to C-type lectin-like receptor-2 (CLEC-2) on platelets, and PDPN/CLEC-2 interaction facilitates blood/lymphatic vessel separation. We previously produced an anti-human PDPN monoclonal antibody (mAb), clone NZ-1 (rat IgG2a, lambda) and its rat-human chimeric mAbs (NZ-8/NZ-12), which neutralize PDPN/CLEC-2 interactions and inhibit platelet aggregation and cancer metastasis. In this study, we first developed a humanized anti-human PDPN mAb, named as NZ-27. We further produced a core-fucose-deficient version of NZ-27, named as P1027 and a core-fucose-deficient version of NZ-12, named as NZ-12f. We investigated the binding affinity, antibody-dependent cellular cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), and antitumor activity of P1027 and NZ-12f. We demonstrated that the binding affinities of P1027 and NZ-12f against LN319 (a human glioblastoma cell line) are 1.1 × 10−8 and 3.9 × 10−9 M, respectively. ADCC reporter assays demonstrated that NZ-12f shows 1.5 times higher luminescence than P1027. Furthermore, NZ-12f showed 2.2 times higher ADCC than P1027, whereas both P1027 and NZ-12f showed high CDC activities against LN319 cells. Using LN319 xenograft models, P1027 and NZ-12f significantly reduced tumor development in an LN319 xenograft model compared with control human IgG. Treatment with P1027 and NZ-12f may be a useful therapy for patients with PDPN-expressing cancers.
Introduction
A 36-kDa type I transmembrane O-glycoprotein, podoplanin (PDPN)/Aggrus/T1α/gp36, is a platelet aggregation-inducing factor. It is expressed in many cancers, including malignant brain tumors, mesothelioma, and several squamous cell carcinomas.(1–10) Expression of PDPN is associated with cancer metastasis, epithelial–mesenchymal transition, malignant progression,(11–18) and clinical outcome.(19–21) Only a small and phenotypically distinct subset of cells in solid tumors are considered tumor-initiating cells (TICs) that are resistant to conventional therapies and responsible for relapse.(22) Therefore, targeting TICs could be useful for cancer therapy.(23) PDPN is reported to be a TIC marker(24); thus, immunotherapy using specific antibodies against PDPN may eradicate TICs in cancers.
We previously produced a rat anti-human PDPN monoclonal antibody (mAb), NZ-1.(5) NZ-1 is highly specific and sensitive, and displays high binding affinity against PDPN.(25,26) NZ-1 inhibited tumor cell-induced platelet aggregation and cancer metastasis.(12) NZ-1 is highly internalized into glioma cells, and is well accumulated into tumors in vivo, making NZ-1 a suitable candidate for therapy against malignant gliomas.(27) We constructed a single-chain antibody variable region fragment using NZ-1 (NZ-1-scFv).(28) NZ-1-scFv was then fused to Pseudomonas exotoxin A (NZ-1-scFv-PE38). NZ-1-scFv-PE38 demonstrated significant activity against glioblastoma and medulloblastoma cells, and induced tumor growth delay in D2159MG and D283MED using in vivo tumor models.
We previously produced rat-human chimeric anti-PDPN antibodies (NZ-8 and NZ-12) from NZ-1.(29,30) Light chains of NZ-8 and NZ-12 are human kappa and human lambda, respectively. NZ-12 possesses higher antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) compared with NZ-8.(30) NZ-12, together with human natural killer (NK) cells, showed antitumor effects against malignant pleural mesothelioma orthotopic xenograft mice.(31) Furthermore, radioimmunotherapy (RIT) with 90Y-labeled NZ-12 significantly suppressed tumor growth in an NCI-H226 mesothelioma xenograft mouse model, prolonging survival without loss of body weight or obvious adverse effects.(32)
Many studies have revealed that core-fucose-deficient antibodies are known to show higher ADCC activities.(33,34) Similarly, we previously produced a core-fucose-deficient version of anti-podocalyxin mAb (47-mG2a), which was named as 47-mG2a-f, to augment its ADCC activities.(35) As expected, 47-mG2a-f exhibited stronger ADCC activities than 47-mG2a against OSCC cell lines such as HSC-2 and SAS cells, leading to higher antitumor activities in those xenograft models. Furthermore, we recently developed a core-fucose-deficient mouse-canine chimeric anti-dog PDPN mAb (P38B), named as P38Bf.(36) We compared ADCC between P38B and P38Bf against Chinese hamster ovary (CHO)/dPDPN cells. Although both P38B and P38Bf revealed high ADCC activities against CHO/dPDPN cells, P38Bf demonstrated significantly higher ADCC compared with P38B, especially at low concentrations, indicating that core-fucose-deficient mAbs are very useful for increasing ADCC activities.
In this study, we produced a core-fucose-deficient version of a humanized anti-human PDPN mAb (NZ-27), named as P1027 and a core-fucose-deficient version of rat-human chimeric anti-human PDPN mAb (NZ-12), named as NZ-12f, and compared their binding affinity, ADCC/CDC activities, and antitumor activity.
