
Abstract
The gastrointestinal tract is lined by a single layer of epithelial cells that secrete mucus toward the lumen, which collectively separates the immune sentinels in the underlying lamina propria from the intestinal microflora to prevent aberrant immune responses. Inflammatory bowel disease (IBD) describes a group of autoimmune diseases that arise from defects in epithelial barrier function and, as a consequence, aberrant production of inflammatory cytokines. Among these, interleukin (IL)-6, IL-11, and IL-22 are elevated in human IBD patients and corresponding mouse models and, through activation of the JAK/STAT3 pathway, can both propagate and ameliorate disease. In particular, cytokine-mediated activation of STAT3 in the epithelial lining cells affords cellular protection, survival, and proliferation, thereby affording therapeutic opportunities for the prevention and treatment of colitis. In this review, we focus on recent insights gained from therapeutic modulation of the activities of IL-6, IL-11, and IL-22 in models of IBD and advocate a cautionary approach with these cytokines to minimize their tumor-promoting activities on neoplastic epithelium.
Introduction
I
The human gastrointestinal (GI) tract extends from the oral cavity to the rectum and includes the small intestine and colon. A single layer of the intestinal epithelial cell (IEC) line, the GI tract, creates a physical barrier, which separates luminal bacteria from the host's immune system. An estimated 1013 bacteria from over 160 different species colonize the 30 m2 absorptive surface of the GI tract in humans (Qin and others 2010; Helander and Fändriks 2014). The general acceptance by the scientific community that immune responses to specific microbial populations contribute to the pathogenesis of IBD has not only led to a renewed interest in the role of the epithelial barrier in the host defense response, but also triggered an interest in the role of the microbiome. In IBD patients, a significant reduction in microbial diversity has been observed, including a reduction of the Firmicutes genus Clostridium leptum in CD patients. Meanwhile, the colitogenic species from the Streptococcus and Haemophilus phyla is only found in IBD patients (Manichanh and others 2006; Palm and others 2014). However, not all microbial species are considered pathogenic, with excessive abundance of the Firmicutes member Faecalibacterium prausnitzii being linked to reduced disease recurrence following ileal resection in CD patients (Sokol and others 2008). The increased recognition by the scientific community that immune responses to specific microbial populations contribute to the pathogenesis of IBD has led to a renewed interest in the role of the epithelial barrier in the host defense response.
IBD is associated with chronic inflammation of the bowel, which is thought to develop, in part, from bacteria breaching the epithelial barrier, invading the body, and triggering a perpetuating inflammatory response. A combination of highly specialized IECs and a variety of immune cell populations are critical for maintenance of the integrity of the mucosal GI barrier. Most important among the IECs are goblet cells that secrete mucins, including mucin-2, which comprises the major component of the mucus layer that physically limits the access of commensal bacteria to IECs (Johansson and others 2008). Meanwhile, Paneth cells sit adjacent to IEC stem cells at the base of intestinal crypts and secrete antimicrobial peptides, including α-defensins and lysozyme (Wehkamp and others 2004). Specialized epithelial microfold (M) cells are situated above lymphoid follicles, such as Peyer's patches in the small intestine, and allow immune cells to sample luminal antigens (Mabbott and others 2013). In addition, dendritic cells (DCs) can penetrate through tight junctions between IECs to directly sample luminal antigens and process them for subsequent presentation to T and B cells by antigen presentation (Rescigno and others 2001). Intraepithelial lymphocytes (IELs), conventional T cells, and γδT cells alongside other immune cells found within the gut or mucosal-associated lymphoid tissue (GALT and MALT, respectively) can secrete cytokines in response to microbial exposure (Wang and others 2014). Innate lymphoid cells (ILCs) are present at mucosal surfaces, including the GI tract, and, analogous to the adaptive T helper cell groups (Th1, Th2, and Th17), have been functionally classified as ILC1, ILC2, and ILC3 (Spits and others 2013). Through the production of cytokines that induce expression of antimicrobial genes, ILC3 cells, similar to Th17 cells, are of particular importance for the maintennace of intestinal homeostasis during infection and inflammation (Mielke and others 2013; Qiu and others 2013).
CD is characterized by transmural inflammation that may involve the entire GI tract, although development of disease in the terminal ileum, cecum, colon, and perianal area is most common (Baumgart and Sandborn 2012). In contrast, UC is characterized by superficial inflammation affecting the mucosa and submucosa in the colon, with crypt abscesses, pseudopolyps, and ulcers commonly observed (Ordás and others 2012). Genome-wide association studies (GWAS) revealed a number of polymorphisms in the JAK/STAT signaling pathway that are associated with IBD susceptibility. Among these, polymorphisms in the Janus Kinase (JAK) family of cytokine receptor-associated cytoplasmic tyrosine kinases, JAK2 (rs10758669) and TYK2 (rs280519, rs2304256), as well as in the gene encoding the Jak target protein, Signal Transducer and Activator of Transcription 3 (STAT3; rs744166), are associated with CD (Barrett and others 2009). Moreover, the STAT3 polymorphism (rs12948909) is associated with CD and UC, although it still remains unclear how these polymorphisms functionally contribute to the diseases (Anderson and others 2009; Barrett and others 2009; Ferguson and others 2010; Sato and others 2009).
