
Abstract
Purpose:
Pharmacokinetic evaluation of ocular penetration and systemic accumulation of preservative-free bimatoprost 0.01% ophthalmic gel (PFB 0.01% gel).
Methods:
In a preclinical study, pigmented rabbits received a single ocular administration of PFB 0.01% gel (N = 15) or preserved bimatoprost 0.01% or 0.03% ophthalmic solution [PB 0.01% (N = 15) or PB 0.03% (N = 15)]. The aqueous humor, iris, and ciliary body were analyzed for bimatoprost+bimatoprost free acid. In a Phase 1, randomized, open-label clinical study, healthy participants received PFB 0.01% gel (N = 20) or PB 0.01% (N = 20) daily in each eye (Days 1–15). Bimatoprost levels in human plasma were analyzed on Days 1 and 15. All serological analyses used validated methods. Adverse events were collected throughout and ocular assessments were performed on Days 1 and 15.
Results:
In the preclinical study, Cmax (bimatoprost+bimatoprost free acid) for PFB 0.01% gel, PB 0.01%, and PB 0.03% was 50.2, 26.3, and 59.9 ng/mL; AUC0.5–8 h was 134.0 ng·h/mL, 67.0 ng·h/mL, and 148.0 ng·h/mL. In the clinical study, systemic exposure to bimatoprost (AUC0–last) on Days 1 and 15 was lower for PFB 0.01% gel (0.5248 and 0.5645 ng·min/mL) than PB 0.01% (0.8461 and 0.7551 ng·min/mL), with no systemic accumulation of bimatoprost in either group. There were no clinically important differences between groups in ocular or systemic tolerability in the clinical study and no serious adverse events.
Conclusions:
PFB 0.01% gel showed improved ocular penetration compared with PB 0.01%. Systemic absorption was comparable, with a favorable clinical safety profile, supporting PFB 0.01% gel as a potential treatment for glaucoma and ocular hypertension.
Introduction
The preferred first-line treatment for the pharmacological management of glaucoma and ocular hypertension (OHT) is the use of prostaglandin analogs (PGAs). 1 Although PGAs can effectively reduce intraocular pressure (IOP) with daily dosing, they are associated with ocular adverse events (AEs), including conjunctival hyperemia, eye pruritus, eyelid erythema, and eyelash growth.2,3
Preserved bimatoprost 0.03% ophthalmic solution (PB 0.03%) is a PGA that was approved in the United States in 2001 and in Europe in 2002 as Lumigan® 0.03% (Allergan Inc, Ireland), with proven efficacy in lowering IOP in patients with glaucoma and OHT.4–6
Although it showed an acceptable safety profile, its use was associated with reduced tolerability, notably conjunctival hyperemia. This resulted in the development of a new formulation with a lower concentration of bimatoprost, PB 0.01% ophthalmic solution (PB 0.01%), approved in 2010 as Lumigan 0.01% (Allergan Inc., Ireland), shown to have comparable efficacy and improved tolerability, in particular, less frequent and severe conjunctival hyperemia.3,7,8 However, to improve penetration of bimatoprost for the lower dose solution, a 4-fold higher concentration of the preservative benzalkonium chloride (BAK), which also acts to enhance ocular penetration,
9
was included in PB 0.01% (200 ppm BAK vs. 50 ppm in PB 0.03%). Since long-term exposure to BAK is associated with ocular AEs, an increased risk of glaucoma surgery failure, and reduced adherence to treatment regimens,10–12
the higher concentration of BAK included in PB 0.01% may exacerbate or cause ocular surface disease, particularly when used as a long-term daily treatment for glaucoma.
13
This led to the development of a novel preservative-free bimatoprost 0.01% ophthalmic gel (PFB 0.01% gel) formulation, approved as Elymbus® in Europe since 2024 (
In a recent Phase 3 trial, noninferiority in terms of IOP-lowering efficacy along with a more favorable safety profile, in particular a reduced incidence of conjunctival hyperemia, was demonstrated for PFB 0.01% gel compared with PB 0.01%. 14 Here we report preclinical ocular pharmacokinetic data following topical administration of PFB 0.01% gel in comparison with PB 0.01% and PB 0.03%, as well as Phase 1 clinical systemic pharmacokinetic data following ocular administration of PFB 0.01% gel in comparison with PB 0.01%. The aims of these studies were to demonstrate ocular penetration of the gel formulation in an animal model and to evaluate systemic absorption of bimatoprost with this novel formulation.