Materials and Methods
Cell lines
LN319 was purchased from Addexbio Technologies (San Diego, CA). ExpiCHO-S was purchased from Thermo Fisher Scientific, Inc., (Waltham, MA). BINDS-09 (FUT8-knocked out ExpiCHO-S cells) was established in our previous study.(36) LN319 was cultured in Dulbecco's modified Eagle's medium (DMEM) medium (Nacalai Tesque, Inc., Kyoto, Japan), supplemented with 10% heat-inactivated fetal bovine serum (Thermo Fisher Scientific, Inc.) at 37°C in a humidified atmosphere of 5% CO2 and 95% air. ExpiCHO-S and BINDS-09 were cultured following manufacturer's instructions.
Antibodies
For the generation of a rat-human chimeric anti-human PDPN mAb (NZ-12), appropriate VH of rat NZ-1 antibody and CH of human IgG1 were subcloned into pCAG-Neo (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan), and VL of rat NZ-1 antibody and CL of human lambda chain were subcloned into pCAG-Ble vectors (FUJIFILM Wako Pure Chemical Corporation). For creation of a humanized anti-human PDPN mAb (NZ-27), the complementarity determining region (CDR) of VH in the NZ-1 antibody, frame sequences of VH in human Ig, and CH of human IgG1 were subcloned into the pCAG-Neo vector (FUJIFILM Wako Pure Chemical Corporation). The CDR of VL in the NZ-1 antibody, frame sequences of VL in human Ig, and CL of human lambda chain were subcloned into the pCAG-Ble vector (FUJIFILM Wako Pure Chemical Corporation). Antibody expression vectors of NZ-27 and NZ-12 were transfected into BINDS-09 using the ExpiCHO-S Expression System (Thermo Fisher Scientific, Inc.), respectively. Produced mAbs are named as P1027 and NZ-12f, respectively. P1027 and NZ-12f were purified using Protein G Sepharose 4 Fast Flow (GE Healthcare UK, Ltd., Buckinghamshire, England). Human IgG was purchased from Sigma-Aldrich Corp. (St. Louis, MO).
Determination of the binding affinity by flow cytometry
LN319 cells (2 × 105 cells) were resuspended with 100 μL of serially diluted antibodies (P1027 and NZ-12f; 0.006–100 μg/mL) followed by FITC-labeled anti-human IgG (Thermo Fisher Scientific, Inc.). Fluorescence data were collected using a cell analyzer (EC800; Sony Corp., Tokyo, Japan). Dissociation constants (KD) were obtained by fitting binding isotherms using built-in one-site binding models in GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA).
ADCC reporter gene assay
We performed the FcγRIIIa stimulation assay using an ADCC Reporter Bioassay kit from Promega (Madison, WI), following the manufacturer's instructions.(37) LN319 was inoculated into a 96-well white solid plate, 12,500 cells per well. Anti-PDPN mAbs were serially diluted and added to LN319 cells. Engineered, immortalized Jurkat T lymphocytes (Promega) expressing the FcγRIIIa receptor (75,000 cells in 25 μL) were then added and cocultured with antibody-treated target cells at 37°C for 6 h. Luminescence using the Bio-Glo Luciferase Assay System (Promega) was measured with a GloMax luminometer (Promega).
Atibody-dependent cellular cytotoxicity
Human NK cells were purchased from Takara Bio, Inc., (Shiga, Japan). Target cells were labeled with 10 μg/mL Calcein AM (Thermo Fisher Scientific, Inc.) and resuspended in the same medium. Target cells were plated in 96-well plates (1 × 104 cells/well) and mixed with effector cells, anti-PDPN antibodies, or control human IgG. After a 6.5-hour incubation, Calcein AM release to the supernatant from each well was measured. Fluorescence intensity was determined using a microplate reader (Power Scan HT; BioTek Instruments, Winooski, VT) with excitation and emission wavelengths of 485 and 538 nm, respectively. Cytolytic activity (% of lysis) was calculated as % lysis = (E − S)/(M − S) × 100, where E is the fluorescence of combined target and effector cells, S is spontaneous fluorescence of target cells only, and M is maximum fluorescence measured after lysing all cells with a buffer containing 0.5% Triton X-100, 10 mM Tris-HCl (pH 7.4), and 10 mM of EDTA.
Complement-dependent cytotoxicity
Target cells were labeled with 10 μg/mL Calcein AM and resuspended in the same medium. The target cells were plated in 96-well plates (2 × 104 cells/well) and mixed with either anti-PDPN antibodies or control human IgG and 10% of rabbit complement. After a 6.5-hour incubation, the Calcein AM release of supernatant from each well was measured. Fluorescence intensity was determined using a microplate reader (Power Scan HT) with an excitation wavelength of 485 nm and an emission wavelength of 538 nm. Cytolytic activity (% of lysis) was calculated as % lysis = (E − S)/(M − S) × 100, where E is fluorescence of combined target and effector cells, S is spontaneous fluorescence of target cells only, and M is maximum fluorescence measured after lysing all cells with a buffer containing 0.5% Triton X-100, 10 mM Tris-HCl (pH 7.4), and 10 mM of EDTA.
