
Abstract
Dry eye disease (DED) is a common ocular condition that can impair vision and may adversely impact quality of life. Due to the inflammatory nature of this disorder, topical corticosteroids are an effective treatment option, particularly for moderate-to-severe DED when first-line treatments, such as ocular lubricants, are insufficient. Loteprednol etabonate (LE) is a retrometabolically designed corticosteroid with a low propensity to cause corticosteroid-related adverse effects, such as elevated intraocular pressure (IOP). This review was conducted to provide an assessment of published studies on the use of LE for treatment of inflammation associated with DED. Twelve prospective and 2 retrospective studies evaluating LE ophthalmic suspension 0.5% and 2 prospective studies evaluating LE ophthalmic gel 0.5% were identified. LE given as monotherapy or with artificial tears (AT) improved signs of DED, especially among patients with a more pronounced inflammatory component, and also improved DED symptoms compared to baseline and/or control. Treatment with LE before cyclosporine A (CsA) therapy reduced stinging upon CsA initiation and provided more rapid relief of DED signs and symptoms than CsA plus AT alone. In patients with meibomian gland dysfunction, LE alone, or in addition to eyelid scrubs/warm compresses, reduced clinical signs and symptoms, and tear proinflammatory cytokine levels. Overall, LE was safe and well tolerated, with minimal effects on IOP. While larger and longer-term studies are warranted, these data support the use of LE as a safe and effective treatment option for DED.
Introduction
Dry eye disease (DED) or dysfunctional tear syndrome (DTS), clinically referred to as keratoconjunctivitis sicca, is defined by the Tear Film and Ocular Surface Society's Dry Eye Workshop II (TFOS DEWS II) panel as a multifactorial disease of the ocular surface characterized by a loss of homeostasis of the tear film and accompanied by ocular symptoms, such as discomfort and visual disturbance. This condition involves tear film instability and hyperosmolarity, ocular surface inflammation and damage, and neurosensory abnormalities. 1 The central mechanism underlying the pathophysiology of DED is ocular surface water loss leading to hyperosmolar stress, which can result either from reduced lacrimal secretion (aqueous deficient DED) or excessive evaporation from the exposed tear film (evaporative DED). 2 For both these types of DED, a myriad of factors may contribute to the initiation and progression of the disease, both physiological and environmental. Especially in later stages of the disease, both lacrimal deficiency and evaporative loss can contribute to hyperosmolarity of the tear film, further exacerbating the condition. 2 The resulting hyperosmolar stress directly or indirectly may induce inflammation resulting in tissue damage and a loss of epithelial and goblet cells. This further decreases surface wettability leading to local tear instability/early tear film breakup and resultant worsening hyperosmolarity via a vicious circle. 2 Tear hyperosmolarity, loss of lubrication, neurosensory factors, and inflammatory mediators contribute to pain in DED, while tear and ocular surface irregularities cause visual symptoms. 2
Current etiological classification of DED considers the nonmutually exclusive categories of aqueous-deficient dry eye (ADDE) and evaporative dry eye (EDE), 1 with ADDE divided into Sjögren's syndrome (SS) dry eye and non-SS dry eye and EDE comprising lid-related, or intrinsic, EDE, and ocular surface-related EDE. 2 Hybrid subtypes of DED, including components of both ADDE and EDE, have also been described. 2 According to an expert panel from the Cornea, External Disease, and Refractive Society (referred to as the CEDARS DTS panel), the 4 main subtypes/causes of DTS/DED are aqueous deficiency, blepharitis/meibomian gland dysfunction (MGD; evaporative and nonevaporative), goblet cell/mucin deficiency, and exposure-related DTS. 3
The global prevalence of symptomatic DED ranges from 5% to 50%, while studies in which DED was diagnosed primarily based on signs have generally yielded higher estimates and more variable rates of disease. 4 An analysis of data from the 2013 National Health and Wellness Survey, a cross-sectional, population-based survey of U.S. adults 18 years of age and older, reported a prevalence of diagnosed DED of 6.8%, corresponding to 16.4 million people in 20135; an additional 2.5% of participants (corresponding to ∼6 million people) reported symptoms of DED but had not received a diagnosis. 5 Similarly, in a large U.S. database, the overall prevalence of DED between 2003 and 2015, based on diagnostic codes and pharmacy claims, was 5.3%. 6 Both analyses found that the prevalence of DED was higher among women than men and increased with age.5,6 Another large U.S. claims database analysis showed that DED represented the sixth most common ocular condition for which patients sought care. 7 DED can impair vision (ie, difficulty reading, driving at night, performing computer work, and watching television) across all levels of disease severity, although generally greater limitations occur with increasing disease severity. 8 Accordingly, DED is associated with markedly reduced work productivity and work performance as well as with impairment in activities of daily living outside of work. 9 DED also has a substantial negative impact on health-related quality of life, with burden increasing with severity of disease.10,11