Methods
Preclinical study
Study design and animals
Forty-five female Dutch-Belted (pigmented) rabbits aged 2–3 months and weighing 2.0–2.5 kg (supplied by Envigo, Denver, PA, USA) were observed for signs of illness with particular attention paid to their eyes for 5 days before the start of the study. Animals were ear-tagged and also identified in their ears using indelible ink following inclusion. They were housed in pairs in standard cages under identical environmental conditions (temperature 18°C ± 3°C and relative humidity 45%–80%), and rooms were continuously ventilated (15–20 air volumes/h) and had a light- and darkness-controlled cycle (light from 7 am to 7 pm). Animals had free access to food (∼90 g/day of a standard dry pellet diet) and tap water. The general clinical signs and appearance of all animals were observed throughout the study, and all animals were treated according to the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research.
15
Iris Pharma (La Gaude, France) conducted the preclinical study on behalf of
Animals in good health, without visible ocular defects, and within ±20% of the mean body weight were included in the study and randomized in a 1:1:1 ratio to receive PFB 0.01% gel (0.1 mg/g bimatoprost), PB 0.01% (0.1 mg/mL bimatoprost), or PB 0.03% (0.3 mg/mL bimatoprost). All animals received a single ocular topical administration (25 µL) in the upper corneoscleral limbus of each eye on Day 1. In each group, 3 animals were euthanized by an intravenous injection of pentobarbital following sedation at 0.5, 1, 2, 4, and 8 h after administration, providing 6 eyes at each time point in each group. Immediately after euthanasia, the aqueous humor (AH) and iris and ciliary body (ICB) were sampled. Internal standards (bimatoprost-d4 and bimatoprost free acid-d4) were added to the samples and then treated with ethyl acetate. After centrifugation, the organic layer was evaporated under a nitrogen stream, and the dry residue was dissolved in methanol/water and analyzed by a validated rapid resolution liquid chromatography/tandem mass spectrometry (RRLC-MS/MS) method. Bimatoprost and bimatoprost free acid concentrations were then determined from the ocular matrices [lower limits of quantification (LLOQ): 0.63 ng/mL in AH and 1.8 ng/mL in ICB for bimatoprost; 1.3 ng/mL in AH and 3.6 ng/mL in ICB for bimatoprost free acid].
Phase 1 clinical study
Study design and participants
A Phase 1, randomized, open-label, parallel-group study was conducted at a single study site in the United States (ClinicalTrials.gov Identifier: NCT05729594). The study protocol and one protocol clarification regarding the bioanalytical method were approved by an independent ethics committee and the study was performed according to the ethical principles that have their origins in the Declaration of Helsinki and the International Council for Harmonization guidelines for Good Clinical Practice. An informed consent form was signed by each participant before enrollment into the study. The study was conducted between February and April 2023.
Healthy male or female participants aged between 18 and 55 years and with no ocular symptoms and normal ocular examination [far best corrected visual acuity ≤2 in LogMAR (e.g., ≥20/32 Snellen equivalent) and no ocular surface abnormalities] were eligible for inclusion. The main exclusion criteria were as follows: history of ocular trauma, infection, or inflammation in the 3 months preceding the study; ocular pathology such as blepharitis, conjunctivitis, uveitis, or other ocular infection or inflammation; IOP <10 mmHg or >21 mmHg; diabetes; hypersensitivity to any component of PFB 0.01% gel, PB 0.01%, or PB 0.03%; body mass index <18 kg/m2 or >32 kg/m2; women who were pregnant or breastfeeding, or not using adequate contraception; drug or alcohol abuse; planned application of eye makeup during the study, or permanent makeup procedure such as eyelash tinting or eyeliner tattoos within 1 week before the study or planned during the study; and any condition judged by the investigator to be prohibitive to study participation.
Participants were randomly assigned in a 1:1 ratio to receive one drop of PFB 0.01% gel or PB 0.01% in the conjunctival sac of each eye at 3 pm (±1 h) from Day 1 to Day 15. Participants attended the study site on Day 1 (Visit 1) and Day 15 (Visit 2). On these occasions, PFB 0.01% gel or PB 0.01% was administered by study site staff; on Days 2 to 14, the administration was performed by the participant. Punctal occlusion was not performed.
Blood samples (∼6 mL on each occasion) were collected on Days 1 and 15 before administration and at 5 min, 10 min, 15 min, 20 min, 30 min, 1 h, and 3 h postadministration. Serological analysis of bimatoprost was performed using a validated method, which included solid-phase extraction plasma sample preparation and LC–MS/MS (LLOQ: 0.005 ng/mL).