Antitumor activity of anti-PDPN antibodies in xenografts of LN319
Sixteen 6-week-old female BALB/c nude mice were purchased from Charles River and used at 7 weeks of age. LN319 cells (0.3 mL of 1.33 × 108 cells/mL in DMEM) were mixed with 0.5 mL BD Matrigel Matrix Growth Factor Reduced (BD Biosciences, San Jose, CA). Mice were injected subcutaneously in the left flank (100 μL) with the suspension mentioned earlier (5 × 106 cells). After day 1, 100 μg of P1027, NZ-12f, and control human IgG in 100 μL PBS were injected into mice through intraperitoneal injection (i.p.). Additional antibodies were injected on days 8 and 16. Human NK cells (8.0 × 105 cells) were injected around the tumors on days 1, 8, and 16. Twenty-one days after cell implantation, all mice were euthanized by cervical dislocation and tumor diameters and volumes were determined as previously described.(38)
Statistical analyses
All data were expressed as mean ± SEM. Statistical analysis used ANOVA and Tukey–Kramer's test using R statistical (R Foundation for Statistical Computing, Vienna, Austria). p < 0.05 was adopted as a level of statistical significance.
Results
Development of a core-fucose-deficient humanized anti-PDPN mAb (P1027) and a core-fucose-deficient rat-human chimeric mAb (NZ-12f)
In this study, we produced core-fucose-deficient humanized anti-PDPN (P1027) and rat-human chimeric antibodies (NZ-12f) (Fig. 1). Binding affinity of P1027 and NZ-12f against LN319 cells was 1.1 × 10−8 and 3.9 × 10−9 M, respectively, indicating that both mAbs possess high affinity for LN319 cells, but NZ-12f possesses higher binding activity (Fig. 2).
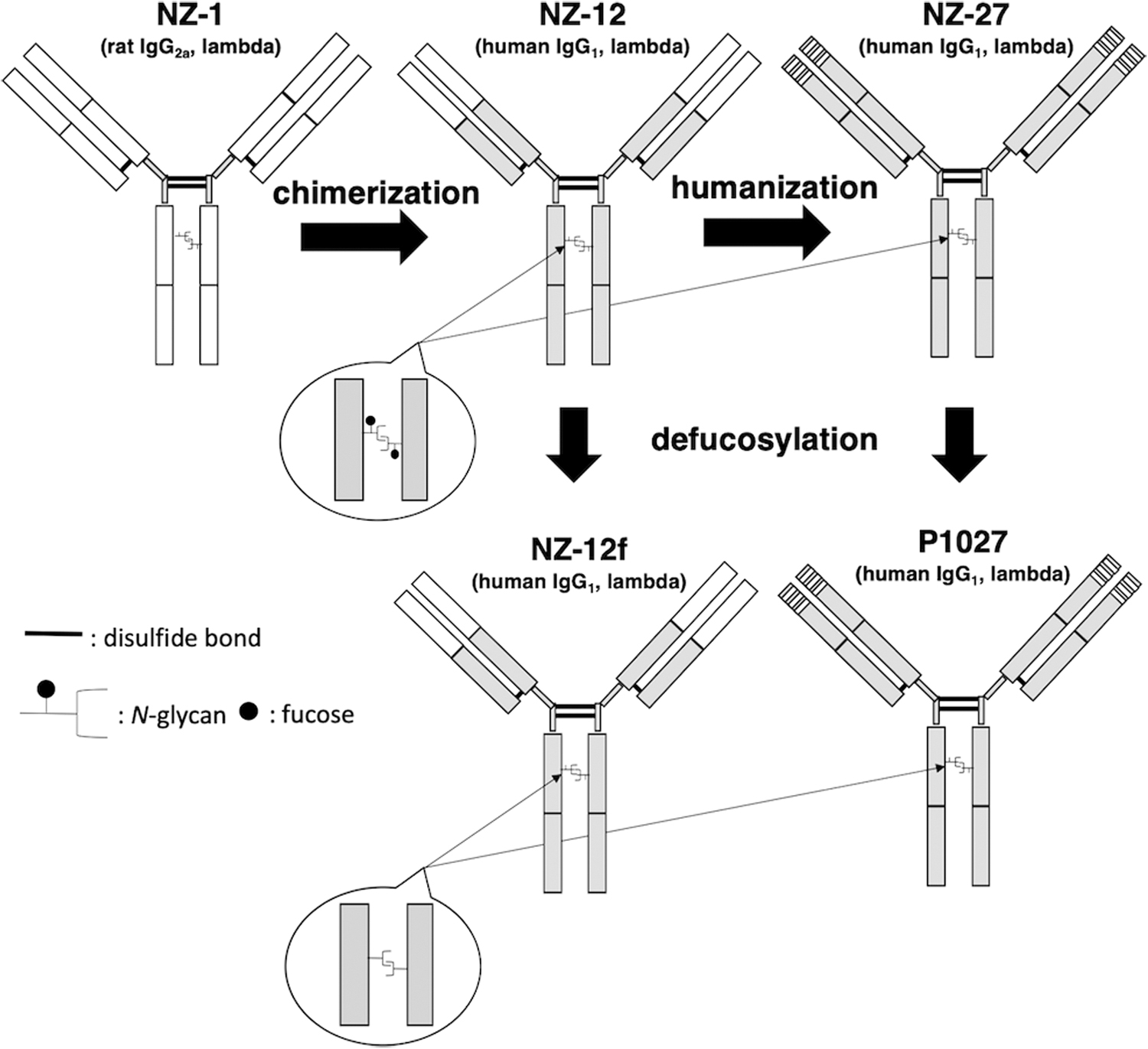
Development of a core-fucose-deficient humanized anti-PDPN mAb (P1027) and a core-fucose-deficient rat-human chimeric mAb (NZ-12f). White box, derived from NZ-1 (rat IgG); gray box, derived from human IgG. mAb, monoclonal antibody; PDPN, podoplanin.
