
Abstract
Abstract
Background:
Lymphovascular invasion (LVI) is an important step in the metastatic cascade; tumor cell migration and adhesion to blood and lymphatic vessels is followed by invasion through the vessel wall and subsequent systemic spread. Although primary breast cancers and melanomas have rich blood vascular networks, LVI is predominately lymphatic in nature. Whilst the adhesion of tumor cells to blood endothelium has been extensively investigated, there is a paucity of information on tumor cell adhesion to lymphatic endothelium.
Methods and Results:
Breast cancer (MDA-MB-231 and MCF7) and melanoma (MeWo and SKMEL-30) cell adhesion to lymphatic (hTERT-LEC and HMVEC dLy Neo) and blood (HUVEC and hMEC-1) endothelial cells were assessed using static adhesion assays. The effect of inflammatory conditions, tumor necrosis factor-α (TNF-α) stimulation of endothelial and tumor cells, on the adhesive process was also examined. In addition, the effects of TNF-α stimulation on tumor cell migration was investigated using haplotaxis (scratch wound) assays. Breast cancer and melanoma cells exhibited higher levels of adhesion to blood compared to lymphatic endothelial cells (p<0.001). TNF-α stimulation of endothelial cells, or of tumor cells alone, did not significantly alter tumor–endothelial cell adhesion or patterns. When both tumor and endothelial cells were stimulated with TNF-α, a significant increase in adhesion was observed (p<0.01), which was notably higher in the lymphatic cell models (p<0.001). TNF-α-stimulation of all tumor cell lines significantly increased their migration rate (p<0.01).
Conclusions:
Results suggest that metastasis resultant from lymphatic vessel-tumor cell adhesion may be modulated by cytokine stimulation, which could represent an important therapeutic target in breast cancer and melanoma.
Introduction
In vitro, endothelial cell monolayers have been used as a model system to investigate tumor–endothelial interactions to mimic interactions that occur in vivo. Current knowledge of tumor–endothelial interaction is largely derived from studies using blood endothelial cells (BEC). These studies use a range of endothelial cells to investigate tumor cell adhesion; however the most widely used cell type are large vein endothelial cells from human umbilical cords (HUVEC).6–11 In addition to HUVEC, human microvascular endothelial cells (hMEC) have also been used to study tumor–endothelial interactions. The pro-inflammatory cytokines, tumor necrosis factor-alpha (TNF-α) and interleukin 1-beta (IL1-β), have been shown to increase adhesion of pancreatic carcinoma cells to hMEC. 12 Adhesion of melanoma cell lines; MeWo, SKMEL-28, JPC298 and HT144 to human dermal microvascular endothelial cells (hDMEC) has also been investigated. 13
In contrast to blood endothelium, mechanisms underlying the adhesion of tumor cells to lymphatic endothelium are still poorly understood. Only a limited number of studies have utilized lymphatic endothelial cells (LEC), primarily due to the lack of molecular tools capable of specifically distinguishing LEC from BEC. Studies investigating the interaction between tumor and lymphatic endothelium have become possible over the last 10 years, following the discovery of lymphatic-specific markers such as VEGFR-3, 14 LYVE-1, 15 Prox-1, 16 and podoplanin/D2-40.17,18 Studies utilizing primary LEC have shown an increase in the attachment of breast cancer cells (MCF7 and MDA-MB-231) following stimulation with anti-CCL-2 antibody. 19 In addition, the adhesion of MCF7 cells to human microvascular lung lymphatic endothelial cells (HMVEC-LLy) was reduced following treatment with an anti-Tn antibody, a common tumor-associated carbohydrate antigen present in most carcinomas. 20
Studies conducted thus far have utilized either BEC or LEC to examine tumor–endothelial adhesion. To the best of our knowledge, no studies have conducted a direct side-to-side comparison of blood versus lymphatic adhesion. In light of the importance of lymphatic invasion in the initial metastatic process, it is important to have a suitable model system to investigate tumor–lymphatic interactions, which is crucial to aid our understanding of metastatic dissemination via this route.
The aim of the current study was to directly compare the adhesion pattern of breast cancer and melanoma cell lines to BEC and LEC models (primary and immortalized cell lines). Furthermore, the stimulatory effect of the cytokine, TNF-α, on tumor cell adhesion to the endothelial cell models, and on tumor cell migration was investigated.
Materials and Methods
Cell lines and culture
Endothelial cells
HUVEC were isolated from human umbilical cords obtained from the Department of Obstetrics, City Hospital, Nottingham, using the collagenase isolation technique as described previously. 21 Cells from three cords were pooled to give a single batch of experimental HUVEC. Cultured flasks were coated with 0.2% gelatin (Sigma, UK) prior to culture. HUVEC were maintained in 37% nutrient mixture F-12 HAM (Sigma), 3.7% 199 media (Sigma), 20% iron supplemented donor calf serum (PAA Laboratories, Austria), 1% sodium bicarbonate (Sigma), 14 mM HEPES (Sigma), 2 mM L-glutamine (Sigma), 1% penicillin/streptomycin (Sigma), 7.5 U/ml heparin (CP Pharmaceuticals, UK), 25 ng/ml epidermal growth factor EGF (Peprotech, UK) and 12.5 ng/ml basic fibroblast growth factor (bFGF; Peprotech). HUVEC were used between passage 2 and 6. Human microvascular endothelial cells (hMEC-1), from ATCC, are an immortalized human dermal microvascular endothelial cell, immortalized using SV40 large T-Ag. 22 Cells were grown in Endothelial Basal Medium, EBM (Lonza, USA), 10% iron supplemented donor calf serum, 1 μg/ml hydrocortisone (Sigma), 10 ng/ml EGF, and 1% penicillin/streptomycin. Cells were used between passage 4 and 18. Human telomerase reverse transcriptase immortalized LEC (hTERT-LEC) were a kind gift from R. Nissato and M. Pepper. 23 Cells were used between passage 27 and 34. hTERT-LEC were cultured in endothelial EBM with EGM-2 bullet kit (Lonza). Neonatal dermal lymphatic microvascular endothelial cells (HMVEC-dLy Neo) (Lonza), are a primary LEC. The medium for this cell line was prepared as hTERT-LEC. Cells were used between passage 4 and 6.