AEs were collected from Days 1 to 15. This period was extended to follow-up on all ongoing AEs after the end of the treatment period, until fully resolved or when medically justifiable to stop further follow-up. The collection of AEs was done by study site staff on Days 1 and 15, and using participant diaries from Days 2 to 14. Vital signs, physical examination, and ocular assessments [visual acuity (Snellen chart), slit lamp examination (blepharitis, eyelid edema, change in iris pigmentation, abnormal eyelash aspect, follicular/papillary conjunctivitis, and other ocular abnormality evaluated on a 0 {none}−3 {severe}-point severity scale), conjunctival hyperemia (McMonnie’s photographic scale, 0–5), corneal fluorescein staining (modified Oxford 7-point scale), and conjunctival staining (modified Oxford 7-point scale)] were performed before administration on Day 1 (baseline) and Day 15.
Test products
In each study, PFB 0.01% gel (
Statistical analyses
The objective of the preclinical study was to compare the bioavailability of bimatoprost and bimatoprost free acid in the target tissues (AH and ICB). All analyses were descriptive. Pharmacokinetic parameters [apparent Cmax (maximum observed concentration), tmax (time to reach Cmax), AUC0.5–8 h (area under the concentration–time curve from time 0.5 to 8 h)] were calculated using Microsoft Excel® software (version 16.69.1) and concentrations were expressed in ng/g or ng/mL of tissue specimen. Sample concentrations below the LLOQ were replaced by “0” for mean and standard deviation (SD) calculations, and AUC was calculated using the linear trapezoidal method using linear interpolation between data points.
The primary endpoint of the clinical study was the plasma concentrations of bimatoprost (bimatoprost free acid was not quantifiable in plasma) at each time point. The following pharmacokinetic parameters were planned to be calculated: AUC0–3 h inf (area under the plasma concentration–time curve from time 0 to 3 h), AUC0–last (area under the plasma concentration–time curve from time 0 to tlast), AUC0–inf (area under the plasma concentration–time curve from time zero to infinity), Clast (last quantifiable observed plasma concentration), Cmax, tlast (time to reach Clast), tmax, λz (apparent terminal elimination rate constant), and t½ (terminal elimination half-life), as well as accumulation ratios (Racc) for Cmax and AUC0–3 h at Day 1 and Day 15. However, λz, t½, and AUC0–inf were not calculated as predefined rules for their calculation were not achieved for more than half of the participants. Secondary endpoints included an evaluation of AEs and ocular signs.
In the clinical study, the pharmacokinetic analyses used the pharmacokinetic analysis set (participants who received at least one dose of PFB 0.01% gel or PB 0.01% without any event and/or a major protocol deviation affecting the pharmacokinetic evaluation and with at least one visit and a complete pharmacokinetic profile (i.e., all pharmacokinetic samples done). The safety set (all participants who received at least one dose of PFB 0.01% gel or PB 0.01%) was used for the secondary endpoints. Demographic and safety data were analyzed using SAS® software version 9.4 (SAS Institute Inc., Cary, NC, USA), and AEs were coded using the Medical Dictionary for Regulatory Abbreviations (MedDRA) version 25.1. Pharmacokinetic data were analyzed using Phoenix® WinNonlin® version 8.1 or higher (Certara Inc., Princeton, NJ, USA) and SAS software version 9.4.
There was no formal sample size calculation for either the preclinical study or the Phase 1 clinical study since no formal statistical hypothesis was tested and all analyses were descriptive except for between-group comparisons of bimatoprost and bimatoprost free acid concentrations in the AH and ICB using the Wilcoxon rank-sum test for the comparison of two samples, with P = 0.05 as the threshold for statistical significance. This nonparametric test, which compares median rather than mean values, was used since the hypothesis of data normality could not be confirmed due to the small sample size. A total of 3 animals (6 eyes) per group per time point for the preclinical study and 20 participants per group (40 participants overall) for the Phase 1 clinical study were considered sufficient to meet the study endpoints.
Results
Preclinical study
A total of 3 animals (6 eyes) for each time point and each treatment (i.e., 15 animals per treatment overall) were evaluated as planned. All animals were female. The evolution of body weight was normal for all animals, and no behavioral changes were observed.
Bimatoprost and/or bimatoprost free acid were quantified from all 3 treatment groups from the earliest time point (0.5 h). The kinetic profiles in both AH and ICB remained similar between the different preparations, displaying the same tmax at 1 h, and levels below LLOQ by 8 h postadministration. However, in AH, median bimatoprost+bimatoprost free acid concentration was significantly higher (P < 0.05) for PFB 0.01% gel versus PB 0.01% at 0.5 and 2 h after administration and for PB 0.03% versus PB 0.01% at 0.5, 1, and 4 h after administration (Fig. 1A), and in ICB, median bimatoprost+bimatoprost free acid concentration was significantly higher (P < 0.05) for PFB 0.01% gel versus PB 0.01% at 0.5 and 4 h after administration and for PB 0.03% versus PB 0.01% at 0.5, 1, and 2 h after administration (Fig. 1B).