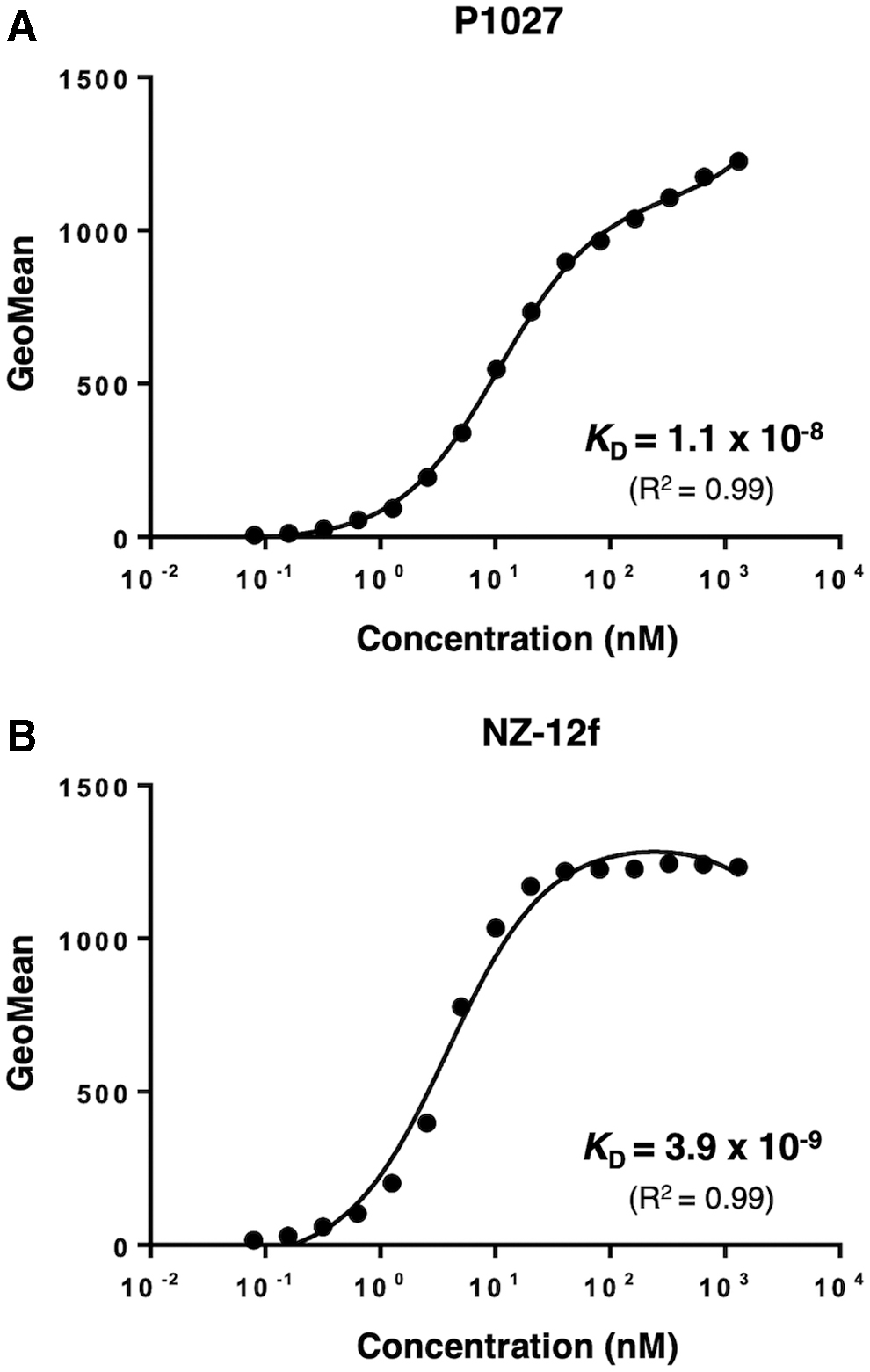
Determination of binding affinity using flow cytometry. LN319 (2 × 105 cells) was resuspended with 100 μL of serially diluted P1027 (
ADCC reporter gene assay
We used LN319 glioblastoma cells as target cells, and compared luminescence between P1027 and NZ-12f. EC50s for P1027 and NZ-12f were 149 and 14.3 ng/mL, respectively (Fig. 3A, B). At a concentration of 3.3 μg/mL, NZ-12f showed 1.5 times higher luminescence than P1027 (Fig. 3C). Thus, NZ-12f might display higher ADCC activity than P1027.