Tumor cells
Breast cancer cell lines; MCF7 (used between passage 23 and 35) and MDA-MB-231 (used between passage 15 and 28) and melanoma cell lines; MeWo (used between passage 14 and 30) and SKMEL-30 (used between passage 18 and 30), from ATCC, were used in this study. MCF7 and SKMEL-30 were maintained in RPMI-1640 (Sigma), 10% iron-supplemented donor calf serum, and 1% penicillin/streptomycin. MDA-MB-231 were maintained in minimal essential medium (MEM) EAGLE (Sigma), 0.1 mM non-essential amino acids (Sigma), 2 mM L-glutamine, 1% penicillin/streptomycin, and 1% iron-supplemented donor calf serum. MeWo were maintained in MEM EAGLE, 10% iron-supplemented donor calf serum, 2 mM L-glutamine, 1% sodium bicarbonate, 0.1 mM non-essential amino acids, and 1 mM sodium pyruvate (Sigma).
Peripheral blood mononuclear cell isolation
Peripheral blood mononuclear cells (PBMC) were isolated from whole blood using density-gradient centrifugation method as described. 24 Blood was collected from a consent healthy donor in a vacutainer tube (BD, UK) containing 170 IU heparin. 2.5 ml histopaque 1119 (Sigma) was added to a 15 ml tube to separate the granulocytes, followed by histopaque 1077 (Sigma) to separate the mononuclear cells. 5 ml of heparinrized blood was layered on top of the histopaque 1119 and histopaque 1077 and centrifuged for 30 min, 670 g, followed by the removal of PBMC at the second layer. Cells were washed twice in washing buffer (0.1% bovine serum albumin in phosphate buffered saline, PBS) and counted by hemocytometer.
Static adhesion assay
Endothelial cells were grown to confluence in a 24-well plate (seeding density; 0.75×105 cells/ml, 1.3×105 cells/ml, 1.3×105 cells/ml, and 1.5×105 cell/ml for HUVEC, hMEC-1, hTERT-LEC, and HMVEC-dLY cells, respectively). Complete media was removed and replaced with growth factor-free media in control wells or growth factor free media supplemented with TNF-α (2.5 ng/ml, 5 ng/ml, 10 ng/ml) for 24 h.
PBMC adhesion to endothelial cells with and without TNF-α stimulation was carried out immediately prior to tumor cell adhesion experiments to assess immune cell–endothelial cell interactions, in addition to acting as a control to demonstrate that the endothelial cells and the cytokine were responding appropriately. PBMC adhesion was assessed by adding 4×105 cells/well for 5 min, as this was identified as the optimal assay time (data not shown). Non-adherent cells were washed from the endothelial layer with washing buffer and assessed visually by counting adherent cells in the central area of the well using phase contrast microscopy (10X magnification).
Tumor cell adhesion was assessed following cell labeling with 1 μM Cell Tracker Green CMFDA (Invitrogen, USA) for 30 min at 37°C. This concentration of Cell Tracker Green was determined not to influence cell viability over 72 h (data not shown). After labeling, tumor cells were resuspended in RPMI-1640 + 10% iron-supplemented donor calf serum. 0.5×105 cells/well were incubated for 35 min at 37°C on the different endothelial cell monolayers. 35 minutes was determined as the optimal adhesion time required for tumor cell adhesion (data not shown).
Non-adherent cells were washed with RPMI-1640 + 10% iron-supplemented donor calf serum. Adherent tumor cells were counted using a fluorescence microscope (Nikon, Japan), in the central area of the well which was marked manually with grid lines on the bottom of the plate. Two fields of view were counted in each well (20X magnification). Representative photomicrographs of tumor cells adhering to endothelium are shown in Figure 1. Experiments were conducted twice, both in triplicate. Results were expressed as the absolute number of cells adhered to the endothelial layer and as the percentage of cells adhered relative to control. In addition to assessing adhesion to control (unstimulated) and TNF-α stimulated endothelial cells, assays were also performed under the following conditions: (1) stimulating only tumor cells with TNF-α (2.5, 5, and 10 ng/ml) and (2) stimulating both endothelial and tumor cells simultaneously with TNF-α (5 ng/ml). In both conditions, stimulations with TNF-α were for 24 hours.