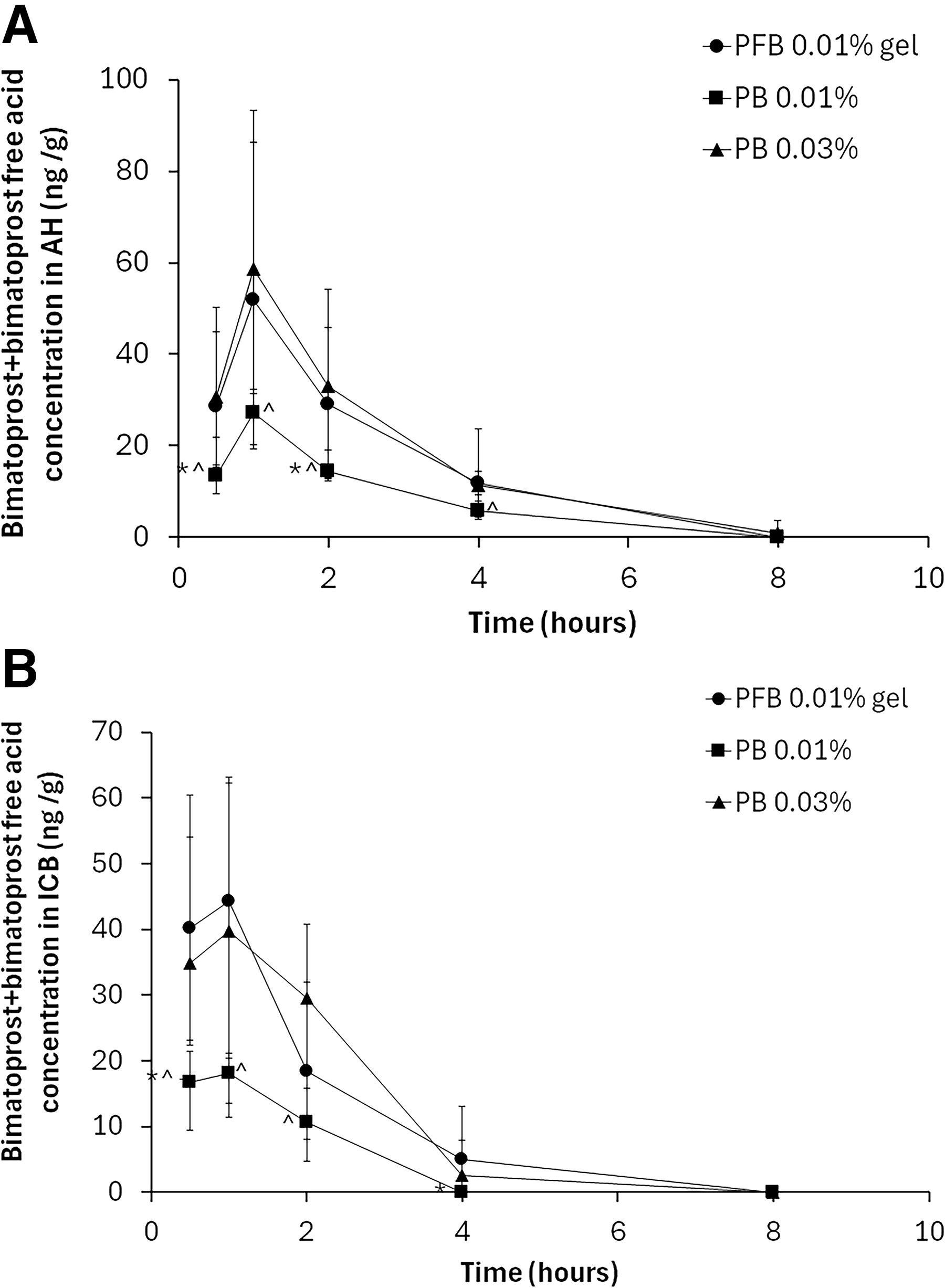
Data are median ± 95% confidence interval concentrations of bimatoprost+bimatoprost free acid. *P < 0.05 for comparison of PB 0.01% versus PFB 0.01%. ^P < 0.05 for comparison of PB 0.01% versus PB 0.03%. n = 3 animals (6 eyes) per time point. AH, aqueous humor; ICB, iris and ciliary body; PB 0.01%, preserved bimatoprost 0.01% ophthalmic solution; PFB 0.01%, preservative-free bimatoprost 0.01% ophthalmic gel.