Tear replacement with ocular lubricants is often used in the initial management of DED and has historically been considered a mainstay of treatment.12,13 Currently 3 prescription medications are approved in the U.S. to treat DED, 2 containing the active cyclosporine A (CsA)14,15 and 1 containing lifitegrast, 16 however, these medications can take weeks or even months to reach their full effectiveness and may cause burning and/or irritation,3,14–19 which can, in turn, affect adherence and persistence.3,14–22 Topical corticosteroids, while not approved by the U.S. Food and Drug Administration (FDA) for treatment of DED, have demonstrated efficacy across a range of ocular inflammatory conditions, including DED, and are often used off label to treat moderate-to-severe DED.12,23 By targeting inflammation, topical corticosteroids may help to break the vicious cycle of inflammatory immune responses in DED. 12 However, the long-term use of ocular corticosteroids is associated with a risk of a number of complications, including ocular hypertension, glaucoma, cataracts, and increased susceptibility to infections.12,23,24
Loteprednol etabonate (LE) is an ocular corticosteroid that was retrometabolically synthesized via modification from an inactive metabolite of prednisolone acetate (PA) and has a 17β-chloromethyl ester at the C-20 position in place of the ketone group found in PA, in addition to a 17α-etabonate moiety.25,26 These modifications preserve the desired pharmacological effect while enabling rapid metabolism to inactive metabolites to limit excess exposure and thus reduce the potential for adverse reactions.27,28 Accordingly, LE has a low risk of clinically significant intraocular pressure (IOP) elevations with both short-term and long-term use and exhibits lower rates of clinically significant IOP elevation compared with other corticosteroids. 29 The incidence of adverse event (AE) reports such as IOP increase, glaucoma, ocular hypertension, and cataracts in association with the use of LE is also reported to be very low. 30
LE was initially formulated as an ophthalmic suspension at concentrations of 0.2% 31 and 0.5%.32,33 LE ophthalmic suspension 0.5% is indicated for the treatment of steroid-responsive inflammatory conditions of the palpebral and bulbar conjunctiva, cornea, and anterior segment of the eye as well as for the treatment of postoperative inflammation following ocular surgery. 32 LE ophthalmic suspension 0.2% is indicated for the temporary relief of signs and symptoms of allergic conjunctivitis. 31 Three additional LE formulations have been approved by the FDA for the treatment of postoperative inflammation and pain following ocular surgery. A nonsettling gel formulation of LE 0.5% was approved in 201234,35 and provides consistent, uniform dosing without the need to shake to resuspend the drug before administration.36,37 The above formulations all have a 4-times daily (QID) dosing regimen.31,32,35 Most recently, 2 formulations which use proprietary technologies and a reduced drug particle size to enhance ocular penetration 38 were introduced; a higher dose suspension formulation with a twice daily (BID) dosing regimen, LE suspension 1%, was approved in 2018,39,40 and in 2019, LE (submicron) gel 0.38% was approved with 3 times daily dosing41,42
Table 1 compares the respective drug concentrations and key excipients of approved suspension and gel formulations of LE. Of note, the vehicle of both LE gel 0.5% and LE (submicron) gel 0.38% contains mucoadhesive polymers, which may potentiate longer contact time with the ocular surface,36,38 and a demulcent/lubricant, which may improve comfort. The LE gel formulations also contain lower concentrations of the preservative benzalkonium chloride (BAK) compared with LE ophthalmic suspension 0.5% and 1% and have a pH close to that of tears,33,36,43 which may be beneficial when treating ocular surface disorders. Pharmacokinetic studies in rabbits demonstrated that LE was present at high concentrations in the cornea and conjunctiva through 24 h following topical administration of LE 0.5% suspension,44,45 LE gel 0.5%, 46 and LE gel 0.38%. 43 In addition, measurable concentrations of LE were detected in tear fluid through 24 h after topical administration of LE gel 0.5% in both rabbits and humans. 46
Comparison of Available Formulations of Loteprednol Etabonate
Alrex® (LE ophthalmic suspension) 0.2%, Bausch + Lomb.
Lotemax® (LE ophthalmic suspension) 0.5%, Bausch + Lomb.
Inveltys® (LE ophthalmic suspension) 1%, Kala Pharmaceuticals.
Lotemax® SM (LE ophthalmic gel) 0.38%, Bausch + Lomb.
Lotemax® (LE ophthalmic gel) 0.5%, Bausch + Lomb.
BAK, benzalkonium chloride; LE, loteprednol etabonate; N/A, information not available; ppm, parts per million.
The TFOS DEWS II staged management and treatment recommendations include a 4-step process for the management of DED (Table 2). These guidelines suggest that repeated short-term pulse therapy of corticosteroids can be an alternative approach (step 2) for patients with moderate-to-severe DED not controlled with other therapies. 12 Long-term corticosteroid therapy is recommended as step 4 in staged management of DED. Similarly, the CEDARS DTS Panel concluded that all ophthalmic corticosteroid formulations are suitable for treatment of inflammation associated with tear insufficiency. 3 Both panels recommend consideration of topical corticosteroids such as LE that pose a lower risk of corticosteroid-related AEs.3,12 In this study, we review published clinical studies on the use of LE for the treatment of DED.
Staged Management and Treatment Recommendations for Dry Eye Disease (Adapted from Jones Et Al. 12 )
Potential variations within the disease spectrum are acknowledged to exist between patients and the management options listed above are not intended to be exclusive. The severity and etiology of the DED state will dictate the range and number of management options selected from 1 or more steps.
One or more options concurrently within each category can be considered within that step of the DED state. Options within a category are not ranked according to importance and may be equally valid.
It should be noted that the evidence available to support the various management options differs and will inevitably be lower for newer management options. Thus, each treatment option should be considered in accordance with the level of evidence available at the time management is instigated.