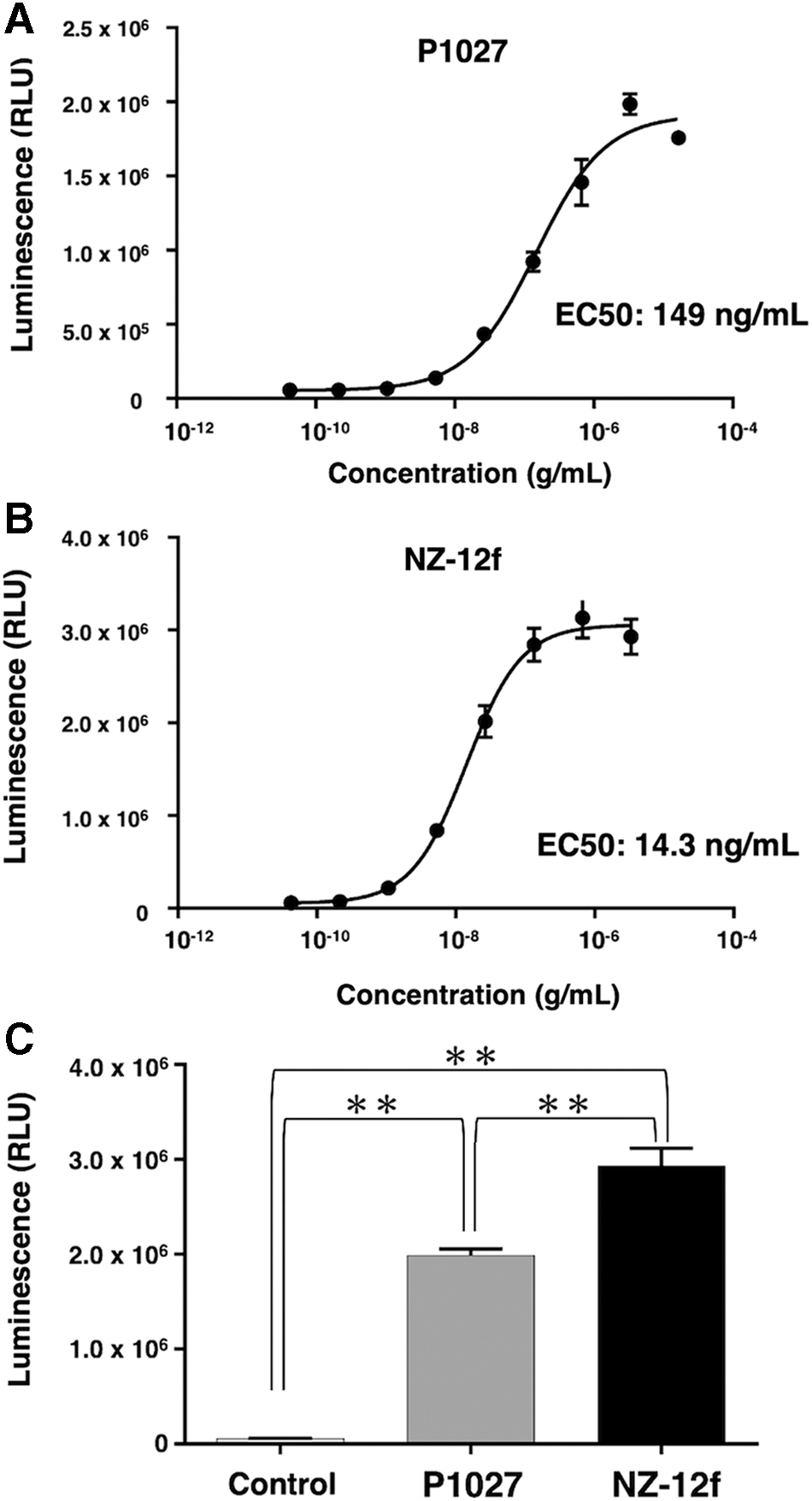
ADCC bioreporter assay.
ADCC and CDC mediated by anti-PDPN antibodies
To apply targeted therapy toward PDPN, we investigated ADCC induction against PDPN-expressing cell lines by anti-PDPN antibodies using human NK cells as effector cells. NZ-12f exhibited ADCC activity (51% cytotoxicity, Fig. 4A) and CDC activity (58% cytotoxicity, Fig. 4B) against LN319 cells; P1027 exhibited ADCC activity (23% cytotoxicity, Fig. 4A) and CDC activity (53% cytotoxicity, Fig. 4B) against LN319 cells, indicating that both NZ-12f and P1027 showed high CDC activities against LN319 cells (P1027 vs. control, p < 0.01; NZ-12f vs. control, p < 0.01; P1027 vs. NZ-12f, n.s.), whereas NZ-12f possesses 2.2 times higher ADCC compared with P1027 (P1027 vs. control, n.s.; NZ-12f vs. control, p < 0.01; P1027 vs. NZ-12f, p < 0.01).