In AH, mean Cmax for bimatoprost+bimatoprost free acid was 50.2, 26.3, and 59.9 ng/mL following a single topical ocular administration of PFB 0.01% gel, PB 0.01%, and PB 0.03%, respectively. In ICB, bimatoprost+bimatoprost free acid mean Cmax was, respectively, 39.5 ng/mL, 17.9 ng/mL, and 42.0 ng/mL. The AUC for each treatment reflected the same trend of concentration–time as observed for Cmax in both AH and ICB (Table 1).
Pharmacokinetic Parameters of Bimatoprost+Bimatoprost Free Acid in Aqueous Humor and Iris Ciliary Body of Dutch-Belted (Pigmented) Rabbits (Preclinical Study)
Data are mean Cmax, tmax, and AUC0.5–8 h.
Number of eyes (n = 3 animals).
AH, aqueous humor; AUC0.5–8 h, area under the AH or ICB concentration–time curve from time 0.5 to 8 h; Cmax, maximum observed AH or ICB concentration; ICB, iris ciliary body; PB 0.01%, preserved bimatoprost 0.01% ophthalmic solution; PB 0.03%, preserved bimatoprost 0.03% ophthalmic solution; PFB 0.01% gel, preservative-free bimatoprost 0.01% ophthalmic gel; tmax, time to reach Cmax.
Maximal concentrations and AUCs of bimatoprost+bimatoprost free acid in AH and ICB were achieved with PFB 0.01% gel, which displayed a 2- to 2.6-fold increase compared with PB 0.01% and remained similar (from 0.9- to 0.95-fold) to PB 0.03%.
Phase 1 clinical study
Participant demographics
A total of 40 participants were enrolled in the Phase 1 clinical study, and all participants completed the study as planned (20 participants in each of the PFB 0.01% gel and PB 0.01% groups).
Demographic and baseline characteristics were similar in the PFB 0.01% gel and PB 0.01% groups, respectively, for age (38.0 and 33.9 years), sex (65% and 75% female participants), ethnicity (25% Hispanic or Latino in each group), race (65% and 55% Black or African American, 30% and 35% White, 5% and 10% Asian), iris color (100% and 90% brown, hazel, or black), BMI (25.27 and 25.15 kg/m2), and IOP (left eye: 17.3 and 16.7 mmHg; right eye: 17.3 and 17.1 mmHg) (Table 2).
Demographic and Baseline Characteristics (Phase 1 Clinical Study)
BMI, body mass index; IOP, intraocular pressure; max, maximum; min, minimum; N, number of participants in group; n, number of participants; SD, standard deviation.
Pharmacokinetics
In both groups, plasma bimatoprost concentrations were low and close to LLOQ (0.005 ng/mL) throughout Day 1 and Day 15. The plasma concentration–time data showed bimatoprost concentrations to be lower in the PFB 0.01% gel group than in the PB 0.01% group on Day 1 (Fig. 2A) and Day 15 (Fig. 2B). In terms of the pharmacokinetic parameters (Table 3), mean Cmax was lower for PFB 0.01% gel than PB 0.01% on Day 1 (0.02147 and 0.03148 ng/mL, respectively) and Day 15 (0.02811 and 0.03147 ng/mL), and median tmax was longer for PFB 0.01% gel (10.0 min on each day) than PB 0.01% (7.5 min on Day 1 and 5.0 min on Day 15). These findings were reflected in the mean AUC0–last data for PFB 0.01% gel and PB 0.01% (0.5248 and 0.8461 ng·min/mL on Day 1 and 0.5645 and 0.7551 on Day 15). For AUC0–3 h, values were also lower for PFB 0.01% gel than PB 0.01% on Day 1 (0.7530 and 1.2072 ng·min/mL) and Day 15 (0.7261 and 1.0919 ng·min/mL). Median tlast was shorter for PFB 0.01% gel (30.0 min on each day) than PB 0.01% (60.0 min on each day). Mean Clast was comparable for each group on each day.