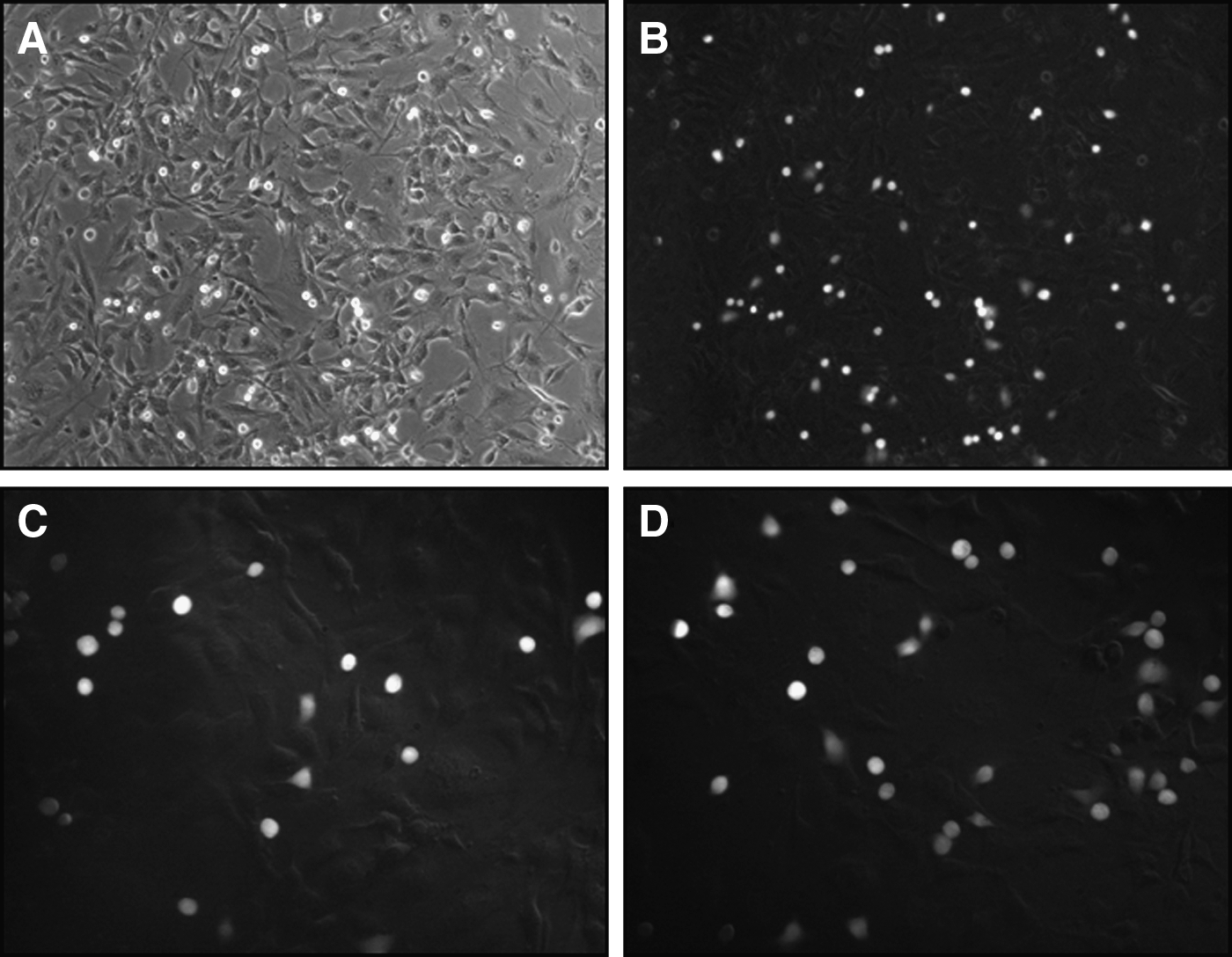
Representative photomicrographs of tumor cell adhesion. MDA-MB-231 cell adhesion to a hMEC-1 monolayer observed under phase contrast microscopy
Migration assay
Haplotaxis (scratch wound) migration assays, as described, 25 were conducted with slight modifications. 5×105 cells/well (MCF7, MeWo, SkMEL-30) or 4×105 cells/well (MDA-MB-231) were plated in a 12-well tissue culture plate and incubated for 48 h to ensure a confluent monolayer. 24 hours post-plating, standard culture media was removed and replaced with growth factor and serum-free media with or without TNF-α (5 ng/ml) for 24 h. Confluent cell monolayers were scratched with a pipette tip to create a wound, devoid of adherent cells. Cell debris was removed by washing twice with PBS and cells cultured in growth factor and serum-free media with 10 μg/ml of mytomycin-c (Sigma), for the duration of the assay, to inhibit cellular proliferation. Wound closure was monitored by photomicrographs at 0, 2, 6, and 24 hours post scratch. The percentage reduction of scratch area at these time points represented the rate of tumor cell migration and was measured using ImageJ 1.43u software (National Institute of Health, USA). Experiments were conducted twice, each in triplicate.
Statistical analysis
Data are represented as mean±standard deviation of two independent experiments performed in triplicate. For PBMC adhesion and tumor cells adhesion under control condition, data are pooled from 28 independent experiments. The Student t-test was used to evaluate variation between control and TNF-α stimulated condition. P<0.05 was used to define significant relationships. Statistical analysis was carried out using GraphPad Prism software version 3.02 (GraphPad Software Inc.).
Results
TNF-α increases PBMC adhesion
PBMC adhesion to unstimulated and TNF-α-stimulated HUVEC, hMEC-1, hTERT-LEC, and HMVEC-dLy Neo was conducted immediately prior to tumor cell adhesion experiments to ensure that endothelial cells were responding as expected. Endothelial cell pre-stimulation with TNF-α for 24 h caused a significant increase in PBMC adhesion compared to the unstimulated controls (p<0.01) (Fig. 2). PBMC adhesion to all endothelial cell models increased with increasing TNF-α concentration and plateaued at 10 ng/ml. PBMC adhesion to TNF-α-stimulated HUVEC was more than that to hMEC-1 but was not significantly different (p>0.05). There was no significant difference in the number of cells adhered to LEC models, hTERT-LEC, and HMVEC-dLY Neo. Adhesion of PBMC was significantly higher to stimulated BEC (HUVEC and hMEC-1) than to LEC (hTERT-LEC and HMVEC-dLy Neo), (p<0.05).