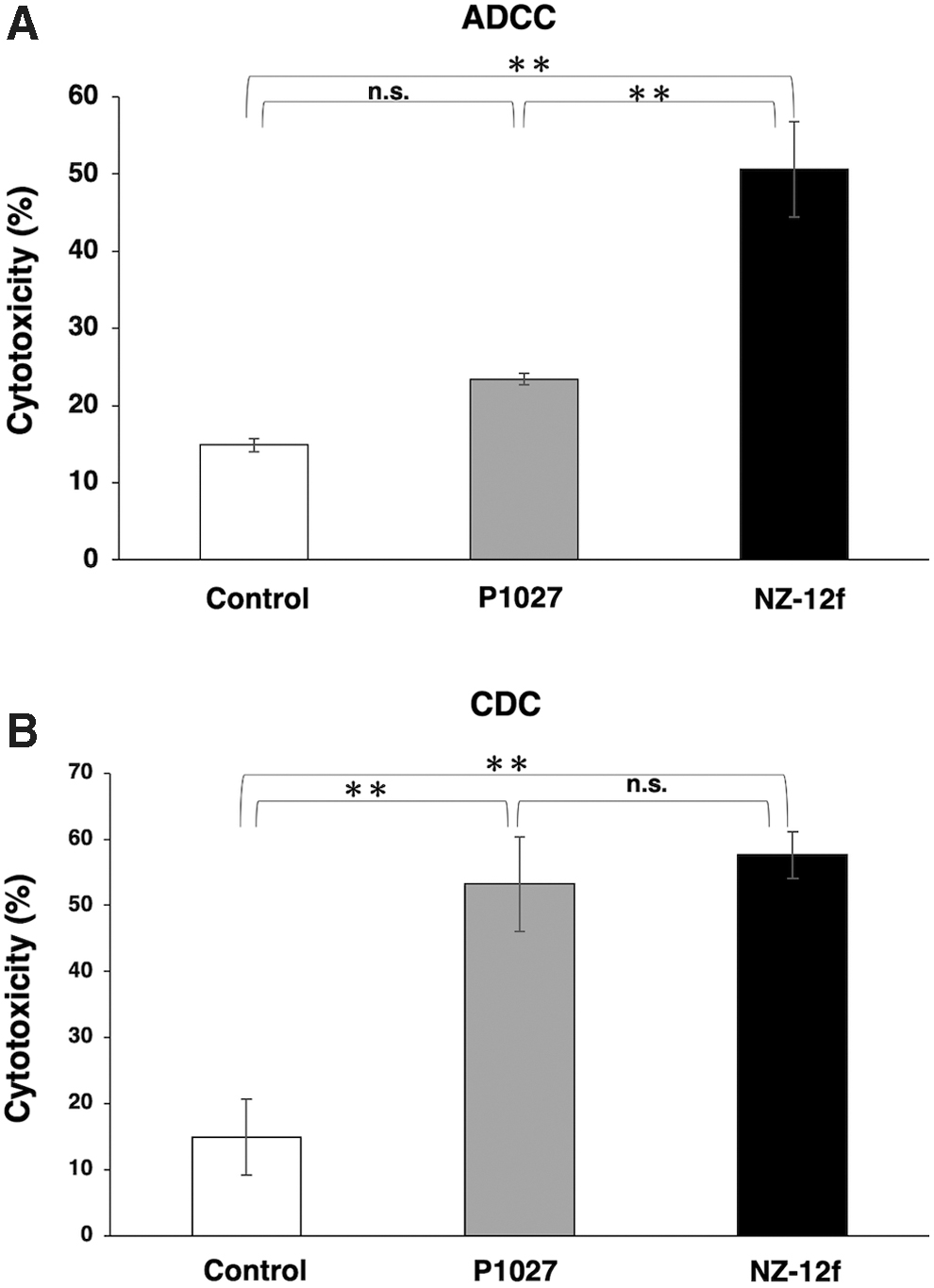
ADCC and CDC activities of anti-PDPN antibodies.
Antitumor activity of P1027 and NZ-12f in LN319 xenografts
Induction of cytotoxicity by P1027 and NZ-12f was investigated in vivo using LN319 cell xenografts in the flanks of nude mice. P1027, NZ-12f, and control human IgG were injected i.p. three times, on days 1, 8, and 16 after cell injection. Human NK cells were injected around tumors on days 1, 8, and 16. Tumor formation was observed in mice in every group. P1027 and NZ-12f treatment significantly reduced tumor development compared with control mice on days 3, 7, 10, 13, 16, and 21 (Fig. 5). No difference between P1027 and NZ-12f was observed at any time. Weights of tumors from P1027 and NZ-12f-treated mice were significantly less than for tumors from IgG-treated control mice (Fig. 6A). No difference in weights of tumors between P1027 and NZ-12f was found. Resected tumors are depicted in Figure 6B. Total body weight was not significantly different among the three groups (Fig. 7A). Mice on day 21 are depicted in Figure 7B. Both P1027 and NZ-12f exert a significant antitumor effect against PDPN-expressing brain tumors.