The use of prescription drugs needs to be considered in the context of the individual patient presentation, and the relative level of evidence supporting their use for that specific indication, as this group of agents differs widely in mechanism of action.
DED, dry eye disease; LFA-1, lymphocyte function-associated antigen 1; MGD, meibomian gland dysfunction.
Methods
Search strategy and criteria for inclusion
A literature search was conducted using BIOSIS Previews from 1993, Medline from 1946, and EMBASE from 1947 through October 2019 (using ProQuest Dialog). Specific search terms were “loteprednol” and “dry eye syndrome” or “dry eye disease” or “keratoconjunctivitis sicca”. In addition, PubMed was searched with no time limitation using the following search terms: “loteprednol” or “dry eye” or “sicca” or “meibomian” or “Sjögren's” or “Sjögren”. Bibliographies of identified references and review articles were also searched for relevant citations. All articles selected for inclusion reported on use for LE for the treatment of DED in human subjects.
Results
The literature search identified 16 prospective studies (12 of LE suspension 0.5%, 2 of LE gel 0.5%) and 2 retrospective studies of LE suspension 0.5%.22,47–61 Of these, 6 studies evaluated LE monotherapy or in combination with artificial tears (AT), 6 studies evaluated LE in combination with and/or compared to CsA, and 4 evaluated LE for the treatment of MGD. Efficacy and safety findings of each study are summarized in Table 3 and summarized by category below.
Clinical Trials of Loteprednol Etabonate for the Treatment of Dry Eye Disease
AE, adverse event; AT, artificial tears; BCVA, best-corrected visual acuity; BID, twice daily; CFS, corneal fluorescein staining; CI, confidence interval; CsA, cyclosporine A; DCD, dendritic cell density; EOD, every other day; FML, fluorometholone; HSCT, hematopoietic stem cell transplantation; IL, interleukin; IOP, intraocular pressure; MMP-9, matrix metalloproteinase-9; OSDI, Ocular Surface Disease Index; QD, once daily; QID, 4 times per day; SANDE, Symptom Assessment in Dry Eye; SNFL, subbasal nerve fiber length; SPEED, Standard Patient Evaluation of Eye Dryness; SS, Sjögren's syndrome; TG-2, transglutaminase-2; TID, 3 times a day; TBUT, tear film breakup time.
LE monotherapy or with AT
Pflugfelder et al. evaluated LE suspension 0.5% for 4 weeks versus vehicle, both instilled QID, for the treatment of the inflammatory component of keratoconjunctivitis sicca in patients with delayed tear clearance. 47 For the primary subjective variable (visual-analog scale [VAS] score for the worst symptom in the worst eye) and the primary objective variable (composite corneal fluorescein staining [CFS] score in the worst eye), changes from baseline to week 4 were significant (P < 0.001) for both LE 0.5% and vehicle, and the difference between LE 0.5% and vehicle was not statistically significant for either variable. 47 Compared with vehicle, LE 0.5% reduced nasal bulbar conjunctival hyperemia at weeks 2 and 4 (P < 0.05) and reduced inferior tarsal conjunctival hyperemia at week 2 (P < 0.05). 