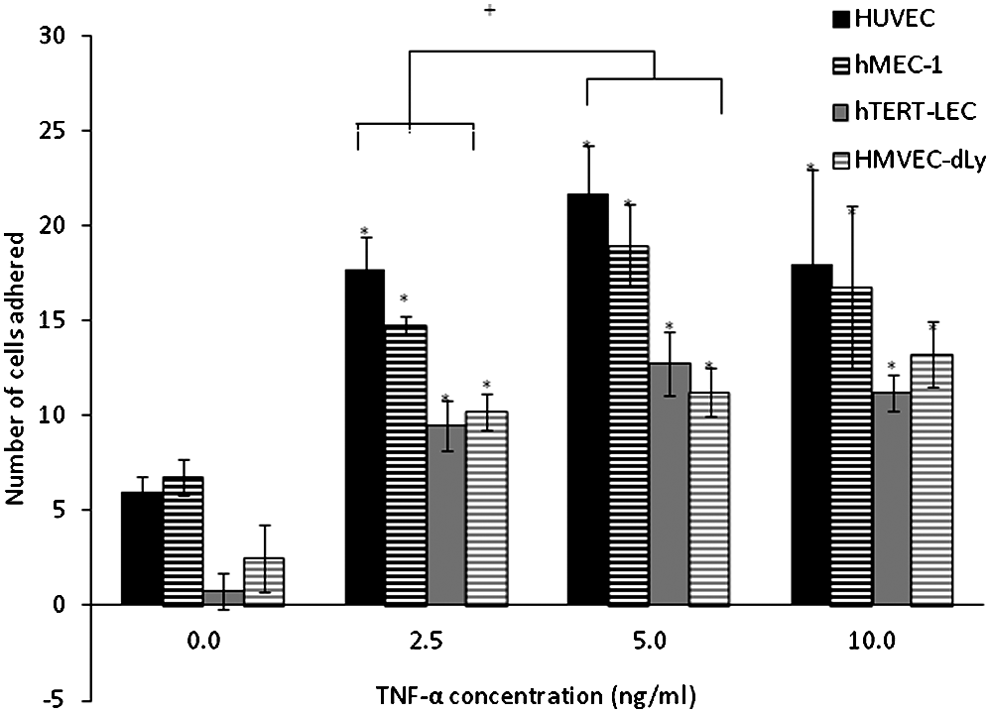
PBMC adhesion to HUVEC, hMEC-1, hTERT-LEC, and HMVEC-dLy Neo under control and TNF-α stimulations. Pre-stimulation of endothelial cells with TNF-α caused a significant increase (*p<0.01) in the number of PBMC adhered to HUVEC, hMEC-1, hTERT-LEC, and HMVEC-dLy Neo compared to the unstimulated controls with the highest adhesion observed to HUVEC. PBMC adhesion to all endothelial cell models increased with increasing concentrations of TNF-α and plateau at 10 ng/ml. Pooled results from 28 independents experiments, each in duplicate (n=56). *significant difference compared to control group, †significant difference between groups, p<0.05.
Tumor cells show preferential adhesion to blood endothelial cells
MCF7, MDA-MB-231, MeWo, and SKMEL-30 showed similar adhesion patterns to all four endothelial cell models under control conditions, however with different affinity (i.e., greater adherence to blood than to lymphatic endothelium) (Fig. 3). Of the four tumor cell lines, MDA-MB-231 cells had the highest affinity towards the BEC, while SKMEL-30 had the highest affinity towards LEC. Both the breast cancer cell lines showed relatively less adherence to the LEC than the BEC in comparison to the melanoma lines. There was no significant difference in the tumor cell adhesion patterns between primary and immortalized cell lines of the same origin: blood and lymphatic.
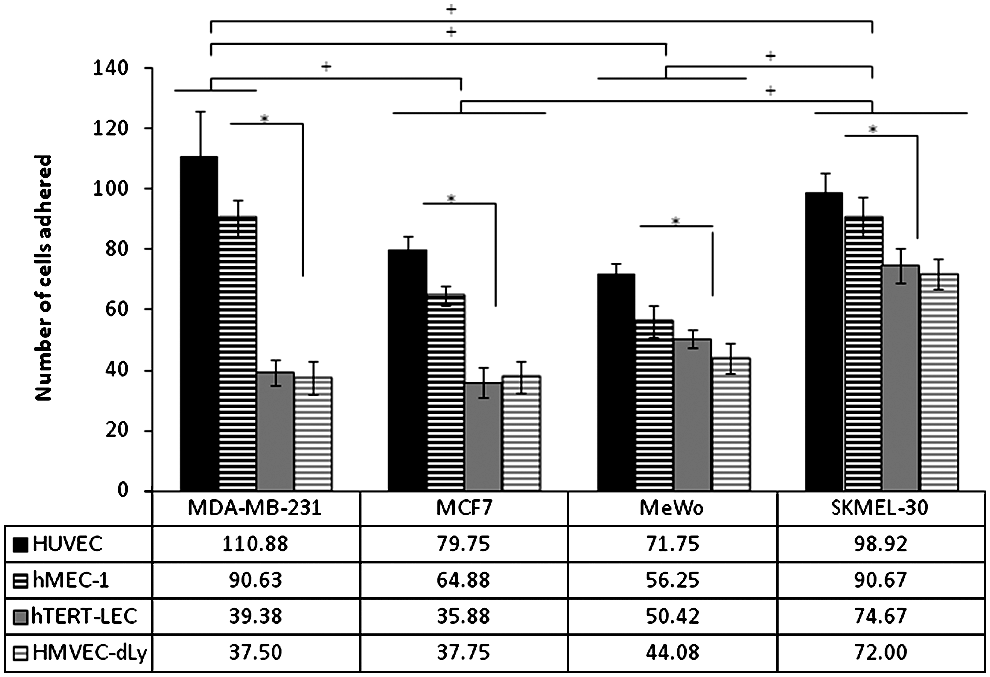
Tumor cell adhesion to HUVEC, hMEC-1, hTERT-LEC, and HMVEC-dLy Neo under control conditions. All endothelial cell models show the same adhesion pattern with the 4 tumor cells, MDA-MB-231, MCF7, MeWo, and SKMEL-30, in which higher adherence to the BEC is observed. The highest tumor cell adhesion is observed to HUVEC opposed to hTERT-LEC and HMVEC-dLy Neo. Pooled results from 28 independents experiments, each in triplicate (n=84). *significant difference within group, †significant difference between groups, p<0.05.
The percentage adhesion of each tumor cell line to TNF-α-stimulated HUVEC, hMEC-1, hTERT-LEC, and HMVEC-dLy Neo was not significantly increased compared to the unstimulated endothelial models (Fig. 4). Pre-stimulation of the tumor cells with TNF-α did not significantly increase their adhesion to BEC models; except for the adhesion of TNF-α stimulated MDA-MB-231 to unstimulated HUVEC (p<0.001). However, pre-stimulation of the tumor cell lines with TNF-α did cause a significant increase of MDA-MB-231, MCF7, and MeWo cells adhesion to hTERT-LEC (p<0.05) (Fig. 5).