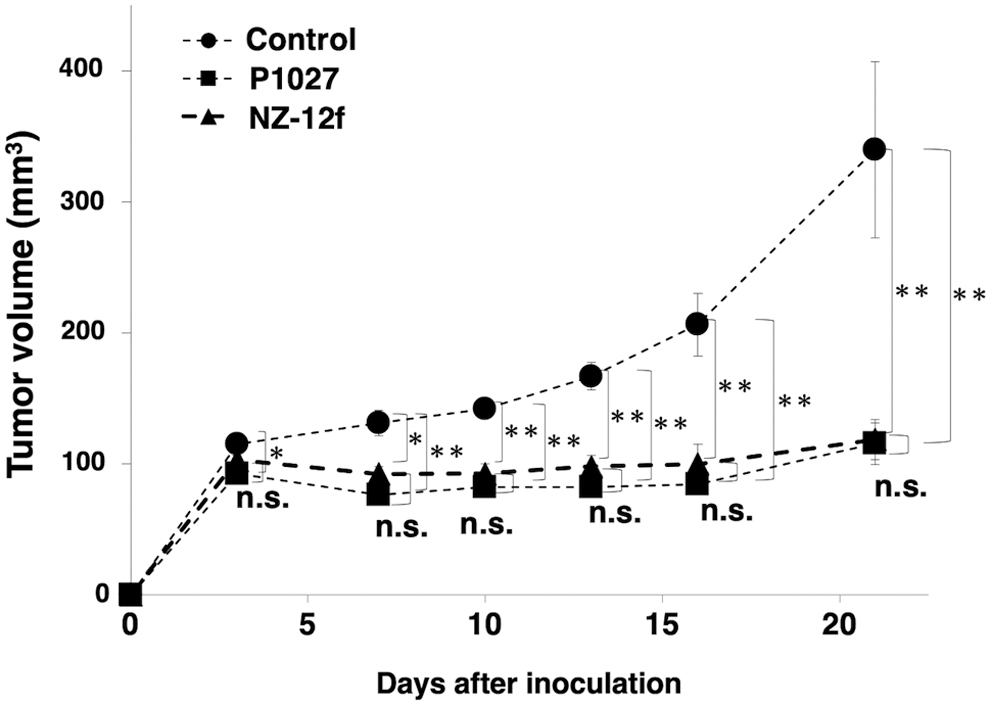
Evaluation of antitumor activity of anti-PDPN antibodies (tumor volume). Tumor volume was measured in mice with LN319 xenografts. Asterisks indicate statistical significance (*p < 0.05, **p < 0.01, n.s.: not significant, Tukey–Kramer's test).
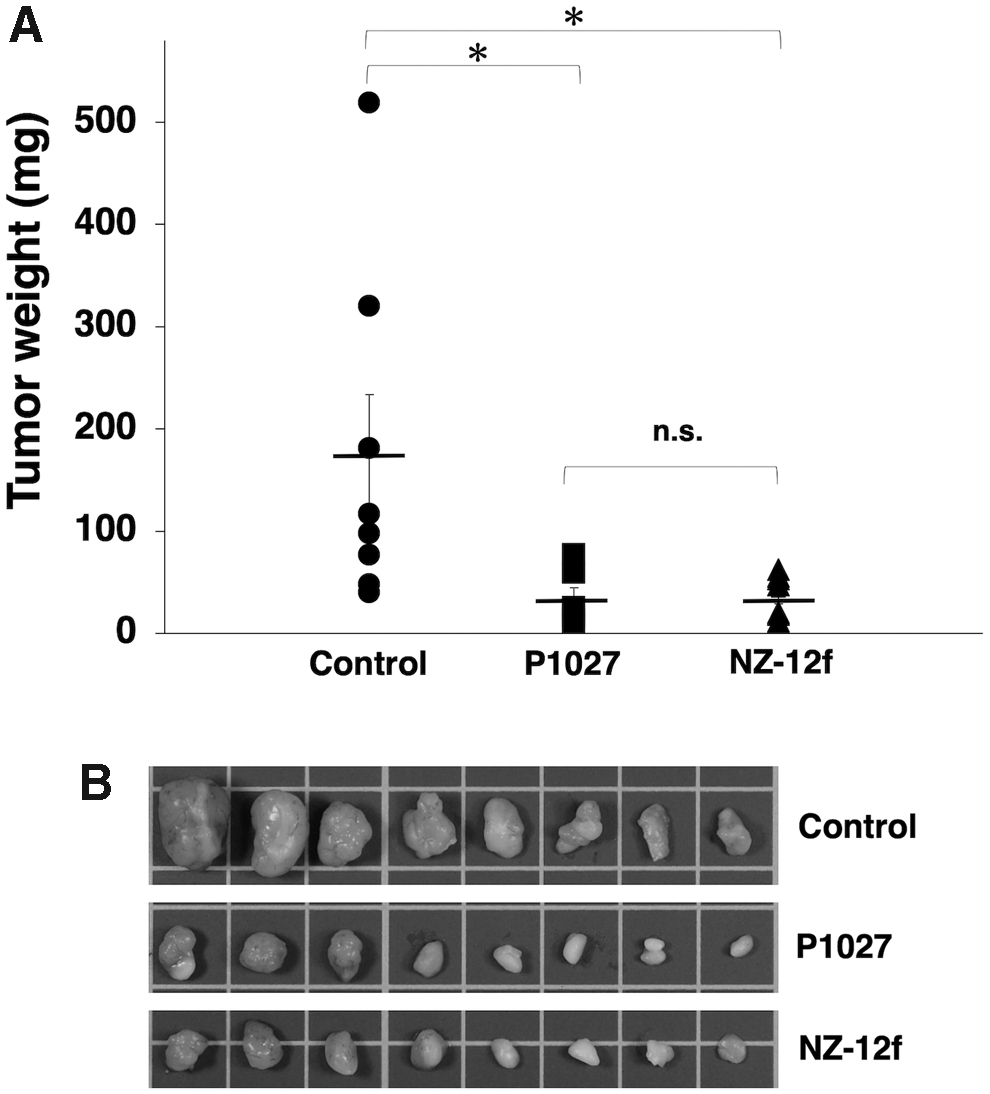
Evaluation of antitumor activity of anti-PDPN antibodies (tumor weight).
Evaluation of antitumor activity of anti-PDPN antibodies (body weight).
Discussion
We previously produced a FUT8-knockout cell line (BINDS-09) from ExpiCHO-S cells.(36) By using BINDS-09, we easily produced core-fucose-deficient antibodies to enhance ADCC activity. In this study, we produced both a core-fucose-deficient humanized anti-PDPN antibody (P1027) and a rat-human chimeric antibody (NZ-12f). Binding affinities of P1027 and NZ-12f against LN319 cells were 1.1 × 10−8 and 3.9 × 10−9 M (Fig. 2), of the affinity of NZ-12f against LN319 is 2.8 times than the affinity of P1027. We next investigated whether this difference of binding affinity affects the ability of antibodies to activate the ADCC reporter gene. The ADCC reporter system consists of an Fc region of an antibody that binds to the FcγRIIIa receptor expressed on an engineered immortalized Jurkat T-lymphocyte cell line.(37) This binding activates gene transcription through the nuclear factor of the activated T cell pathway, inducing the expression of firefly luciferase. The FcγRIIIa stimulation assay can be used to evaluate the ability of an antibody to activate the ADCC reporter gene. NZ-12f displays 1.5 times higher activation of the ADCC reporter gene compared with P1027 (Fig. 3), indicating that binding affinity is associated with the ability of antibodies to activate the ADCC reporter gene. In the ADCC and CDC assays, both P1027 and NZ-12f demonstrated high CDC activities, whereas NZ-12f showed 2.2 times higher ADCC activity compared with P1027 (Fig. 4), which are compatible with results from binding affinity and ADCC reporter assays.