47 A subset analysis was performed in patients with at least a moderate inflammatory component (at least 2 of the following criteria: combined CFS score of ≥10 [Cst criterion], conjunctival injection in at least 1 area of ≥2 [Conj criterion], worst symptom score on VAS of ≥70 [Subj criterion], and redness symptom score on VAS of ≥70 [Red criterion]). In this analysis, LE 0.5% reduced both central CFS (∼33% improvement) and nasal bulbar conjunctival hyperemia in patients meeting Subj/Red or Subj/Conj criteria at week 2 (all comparisons, P < 0.05 versus vehicle). 47 Reduced nasal bulbar conjunctival hyperemia was also evident in patients meeting the Cst/Conj criteria at 2 weeks. Differences between LE 0.5% and vehicle did not reach significance by week 4, with the exception of reduced nasal bulbar conjunctival hyperemia and lid margin injection in patients meeting the Subj/Red criteria (P < 0.05 for both). 47 The authors concluded that LE 0.5% instilled QID may be beneficial in patients who have keratoconjunctivitis sicca with at least a moderate inflammatory component. 47
Villani et al. assessed the efficacy of LE suspension 0.5% (QID for 4 weeks) in patients with moderate-to-severe DED. 48 Ocular Surface Disease Index (OSDI) score, CFS, lissamine green conjunctival staining, dendritic cell density (DCD), and hyperreflective keratocyte density improved from baseline to 4 weeks (P < 0.01 for all). 48 No significant changes were observed in epithelial and keratocyte cell densities, subbasal nerve length, or nerve tortuosity. Patients who demonstrated a Minimal Clinically Important Difference in OSDI scores based on prespecified thresholds of improvement exhibited a decrease in DCD (P < 0.01), whereas those without improvement did not. No differences were observed for the other evaluated signs between patients who responded symptomatically to LE and those who did not. 48
Jung et al. compared a BID dosing regimen of LE suspension 0.5% or fluorometholone (FML) suspension 0.1%, both in combination with AT in Korean patients with severe dry eye associated with SS. 49 In both treatment groups, similar improvements from baseline in tear production (Schirmer test), keratoepitheliopathy, and symptom scores were observed at 6, 12, 18, and 24 months (P < 0.05) and in tear film breakup time (TBUT) at 12, 18, and 24 months (P < 0.05) (Fig. 1). Safety data suggested that LE may have a lower risk of IOP elevation than FML with long-term treatment. 49
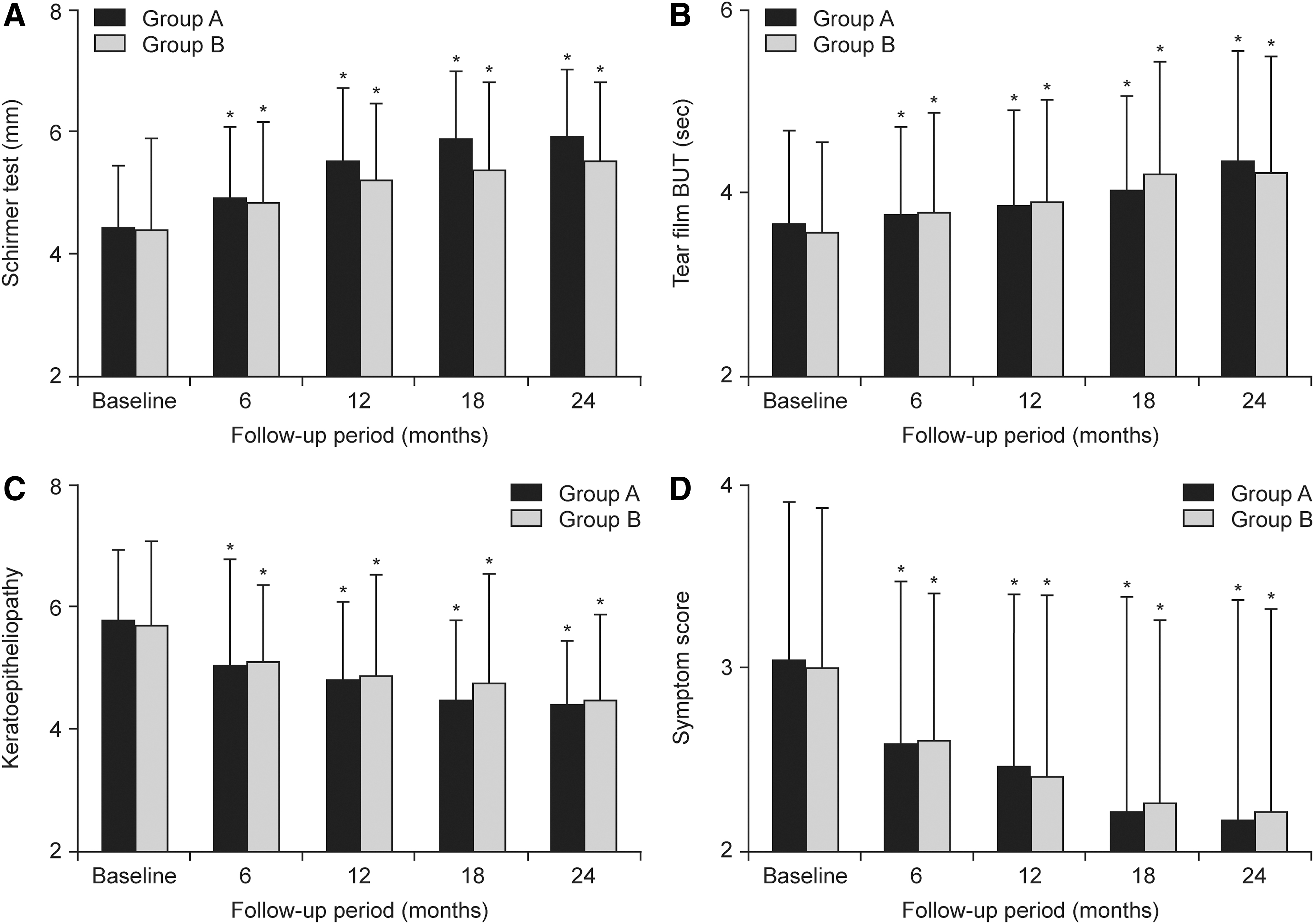
Changes of Schirmer test
In 2 small studies, investigators evaluated the use of low, tapered doses of LE suspension 0.5% in patients with DED. In the first study, patients received LE suspension 0.5% or saline, tapered over 8 weeks. Compared to saline-treated controls, patients receiving LE experienced significant improvements in symptoms and inflammation at 2 and 8 weeks. 50 In the second study, 51 patients instilled LE suspension 0.5% BID for 10 days or LE 0.5% BID for 7 days, with a 5-week taper to 2 days a week thereafter. All patients also received AT. Both groups showed reductions in signs and symptoms of DED compared with baseline (P < 0.001), with patients treated with tapered long-term doses experiencing greater improvements (P < 0.001).