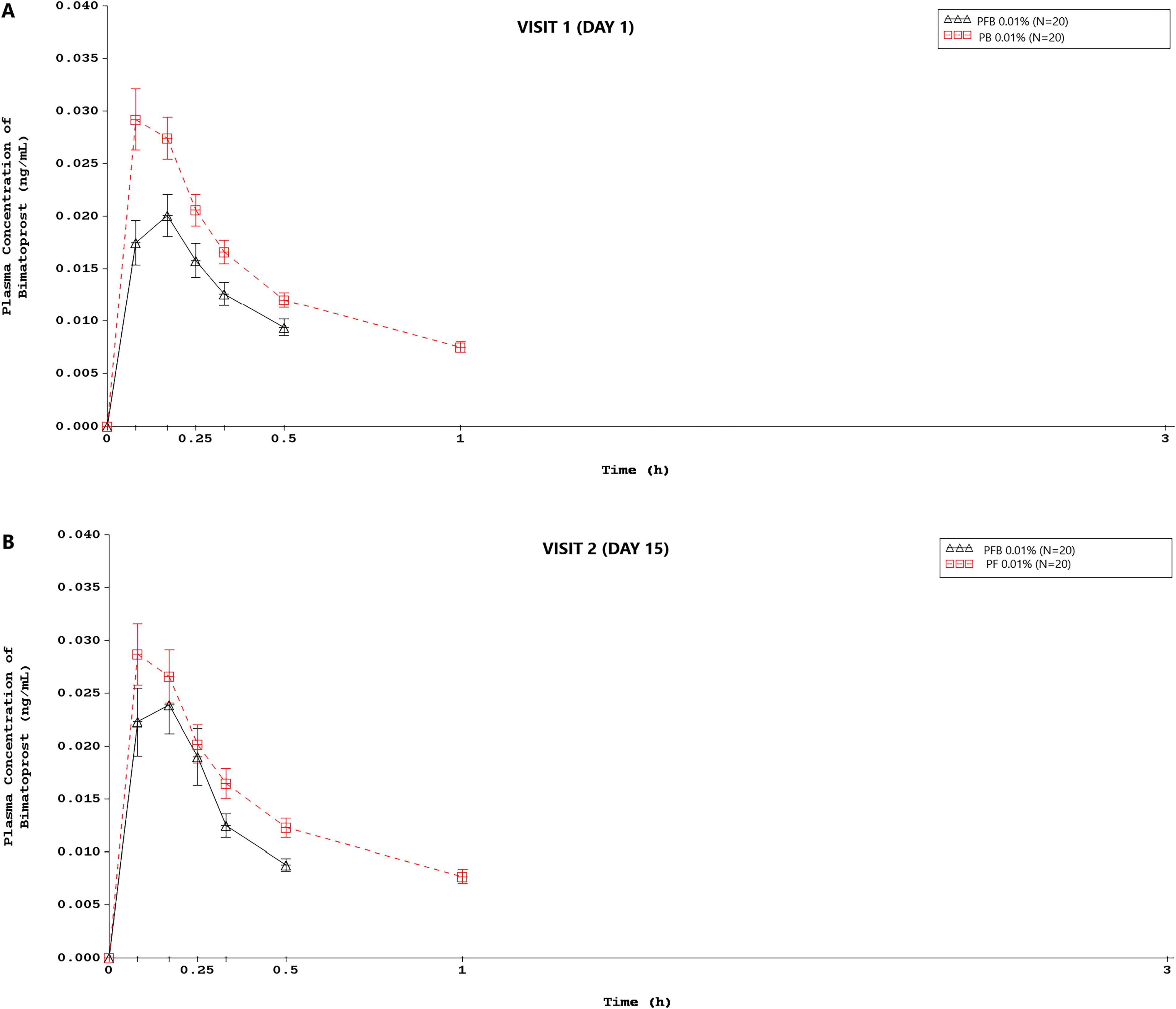
Data are arithmetic mean ± standard error of the mean; N = number of participants in group. Data <LLOQ before the first measurable concentration are replaced by “0.” All data <LLOQ after the first measurable concentration are set to missing. One participant in the PFB 0.01% gel group did not receive the treatment at Day 14 and was excluded from the pharmacokinetic analysis at Day 15. LLOQ, lower limits of quantification.
Pharmacokinetic Parameters of Bimatoprost in Human Plasma (Phase 1 Clinical Study)
One participant did not receive PFB 0.01% gel at Day 14 and was excluded from the pharmacokinetic analysis at Day 15.
AUC0–3 h, area under the plasma concentration–time curve from time zero to 3 h; AUC0–last, area under the plasma concentration–time curve from time zero to tlast; Clast, last quantifiable observed plasma concentration; Cmax, maximum observed plasma concentration; CV%, percent geometric coefficient of variation; min, minimum; max, maximum; Racc, accumulation ratio for Cmax; Racc AUC0–3 h, accumulation ratio for AUC0–3 h; tlast, time to reach Clast.
Accumulation ratios for Cmax and AUC0–3 h were close to 1 except for Racc Cmax for PFB 0.01% gel, which was 1.404, reflecting no overall accumulation of bimatoprost in plasma in either group (Table 3).