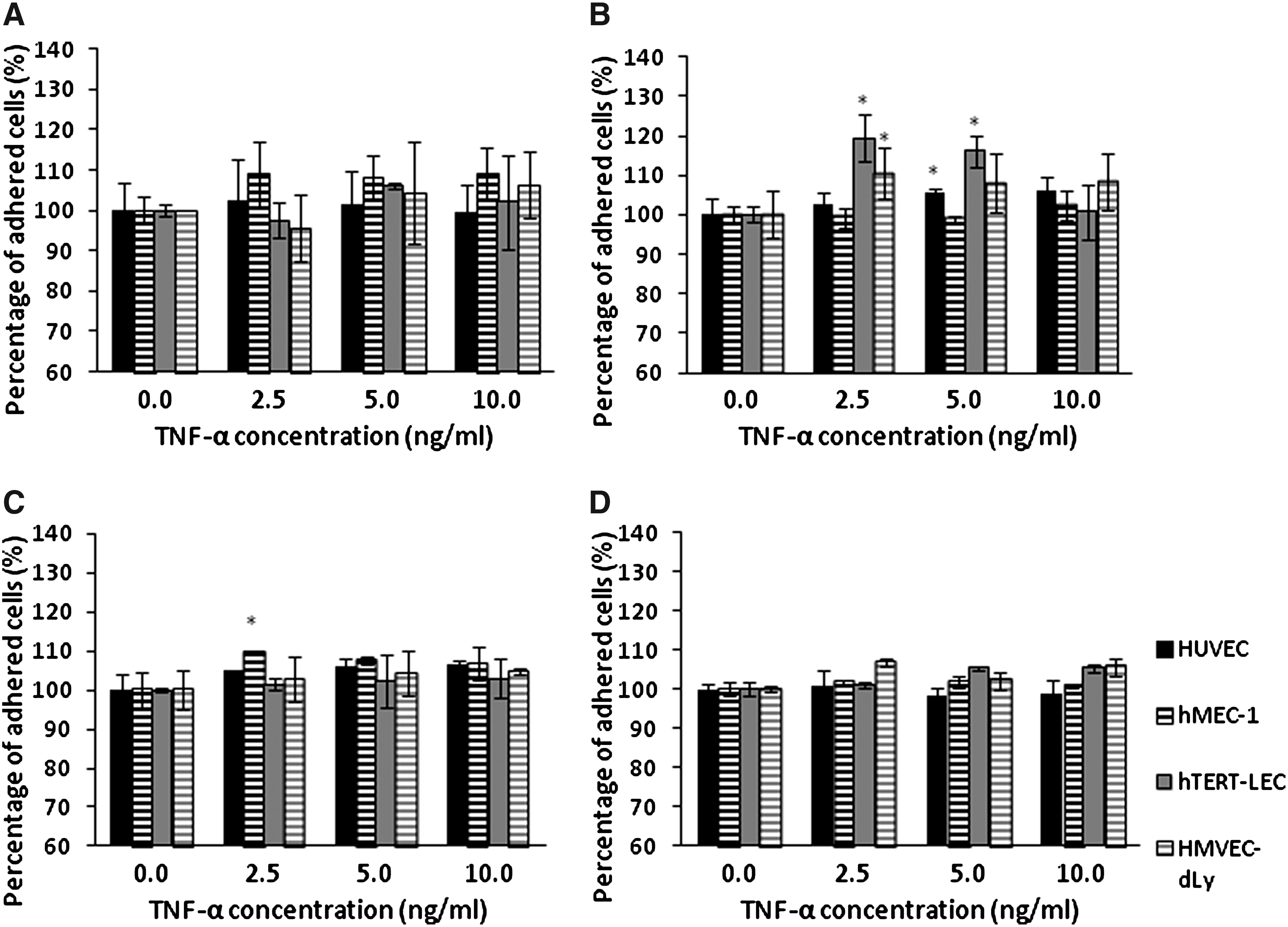
MDA-MB-231
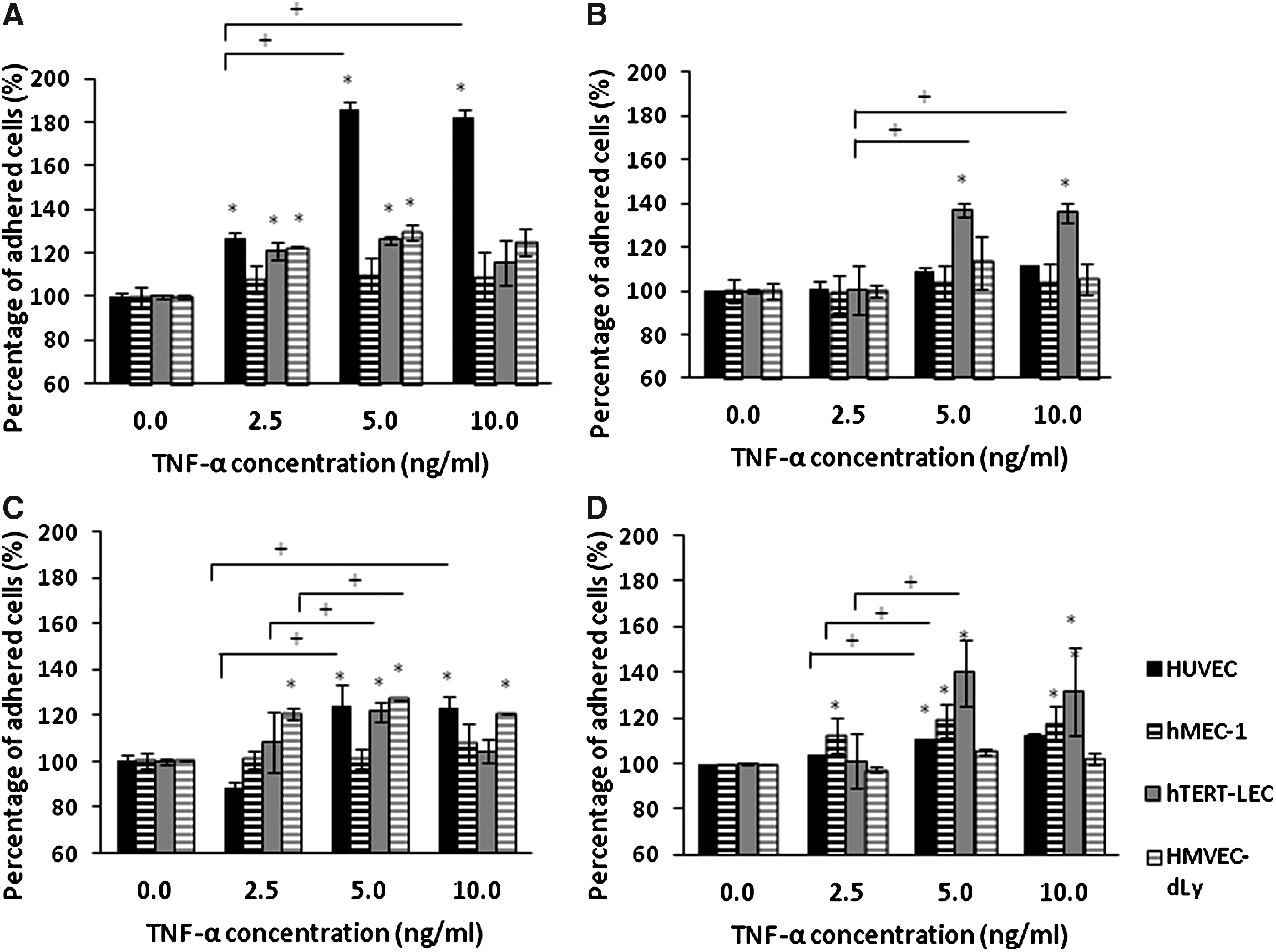
Adhesion of TNF-α stimulated tumor cells; MDA-MB-231
As both breast cancer and both melanoma cell lines showed similar adhesion patterns, with similar affinities, double stimulation experiments using TNF-α were conducted using MDA-MB-231 and MeWo to study the adhesion patterns towards the immortalized endothelial cell lines hMEC-1 and hTERT-LEC. When MDA-MB-231 and the endothelial cells (hMEC-1 and hTERT-LEC) were both simultaneously stimulated with TNF-α, there was a significant increase in tumor cell adhesion to hMEC-1 (p=0.02) and hTERT-LEC (p<0.001) (Fig. 6A). The relative percentage of adhered tumor cells was significantly higher to the LEC compared to the BEC (p<0.05). Similar results were also observed with melanoma where there was a significant increase in adhesion of MeWo to hMEC-1 (p<0.001) and hTERT-LEC (p<0.001); as with the breast line, the percentage adhesion of MeWo cells were also higher in the LEC compared to the BEC (Fig. 6B).