Two enzymes involved in the progression of ocular surface damage in DED are matrix metalloproteinase (MMP)-9, an enzyme involved in the breakdown of extracellular matrix during both physiological and pathological processes, and the proapoptotic enzyme transglutaminase (TG)-2.2,52 The effects of LE suspension on conjunctival expression of these enzymes and on the signs and symptoms of DED 52 were evaluated in patients with mild SS and mild MGD. All patients received saline eye drops as needed and half also received LE 0.5% QID for 15 days. 52 Compared with healthy controls, patients with SS or MGD had elevated levels of both MMP-9 and TG-2 (P < 0.0001 for all). Treatment with LE 0.5% reduced MMP-9 and TG-2 expression compared with baseline and with saline alone in both subgroups (all P ≤ 0.006). Treatment with LE 0.5% also reduced symptom scores and corneal and conjunctival staining compared with baseline, while Schirmer I test scores improved only in patients with SS, and TBUT improved only in those with MGD (all P < 0.05). None of these measures improved significantly with saline in either patient subgroup. 52
LE in combination with and/or compared to CsA
A study in Chinese patients with moderate dry eye 53 evaluated safety and efficacy of 8 weeks of BID treatment with either LE suspension 0.5% or CsA 1%. All the patients also received 0.2% carbomer eye gel (4–6 times/day). Both treatment groups experienced reductions in the overall symptom score versus baseline as early as 2 weeks following initiation of treatment (P < 0.01 for both groups). Greater reductions in overall symptom scores were observed with LE 0.5% versus CsA 1% at 2, 4, and 6 weeks (all P < 0.05) but not at week 8. The authors attributed the difference to less irritation with LE. In both groups, there were similar improvements in CFS at 4, 6, and 8 weeks and goblet cell density at 8 weeks (all P < 0.05 versus baseline). The TBUT increased in the LE group at 8 weeks and in the CsA group at 6 weeks (both P < 0.01 versus baseline) with no significant differences in either parameter between groups at 8 weeks. There was no significant improvement in Schirmer's test in either group.
Boynton et al. compared LE suspension 0.5% to CsA 0.05% for the prophylaxis and treatment of DED after hematopoietic stem cell transplantation (HSCT). 54 Both treatments were instilled BID for 1 month before HSCT and 12 months following HSCT, with no differences between treatment groups in DED incidence or progression over the course of the study. Although the LE group demonstrated a significantly (P = 0.004) smaller increase in CFS and conjunctival lissamine green staining at 3 months and a significantly greater decrease in Schirmer test score at 9 months (P = 0.01), there was no significant difference between groups at 12 months post-HSCT in the average increase in corneal and conjunctival staining from baseline, TBUT, tear osmolarity, change in Schirmer score, best corrected VA, or OSDI.
The safety and efficacy of LE gel 0.5% alone and as induction therapy to CsA 0.05% compared to CsA 0.05% alone were evaluated in patients with mild-to-moderate DED. 55 Patients were treated with LE gel 0.5% BID for 12 weeks, LE gel BID for weeks 1–4, and then CsA 0.05% BID for weeks 3–12, or CsA BID for weeks 1–12. All 3 treatment regimens reduced signs (CFS staining and TBUT) and symptoms (OSDI and Comfort Index) of DED from baseline to week 12 based on 95% confidence intervals. At week 2, greater improvement over baseline was observed with LE gel+CsA versus CsA alone for total OSDI (P = 0.022) and with both LE gel alone and LE gel+CsA versus CsA alone for OSDI visual function domain questions 6–9 (P ≤ 0.041). Compared with CsA alone, LE gel alone produced greater improvement from baseline to week 4 in hyperemia (by keratography) and TBUT (P ≤ 0.04). However, at week 12, no significant differences between LE gel alone or LE gel+CsA compared to CsA alone were noted on any signs or symptoms of DED.
Three studies evaluated LE suspension 0.5% in combination with CsA for the treatment of DED.22,56,57 In the first study, signs, symptoms, and tolerability were compared in patients with mild-to-moderate DED treated with either LE 0.5% BID for 2 to 16 months before initiation of concomitant CsA therapy or CsA alone. 56 Fewer of the patients who received LE induction therapy developed severe stinging (P < 0.02) or discontinued treatment due to severe stinging compared to patients treated with CsA alone (P < 0.04). At 3 months, LE pretreatment was associated with significant improvement in objective signs of DED.
The second study compared induction therapy with LE suspension 0.5% QID followed by CsA/LE, both BID, to AT QID followed by CsA/AT BID in patients with mild-to-moderate DED. The LE or AT was instilled alone for 2 weeks followed by an additional 6 weeks in combination with CsA. 22 While symptoms significantly improved compared to baseline in both treatment groups, the LE group experienced earlier onset of improvement (Fig. 2). Schirmer's test scores improved (P < 0.05) in the LE group but not the AT group. Global self-assessment scores, CFS, and required AT use improved from baseline in both groups (all P < 0.05), with no significant differences between groups, although only the LE group exhibited significant improvements in central, inferior, and cumulative CFS. Lissamine green staining decreased in both groups, but LE was more effective in decreasing lissamine green staining. In terms of tolerability, LE 0.5% reduced stinging associated with CsA at 1 month (P < 0.05), but not at 2 months. There were no significant differences between treatment groups for patient's ratings of overall burning, redness, blurred vision, or improvement in eye comfort at 1 and 2 months.