Safety
The overall safety profile was comparable in both groups. For ocular AEs, 21 AEs were reported by 11 (55.0%) participants in the PFB 0.01% gel group, and 16 AEs were reported by 10 (50.0%) participants in the PB 0.01% group (Table 4). The most common ocular AEs in the PFB 0.01% gel group were (conjunctival and ocular) hyperemia [9 (45.0%) participants], and dry eye, eye pruritus, and blurred vision [all experienced by 3 (15.0%) participants]. In the PB 0.01% group, the most common were (conjunctival and ocular) hyperemia [5 (25%) participants], and dry eye and eye pruritus [all experienced by 2 (10.0%) participants]. All, except one ocular AE (reduced visual acuity in 1 participant in the PB 0.01% group), were considered to be related to the study treatment. Four (20.0%) participants in the PFB 0.01% gel group and 2 (10.0%) participants in the PB 0.01% group reported at least one systemic AE, mainly respiratory, thoracic, and mediastinal disorders [oropharyngeal pain (3 participants), cough, dry throat, and headache (1 participant each) in the PFB 0.01% gel group; throat irritation (2 participants) in the PB 0.01% group]. All systemic AEs except single episodes of cough and headache were considered to be related to the study treatment. There were no serious AEs throughout the study, and no AE led to treatment discontinuation in either group.
Ocular Treatment-Emergent Adverse Events (Phase 1 Clinical Study)
AE, adverse event.
For conjunctival hyperemia, 8 (40%) participants (PFB 0.01% gel) and 3 (15%) participants (PB 0.01%) experienced worsening at Day 15 (mean ± SD maximal worsening: 0.6 ± 0.9 in the PFB 0.01% gel group and 0.2 ± 0.4 in the PB 0.01% group). Corneal and conjunctival staining worsened in 9 (45%) participants (PFB 0.01% gel) and 6 (30%) participants (PB 0.01%) (mean ± SD maximal worsening: 0.43 ± 0.49 in the PFB 0.01% gel group and 0.15 ± 0.56 in the PB 0.01% group).
Discussion
Bimatoprost is the most potent PGA prescribed for the treatment of glaucoma and OHT,16–18 but conjunctival hyperemia associated with chronic use of high-concentration bimatoprost formulations as well as ocular surface problems associated with the inclusion of BAK in preserved formulations, including tear film instability, conjunctival squamous metaplasia and apoptosis, and corneal epithelium disruption, presents barriers to its use.13,19,20 PB 0.01%, developed to counter the problems associated with higher bimatoprost concentrations, contains an increased concentration of BAK to enhance bimatoprost penetration, which is in turn associated with an increased incidence of ocular surface problems. Consequently, clinical development of a novel, preservative-free, low-concentration bimatoprost eye gel (PFB 0.01% gel) was initiated to create an ocular hypotensive medication with comparable efficacy to the marketed PB 0.01%, without the addition of BAK. The gel formulation was developed to increase residence time on the ocular surface compared with an ophthalmic solution such as PB 0.01%, and therefore increase ocular penetration of bimatoprost without the need for a preservative.21,22 In this way it has been possible to maintain ocular penetration and therefore efficacy of bimatoprost 0.01%, while eliminating BAK to avoid tolerability problems associated with the preservative.
To compare the ocular penetration of the new gel formulation to two existing ophthalmic solutions (PB 0.01% and PB 0.03%), an animal model was used to be able to evaluate intraocular drug concentrations. The rabbit is a widely used and suitable animal model for ocular pharmacokinetic studies, with a similar eye size to humans, 23 and Dutch-Belted pigmented rabbits have been used previously in pharmacokinetic studies for a range of glaucoma medications.24–27 The preclinical data that we describe in Dutch-Belted pigmented rabbits showed rapid absorption of bimatoprost into the eye following a single-dose administration of PFB 0.01% gel, with substantially increased overall exposure compared with PB 0.01% and closer to PB 0.03%. These findings are supported by a large Phase 3 clinical trial in patients with open-angle glaucoma (OAG) or OHT, where PFB 0.01% gel and PB 0.01% showed similar efficacy, and noninferiority between groups was demonstrated for the change from baseline in IOP after 12 weeks of treatment. Importantly, in this Phase 3 study, local ocular tolerability, particularly conjunctival hyperemia and corneal fluorescein staining, eye irritation/burning, and eye dryness were better for the PFB 0.01% gel than PB 0.01% over the 12-week period of the study. 14 Of note, AEs typically associated with BAK (irritation/burning, dry eye, eye pruritus) 28 were observed at a higher frequency in the PB 0.01% group compared with the PFB 0.01% gel, and in terms of global local tolerance there was a statistically significant between-group difference favoring PFB 0.01% gel at week 12. 14