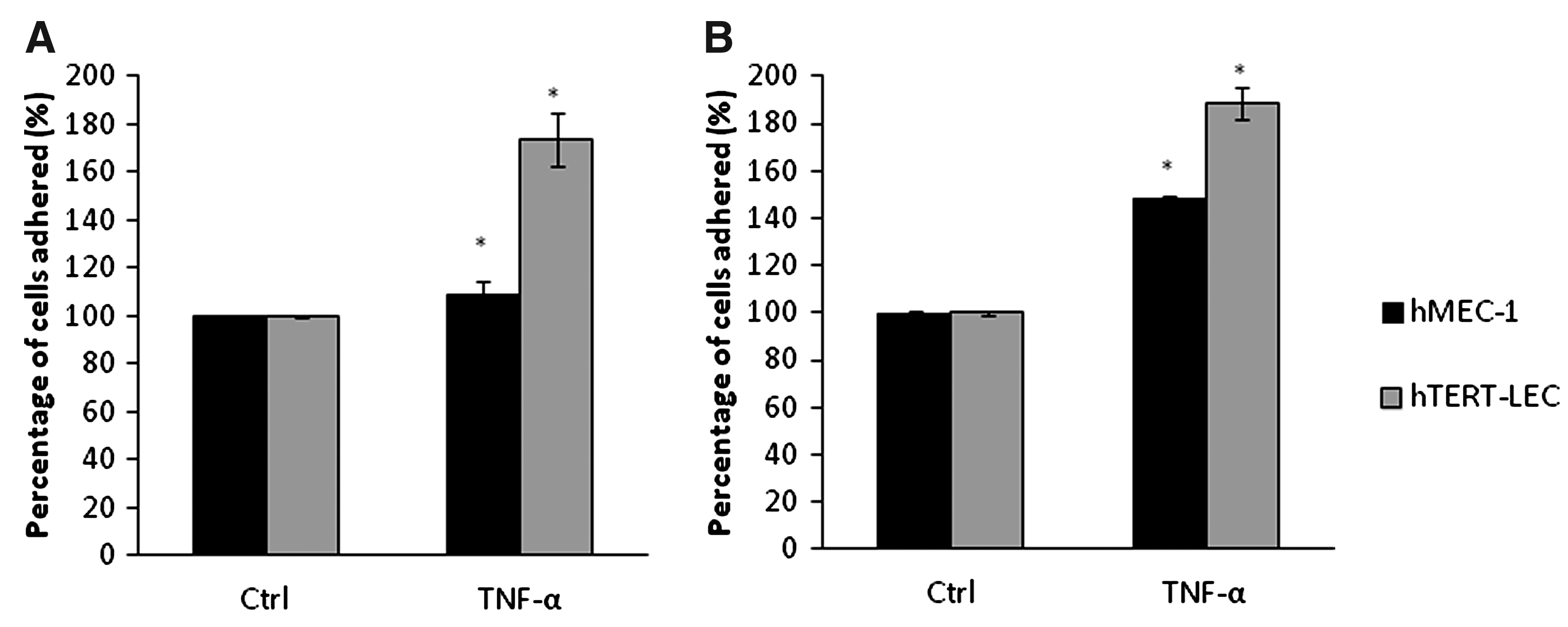
The effect on TNF-α double stimulation on the adhesion of MDA-MB-231
TNF-α-stimulation increases the migratory ability of tumor cells
The effect of TNF-α upon tumor cell migration was also studied. Tumor cell migration is a prerequisite for metastasis in addition to tumor cell–endothelial cells adhesion. The migration of all four tumor cell lines was increased following exposure to TNF-α, which was particularly noticeable with MDA-MB-231 cells. MDA-MB-231 stimulated with TNF-α had a significantly higher migration rate compared to the unstimulated control at all 2, 4, 6, and 24 hours post wounding (p<0.001) (Fig. 7A). MCF7, MeWo, and SKMEL-30 also showed significantly higher migratory ability at 24 hours post wounding; p values<0.01,<0.01, and 0.014, respectively (Fig. 7B–7D).