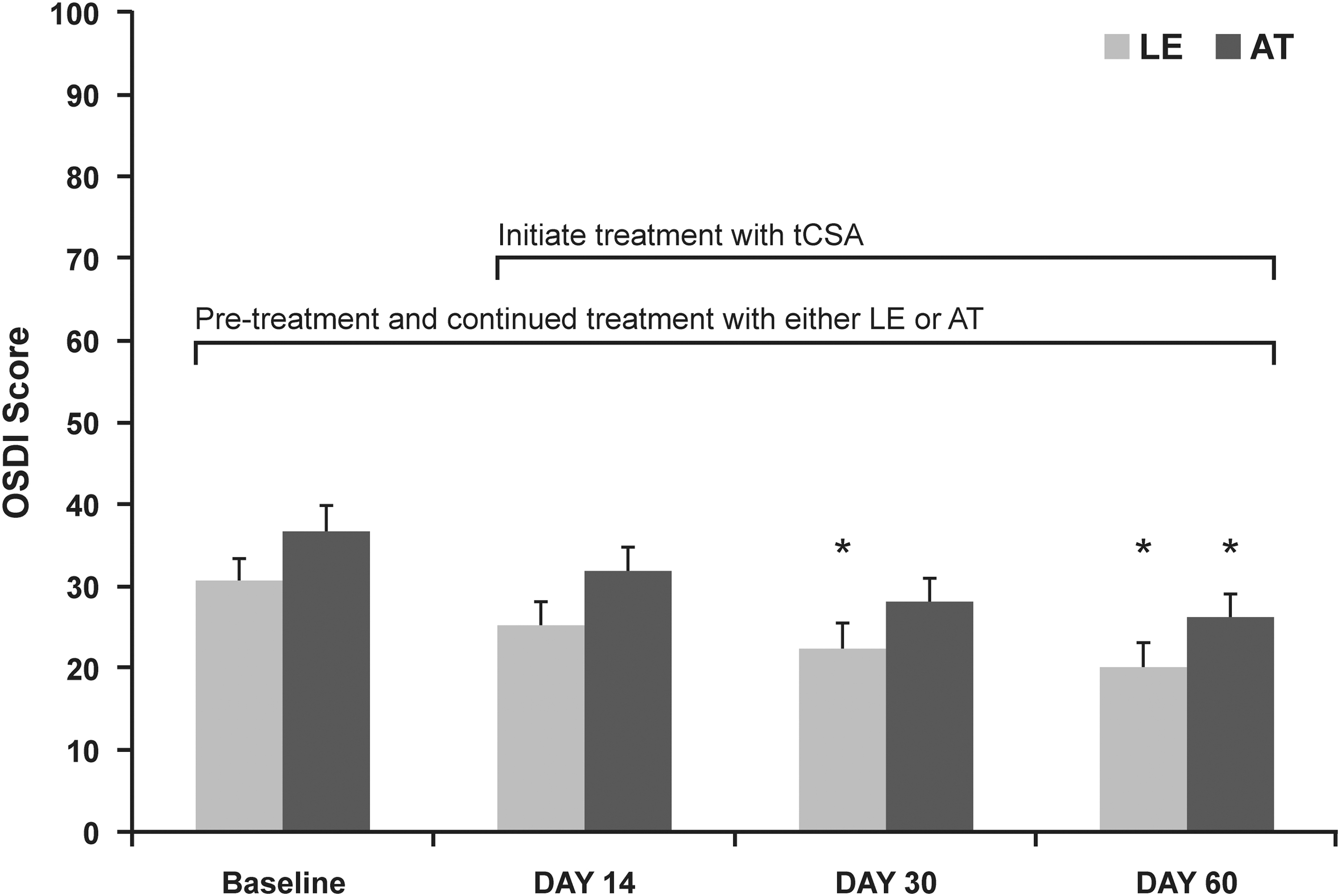
Mean ± SEM OSDI scores for LE+tCsA− (n = 57) and AT+tCsA− (n = 55)-treated patients at baseline and at each of the study visits. 22 OSDI scores for LE+tCSA− and AT+tCsA-treated patients at baseline and at each day of the study. *Statistically significant versus baseline, P < 0.05.
The efficacy of CsA alone versus CsA plus LE suspension 0.5% were further evaluated in a study in Indian patients with moderate DED. 57 Patients received CsA BID for 3 months with or without LE suspension 0.5%, QID for 2 weeks, tapered to BID over the following 6 weeks. All patients also received AT, and signs and symptoms of DED were evaluated at 2 and 6 weeks and at 3 and 6 months. There was a greater improvement in OSDI score, TBUT, CFS, and conjunctival lissamine green staining with LE+CsA versus CsA alone at 3 and 6 months (P ≤ 0.03 for all). Schirmer scores were greater in the LE+CsA group versus the CsA alone group from 2 weeks onward (P ≤ 0.008 for all).