In our previous studies, antitumor effects were observed after treatment of malignant pleural mesothelioma orthotopic xenograft mice with NZ-12 and human NK cells.(31) Furthermore, RIT with 90Y-labeled NZ-12 significantly suppressed tumor growth in an NCI-H226 xenograft mouse model, and prolonged survival without obvious adverse effects.(32) Although P1027 showed lower binding affinity and lower ADCC activities to LN319 cells, antitumor activity of P1027 for LN319 xenografts was equal to that of NZ-12f (Figs. 5 and 6). We further conducted additional in vivo experiments using three times injection of 10 μg antibodies to assess whether the antitumor activities were saturated when three times injection of 100 μg was employed. However, we observed no difference of antitumor activities between P1027 and NZ-12f using three times injection of 10 μg antibodies (data not shown), suggesting that antitumor activities were exerted mainly by CDC because both P1027 and NZ-12f showed high CDC activities against LN319 cells (Fig. 4B). We need to further perform in vivo experiments using the other xenograft models, such as lung cancers and malignant mesotheliomas to confirm that humanized anti-PDPN mAbs may be effective for patients with PDPN-expressing cancers.
Footnotes
Acknowledgment
We thank Ms. Akiko Harakawa for technical assistance in animal experiments.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research was supported in part by AMED under Grant Numbers: JP20am0401013 (Y.K.), JP20am0101078 (Y.K.), JP20ae0101028 (Y.K.), and JP20bm1004001 (Y.K.), and by JSPS KAKENHI Grant Numbers 20K16322 (M.S.), 17K07299 (M.K.K.), and 19K07705 (Y.K.).