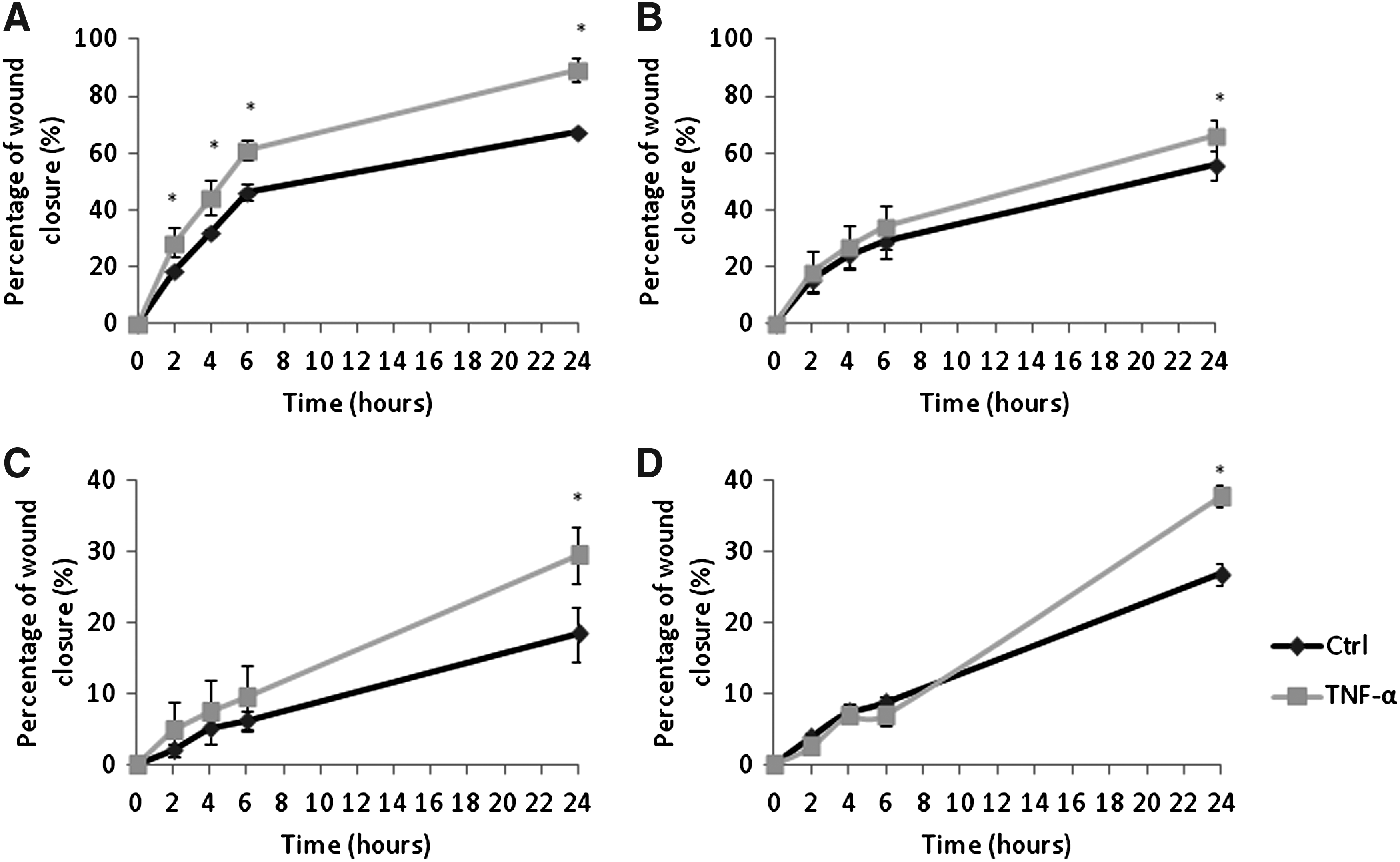
The effect of TNF-alpha on MDA-MB-231
Discussion
In the present study, the adhesion pattern of breast cancer and melanoma cell lines to BEC and LEC models was compared. PBMC adhesion to unstimulated and TNF-α-stimulated HUVEC, hMEC-1, hTERT-LEC, and HMVEC-dLy Neo was also assessed. Adhesion of PBMC was greater to BEC than LEC under control conditions. As PBMC were of blood origin, it may be that they are programmed to interact more with BEC barrier compared to LEC barrier. PBMC adhesion to TNF-α stimulated endothelial cells was significantly higher than the unstimulated controls; 26 which may be due to increased expression of adhesion molecules such as ICAM-1 and VCAM-1 in inflammed HUVEC and human dermal LEC in order to recruit immune cells to the site of inflammation.27–29 The adhesion of PBMC to the endothelial models was used as internal experimental control prior to the assessment of tumor cell adhesion to ensure that both cytokine and endothelial cells were behaving as expected.