LE for the treatment of MGD
A study investigated the effect of LE suspension 0.5% on tear proinflammatory cytokines and clinical outcomes in patients with moderate or severe MGD. 58 Patients received LE 0.5% QID or no corticosteroid for 2 months; all patients also administered eyelid scrubs/warm compresses. At 1 month, LE treatment decreased tear levels of the cytokines interleukin (IL)-6, -8, and -1β (all P ≤ 0.03), while there was no effect in the control group; IL-8 levels were also lower in the LE group compared to control (P = 0.034). Mean tear levels of IL-6 and IL-8 decreased over 2 months in both groups (P < 0.05), while IL-1β decreased in the LE group only (P < 0.05); at 2 months, mean levels of IL-7 were lower in the LE group than the control group (P = 0.03). Mean TBUT increased over time in both treatment groups, with a larger change in TBUT from baseline to month 1 in the LE group (P = 0.012 versus control). With the exception of corneal staining in the control group, all staining scores improved over time (P < 0.001), with greater improvements in the LE group compared to control at 1 and 2 months (CFS, P = 0.009 and 0.018, respectively; conjunctival fluorescein staining, P = 0.007 and P < 0.001, respectively; DEWS staining score, P = 0.001 and P < 0.001, respectively; Oxford staining score, P = 0.001). 58 Both groups demonstrated improvement in lid margin abnormality and meibum quality (P < 0.001), although the LE group had greater improvement in lid margin abnormality at 2 months and meibum quality at 1 and 2 months compared to baseline (P ≤ 0.02). 58 Mean OSDI in both groups showed similar improvement over time (P < 0.001). Compared with control, the LE group experienced greater improvement in expressibility, ocular irritation score, and MGD stage (all P < 0.05). 58
The effect of LE suspension 0.5% on clinical outcomes and tear cytokine levels in patients diagnosed with obstructive MGD in their blind eye wearing a unilateral ocular prosthesis was evaluated in a case series. 59 Patients instilled LE 0.5% QID and performed eyelid scrubs with warm compresses BID in their prosthetic eye for 2 months. Tear levels of all cytokines (IL-6, interferon-γ, monocyte chemotactic protein-1, IL-8, tumor necrosis factor-α, and IL-1β) decreased after 2 months (P < 0.001 for each). Improvements were observed for lid margin abnormality score, meibomian gland expression, meibography scores of the lower eyelid, and ocular symptom scores (P ≤ 0.037 for all), although meibography scores of the upper eyelid were unchanged.
Kheirkhah et al. evaluated the effect of LE suspension 0.5%, LE suspension 0.5%/tobramycin 0.3%, or AT instilled BID bilaterally for 4 weeks in patients with MGD-related DED with low or near-normal baseline corneal subbasal nerve fiber length (SNFL). 60 Among LE-treated patients, there was no improvement in signs or symptoms of DED in patients with low SNFL, but patients with near-normal SNFL exhibited improvements in Symptom Assessment in Dry Eye (SANDE) severity (P = 0.04) and CFS (P = 0.01). None of the assessed signs and symptoms of DED improved in the LE/tobramycin groups. In the AT group, improvement at week 4 was only observed for the Schirmer's test (P = 0.04) in patients with low SNFL, whereas patients with near-normal SNFL demonstrated improvement in OSDI, SANDE frequency and severity scores, and CFS (all P ≤ 0.04). Mean SNFL did not change after 4 weeks in the AT group, but increased in the LE and LE/tobramycin groups (P = 0.04 for both). The authors concluded that significant improvement in symptoms and CFS were generally evident only in patients with near-normal SNFL and suggested that SNFL may help explain variations in response to therapy. In addition, the authors suggested that the LE and LE/tobramycin regimens (BID) may have been inadequate.
The effect of LE gel 0.5% in patients with MGD was investigated in patients treated with LE gel BID bilaterally for 30 days. 61 After 30 days, TBUT increased by 44.3% (P = 0.005), while corneal staining, conjunctival staining, and MGD signs decreased (by 52%, 47.5%, and 31.9%, respectively; all P ≤ 0.006). Mean OSDI and Standard Patient Evaluation of Eye Dryness scores improved by 32.8% and 28.5%, respectively (both P < 0.004). Schirmer score and tear osmolarity remained unchanged throughout the study.
Discussion
Ocular surface inflammation and, in some conditions, secretory components of the lacrimal system, are believed to play a central role in the pathogenesis of DED, and inflammation is universal across all subtypes of DED. 3 Accordingly, topical corticosteroids are often used to treat the inflammation associated with moderate-to-severe DED.12,23 LE is an effective ophthalmic corticosteroid with a reduced risk of corticosteroid-related AEs, such as increased IOP and cataract formation. 29 While LE has been shown to penetrate efficiently into the aqueous humor following topical ocular instillation of either the suspension or gel formulations, the highest concentrations of LE are consistently achieved on the ocular surface (cornea and conjunctiva).43–46
As attested herein, multiple studies have evaluated LE suspension and gel formulations for the treatment of DED stemming from various causes and have also investigated its efficacy as induction therapy in reducing stinging associated with topical CsA in DED. In the 14 reviewed LE suspension 0.5% studies, dosing regimens were BID (5 studies),49,53,54,56,60 QID (6 studies),47,48,52,57–59 QID (induction therapy for CsA) followed by BID (in conjunction with CsA; 1 study), 22 or tapered from BID to every other day (2 studies)50,51 and ranged from 15 days 52 to 2 years. 49 In the 2 studies evaluating LE gel 0.5%, the drug was instilled BID, with treatment duration of 30 days in one study and 12 weeks in the other study. 55
Results from the 6 studies that evaluated LE suspension 0.5% monotherapy or LE suspension in addition to AT indicate that LE improves signs of DED and is safe and well tolerated, without clinically significant IOP elevations.47–52 Treatment with LE suspension 0.5% improved DED symptoms from baseline in all studies.47–52 In addition, in the 2 studies that compared LE with vehicle/saline, LE showed improvement, although in 1 study this was in a subset of patients with higher levels of inflammation.47,50 Therefore, the effect of LE suspension 0.5% may be greater in patients with a more pronounced inflammatory component of DED. 47 Furthermore, LE reduced conjunctival levels of 2 enzymes linked to inflammation (MMP-9) and apoptosis (TG-2) in patients with SS or MGD, suggesting that LE may target multiple pathological mechanisms in DED. 52 When instilled BID in combination with AT in patients with SS over 2 years, LE suspension 0.5% demonstrated comparable efficacy to FML, but was potentially associated with a lower risk of IOP. 49 Results of 2 small studies that evaluated the efficacy and safety of low, tapered doses of LE suspension 0.5% are promising, and this approach warrants further study.50,51
Results of 3 studies suggest that pretreatment with LE suspension 0.5% may reduce stinging upon initiation of CsA therapy.22,56,57 As well, results of 2 prospective studies suggest that induction therapy with LE suspension 0.5% before CsA treatment may provide more rapid or greater relief of DED signs and symptoms than CsA and AT.22,57 Studies comparing LE suspension 0.5% with CsA support the safety of LE 0.5% and suggest that LE 0.5% may be as effective as CsA for treating DED as well as DED following HSCT.53,54 One study evaluating LE gel 0.5% BID alone or as induction therapy to CsA versus CsA alone for inflammation associated with DED found that all 3 treatment regimens reduced signs and symptoms of DED over 12 weeks of treatment. 55 Although early advantages were noted for LE gel 0.5% alone or LE gel 0.5% plus CsA versus CsA alone on some outcomes, there were no significant differences between treatment groups at 12 weeks. All treatments appeared safe and well tolerated. Results of this study support further investigation of LE gel 0.5% for the treatment of DED, alone or in combination with CsA.