It is important to demonstrate that there is no impact on systemic absorption or accumulation of bimatoprost by the new gel formulation that could potentially arise from the prolonged ocular residence time. Consequently, the Phase 1 clinical study was performed in healthy participants to evaluate the systemic pharmacokinetics following daily ocular instillation of PFB 0.01% gel compared with PB 0.01% over 15 days, with safety assessed as secondary endpoints. In this Phase 1 study, bimatoprost was rapidly available in plasma (within 10 min postdose) for both formulations, and systemic exposure was comparable between the two treatments. Importantly, there was no evidence of systemic accumulation of bimatoprost in either group. In a Phase 2 trial that used the same gel formulation but also included timolol, systemic concentrations of bimatoprost were below the limit of detection (0.100 ng/mL) after 12 weeks of once-daily administration. 29
The safety data from the Phase 1 study that we report showed a similar incidence of AEs between PFB 0.01% gel and PB 0.01%, with most ocular AEs being considered treatment-related in each group. It was noted that more conjunctival hyperemia was reported for PFB 0.01% gel (8 [45%] participants) than for PB 0.01% (5 [25%] participants), which is in contrast with the findings of the Phase 3 study described above in which the tolerability profile of PFB 0.01% gel was better than PB 0.01%. However, the two studies were conducted in different settings. The Phase 1 study was in 40 healthy participants and conducted over a relatively short period (15 days), whereas the Phase 3 study was in 485 OAG and OHT patients over a relatively longer period (12 weeks). Considering the difference in study populations, the effect of bimatoprost in the healthy participants included in the Phase 1 study may be more pronounced with an eye gel, which has a longer residency time compared with an eye-drop solution. In addition, it is likely that the short treatment period in the Phase 1 study was insufficient to highlight the better ocular tolerability that would be expected for a preservative-free formulation compared with a BAK-preserved formulation. In contrast, the adverse effects of BAK would be expected to be more apparent in a patient population such as that in the Phase 3 study (glaucoma or OHT patients who had used a PGA medication daily for at least 6 months before study entry), as the ocular intolerability of BAK is generally seen over the longer term. Therefore, the safety results of the Phase 3 study are considered to be more reflective of the real-world situation.
The strengths of the data presented in this article are the inclusion of both preclinical ocular pharmacokinetics and systemic clinical pharmacokinetics, which together provide a comprehensive pharmacokinetic evaluation of PFB 0.01% gel in comparison with PB 0.01%. Limitations of the preclinical study include the single-dose, rather than multiple-dose design to investigate the effect of chronic dosing on ocular pharmacokinetics. For the Phase 1 clinical study, the healthy population, small sample size, and short treatment period are limiting factors in terms of the safety assessment. As described, real differences in ocular tolerability due to the presence of BAK may only become apparent after longer periods of chronic dosing in patients, and the study period of 15 days was considered sufficient to meet the pharmacokinetic endpoints of this Phase 1 study.
Conclusions
We have shown that a novel preservative-free gel formulation of 0.01% bimatoprost shows higher bioavailability in rabbit ocular tissue than preserved 0.01% bimatoprost solution, which contains a high concentration of BAK. The overall systemic pharmacokinetics in humans was comparable for both products. Combined with the favorable tolerability results from a Phase 3 study, PFB 0.01% gel presents a new and valuable tool in the treatment of glaucoma and OHT.
Footnotes
Acknowledgments
The authors thank the Phase 1 study volunteers for their participation. The authors would also like to acknowledge Mrs. Grissel Flores, the principal investigator of the Phase I clinical trial, and Karen Viaud and Sylvia Ghione of Iris Pharma. Dr. Andrew Lane (Lane Medical Writing) provided medical writing assistance in the preparation and development of the article in accordance with the European Medical Writers Association guidelines and Good Publication Practice and was funded by Laboratories Théa.
Data Sharing
All individual data that underlie the results reported in this article as well as the study protocols will be made available to researchers who provide a methodologically sound proposal by application to the corresponding author.
Authors’ Contributions
C.E.: Conceptualization and writing—review and editing. F.T.: Conceptualization and writing—review and editing. H.J.: Conceptualization and writing—review and editing. F.A.: Methodology, investigation, and writing—review and editing. S.N.: Methodology, data curation, formal analysis, validation, and writing—review and editing. F.J.M.-N.: Writing—review and editing. I.S.: Conceptualization and writing—review and editing.
Author Disclosure Statement
C.E.: Consulting and/or presentations from/at AbbVie, Santen,
Funding Information
The studies reported in this article were funded by