The breast cancer, MDA-MB-231 and MCF7, and the melanoma cell lines, MeWo and SKMEL-30, showed different adhesive affinity towards the endothelial cell models, which demonstrated the heterogeneous nature of tumor cell adhesion. 30 Of the four tumor cell lines, MDA-MB-231 had the highest affinity towards all endothelial cell models, which may be related to the highly invasive and aggressive nature of this tumor cell line.31,32 Although the four tumor cell lines showed differences in terms of adhesive potential, they all showed preferential adhesion to BEC opposed to the LEC. This is interesting in light of observations in breast cancer and melanoma that show vascular invasion in these tumor types is principally of lymphatic vessels.2,33,34
Experiments comparing the adhesion of a panel of breast cancer and melanoma cells to both BEC and LEC have not previously been reported. However, the preferential adhesion of colon carcinoma cells, HT29, to lung microvascular endothelial cells (HMVEC), as opposed to large vein endothelial cells (HUVEC) has been observed. 12 This is in contrast to the current observations made using breast cancer and melanoma cells, in which preferential adhesion of all cell lines, MDA-MB-231, MCF7, MeWo, and SKMEL-30 was observed to HUVEC rather than hMEC-1. These results reflect the diverse nature of endothelial cells arising from different vascular beds.35,36
There was no difference in tumor cell adhesion to primary lymphatic cells (HMVEC-dLy Neo) or immortalized lymphatic cells (hTERT-LEC), suggesting that despite the immortalization procedure, hTERT-LEC retain a phenotype similar to the primary cells from which they were derived. 23 It seems, therefore, that hTERT-LEC is a useful in vitro model to study lymphatic–endothelial interactions, which can overcome the problem of the short passage window of primary lymphatic cells.
The preferential adhesion of cancer cells to BEC rather than LEC is, in light of results from patient samples,2,3 somewhat surprising. This may be due to, in tumors, cells being exposed to inflammatory conditions and associated cytokines. TNF-α is a multifunctional cytokine that is known to stimulate angiogenesis, growth, proliferation, and invasion of tumor cells via various signaling pathways such as the NF-КB and MAPK pathway.37,38 Tumor cell adhesion to TNF-α-stimulated HUVEC, hMEC-1, hTERT-LEC, and HMVEC-dLy Neo did not vary significantly when compared to the unstimulated endothelium. Similar results have been described, in which no significant changes were observed in the adhesion of melanoma cell lines; WM9, WM239, and WM119 to unstimulated and TNF-α-stimulated HUVEC. 39 However, others have shown that tumor cell adhesion increases with TNF-α-stimulation. Colon cancer cells, HT29s, adhere more to TNF-α stimulated HUVEC and lung microvascular endothelial cells. The adhesion of breast cancer cells MDA-MB-231 to HUVEC has been shown to increase approximately six-fold following TNF-α-stimulation compared to the unstimulated counterpart. 7 No such increase was observed in the current study, using the same cell models; however both the TNF-α concentrations and the incubation time differs between this and previously published studies.
As the two breast (MDA-MB-231 and MCF7) and the two melanoma (MeWo and SKMEL-30) cell lines showed similar adhesion patterns to BEC and LEC, experiments were conducted to examine the effect of TNF-α stimulated MDA-MB-231 and MeWo cells in influencing the adhesion towards TNF-α activated hMEC-1 and hTERT-LEC. The significant increase in tumor cell adhesion when both the tumor and the endothelial cells were simultaneously stimulated with TNF-α suggests that dual activation with TNF-α might be necessary to promote tumor–endothelial cell adhesion. In tumor cells, TNF-α acts through NF-КB induction of chemokine receptor CXCR4 and upregulation of molecules such as the monocyte chemoattractant protein-1 (MCP-1), IL-8, and ICAM-1. In endothelial cells, TNF-α upregulates various receptors such as lectin-like oxidized low-density lipoprotein receptor 1 (ox-LDL receptor 1) or LOX-1.40–43 Interestingly, with TNF-α stimulation, the adhesion of both MDA-MB-231 and MeWo was higher to the lymphatic endothelial model (hTERT-LEC), as opposed to the blood endothelial cells (hMEC-1).
The migration of tumor cells is a prerequisite for metastatic formation in addition to tumor–endothelial adhesion. Therefore, we sought to study whether TNF-α had any effect upon tumor cell migration. TNF-α stimulation altered the migratory ability of MDA-MB-231, MCF7, MeWo, and SKMEL-30, which was observed at numerous time points. These observations are in agreement with other studies showing that migration of melanoma and cervical cancer cell lines (HBL and HW756) was increased with TNF-α-stimulation. A maximal effect on cell migration on these cell lines was observed between 20 and 24 hours,41,44 during which an increase in ICAM-1 45 and integrin 46 expression on the tumor cells was observed.
In summary, translational Immunohistochemical-based investigations, using human breast tumors and melanomas, have previously demonstrated that lymphatic vessels play an important role in the initial metastatic process from the primary tumour. We have shown that TNF-α-stimulation of both tumor and endothelial cells causes a significant increase in tumor cell adhesion, of which the highest increase is observed on LEC models. Suitable in vitro models are required to fully elucidate the mechanism(s) regulating lymphovascular invasion, which in some tumor types occurs more frequently to lymphatics than invasion of blood vessels. The current study focuses on the adhesion of tumor cells to blood and lymphatic endothelium, and the migration of tumour cells, following cytokine stimulation. It will be interesting, in future work, to examine if such increased migration and endothelial adhesion is paralleled by altered transmigration of tumor cells across blood and lymphatic endothelium to determine if this is also preferentially altered with lymphatic endothelial models following cytokine stimulation. Current results suggest that metastasis resultant from lymphatic vessel–tumor cell adhesion may be modulated by cytokine stimulation, which could represent an important therapeutic target in breast cancer and melanoma.
Footnotes
Author Disclosure Statement
The authors received funding from the British Skin Foundation (Grant number S401) and the Breast Cancer Campaign (Grant number 2011NovSP25) for this work. S. Safuan was also funded by the Ministry of Higher Education Malaysia and the Science University Malaysia. S. Storr was also supported by the Davies Memorial Trust.