Regarding MGD, findings of 2 studies suggest that LE suspension 0.5% QID and eyelid scrubs with warm compresses are beneficial in MGD, including in prosthetic eye wearers.58,59 Furthermore, treatment with LE suspension 0.5% was associated with decreases in levels of tear cytokines, likely related to pharmacological efficacy. 59 Results of a single open-label study of LE gel 0.5% BID also support the efficacy of LE for improving the signs and symptoms of dry eye associated with MGD. 61
Limitations of studies of LE in DED to date include the challenges of DED studies with any treatment, such as the heterogeneity of the condition, the lack of correlation between signs and symptoms, confounding variables (eg, demographics, geographic/environmental factors, ocular or systemic diseases, concomitant medications) that may differentially affect treatment groups, and the difficulty in determining whether observed improvement represents a true treatment effect.62,63 Spontaneous improvement/regression to the mean and nontreatment-related improvements associated with participating in a clinical study (“placebo effects”) may contribute to perceived improvement, especially in subjective symptoms.62,63 Indeed, differentiating between treatment effects and a placebo effect in vehicle-controlled studies is complicated by high placebo response rates in clinical studies of DED and by the potentially therapeutic effects of the vehicle (often essentially an AT) itself.62,63 These limitations may make it especially difficult to observe a benefit of a study medication over vehicle in DED studies and may explain the lack of differentiation between LE and vehicle for some of the outcomes in several studies. Finally, while a number of the studies used these measures or components of them, there is also a need to more consistently evaluate the symptoms of DED with validated instruments, such as the OSDI and the DEQ-5. 64
It should be noted that most of the studies to date evaluated the safety and efficacy of LE suspension 0.5%. However, suspension formulations may be less convenient and possibly less comfortable to instill than gel formulations, which do not require shaking, have a lower concentration of BAK, and have a more physiological pH.33,36,38 In addition to preventing settling, the polycarbophil polymer in current LE gel formulations also prolongs ocular surface retention and contains demulcent/lubricants, which may improve comfort.33,36,38 The reduction in BAK from 0.01% in the suspension formulation to 0.003% in the gel formulations 36 is important given concerns with use of this preservative in DED patients, especially for those who may be on multiple BAK-containing medications long-term.12,65 While LE is also available formulated in a BAK-free ointment, there are no studies to date which have evaluated the ointment for DED. Therefore, larger studies of longer duration are warranted to further assess the safety and sustained efficacy of the newer gel/ointment formulations of LE in DED. In addition, a low-dose suspension formulation of LE (KPI-121 0.25%; Kala Pharmaceuticals, Waltham, MA) with QID dosing is currently under development/evaluation for temporary relief of the signs and symptoms of DED. 66
In conclusion, the available studies suggest that LE is a safe and effective treatment for DED, with low risk of elevated IOP or cataract formation. Larger and longer-term controlled studies, particularly with the newer gel formulations, are warranted to further explore the therapeutic value of LE for this condition.
Footnotes
Acknowledgment
Writing and editorial support was provided by Churchill Communications (Maplewood, NJ).
Author Disclosure Statement
K.B. is a consultant for Alcon, Allergan, Bausch + Lomb, a division of Bausch Health US, LLC, Eyevance Pharmaceuticals, Kala Pharmaceuticals, Novartis, and Sun Pharmaceutical Industries Ltd. J.K. is a consultant for Allergan, EyePoint Pharmaceuticals, Inc., Novartis, and Ocular Science; he served on an ad board with Eyevance Pharmaceuticals, Kala Pharmaceuticals, and Ocular Therapeutix. P.M. is a consultant for Alcon, Allergan, Bausch + Lomb, a division of Bausch Health US, LLC, Bio-Tissue, Inc., Dompé, Eyevance Pharmaceuticals, Kala Pharmaceuticals, Novaliq, Novartis, and Sun Pharmaceutical Industries Ltd. A.R. is a consultant for Alcon, Allergan, Bausch + Lomb, a division of Bausch Health US, LLC, Kala Pharmaceuticals, Novartis, and Sun Pharmaceutical Industries Ltd.
Funding Information
Funded by Bausch Health US, LLC.