STAT3 Activation in IBD
Constitutive activation of STAT3 in intestinal and peripheral CD4+ T cells has been observed in patients with CD, while in UC the lamina propria mononuclear cells and nonimmune colonic epithelial cells show excessive accumulation of activated STAT3 (Lovato and others 2003; Mudter and others 2005; Li and others 2010). The levels of activated STAT3 were also significantly higher in IECs from patients with active UC compared with patients with inactive disease or healthy controls, and these levels correlated with the severity of colitis (Li and others 2010). STAT3 is classically associated with mediating cellular survival of IECs, suggesting that the cell population in which STAT3 is constitutively active may dictate the pathology of IBD. A number of studies using mouse models suggest that epithelial Stat3 activation is essential for the maintenance of GI barrier integrity. This has been best demonstrated by the observation that mice with IEC-specific Stat3 deficiency (Stat3 ΔIEC) or IEC-specific deficiency in suppressor of cytokine signaling (Socs)-3, a negative regulator of Stat3 activation (Socs3 ΔIEC), are highly susceptible to IEC injury in the colon following administration of the luminal irritant Dextran Sulfate Sodium (DSS)(Tebbutt and others 2002; Rigby and others 2007; Bollrath and others 2009; Pickert and others 2009). Other studies have shown that the deletion of Stat3 in neutrophils and macrophages led to the spontaneous development of enterocolitis, suggesting a protective role for Stat3 in multiple cell subsets within the GI mucosa (Takeda and others 1999; Alonzi and others 2004). In line with this hypothesis, Gp130 Y757F mice, which harbor a gp130 knock-in substitution mutation to prevent Socs3-dependent negative regulation and associated systemic ligand-dependent Stat3 hyperactivation, were resistant to acute DSS-induced epithelial damage (Tebbutt and others 2002). In response to repeated DSS challenges, however, Gp130 Y757F mice develop colitis that progresses through to adenoma formation (Putoczki and others, unpublished). It is likely that in these mice excessive, systemic Stat3 activation may favor emergence of a pathogenic cell population because reconstitution of Stat3-deficient CD4+CD45RBhi T cells prevented colitis that was observed following transfer of Stat3-proficient cells, and this was associated with a skewing toward nonpathogenic Treg cells rather than the colitogenic Th17 phenotype (Durant and others 2010).
Activation of STAT3 by the IL-6 Cytokine Family in IBD
The Interleukin (IL)-6 family of cytokines comprises 10 members: IL-6, IL-11, ciliary neurotrophic factor (CNTF), cardiotrophin-1 (CT-1), cardiotrophin-like cytokine (CLC), leukemia inhibitory factor (LIF), neuropoietin (NP), oncostatin M (OSM), IL-27, and IL-31. These cytokines share a 4-helix bundle protein structure and signal through a complex of nonsignaling α-receptors (IL-6Rα, IL-11Rα, CNTFRα) that permit interaction with signal-transducing β-receptors (gp130, LIFR, OSMR, IL-27RA, IL-31RA). The unifying feature of this family is the shared use of the transmembrane β-receptor, gp130, either as a homodimer in the case of IL-6 or IL-11 or as a heterodimer used by the other family members. An exception to this is IL-31, which does not utilize gp130, but instead signals through an IL-31RA/OSMR heterodimer.
Upon engagement of their α-receptors, IL-6 and IL-11 mediate formation of a hexameric complex consisting of two ligands, two α-receptors, and two gp130 molecules (Fig. 1). Subsequent phosphorylation of tyrosine residues, by constitutively associated JAKs, on the cytoplasmic tail of gp130 enables activation of the JAK/STAT, MAPK/ERK, and PI3K/AKT pathways (Ernst and others 2014). Since gp130 expression is ubiquitous, cellular responses to IL-6 and IL-11 are dependent on tissue-restricted expression of IL-6Rα and IL-11Rα. While membrane-bound IL-6Rα is expressed by nonimmune cells, including IECs, hepatocytes, neutrophils, monocytes, macrophages, and other immune cells, a soluble sIL-6Rα is generated by proteolytic cleavage of membrane-bound IL-6Rα to allow for trans-signaling in any cell that expresses gp130 (Rose-John 2012). IL-11Rα is present in 2 isoforms, with the most common, IL-11Rα1, being found on epithelial cells of the GI tract, prostate, ovary, endometrium, and other organs, as well as on macrophages, T cells, and hematopoietic cells (Takagi and others 1999; Bozza and others 2001; Campbell and others 2001; Lai and others 2001; Cork and others 2002; Zurita and others 2004; Sales and others 2010; Putoczki and others 2013). Although, when artificially truncated, IL-11Rα can function as a soluble receptor, this form does not appear to occur naturally (Putoczki and Ernst 2010).
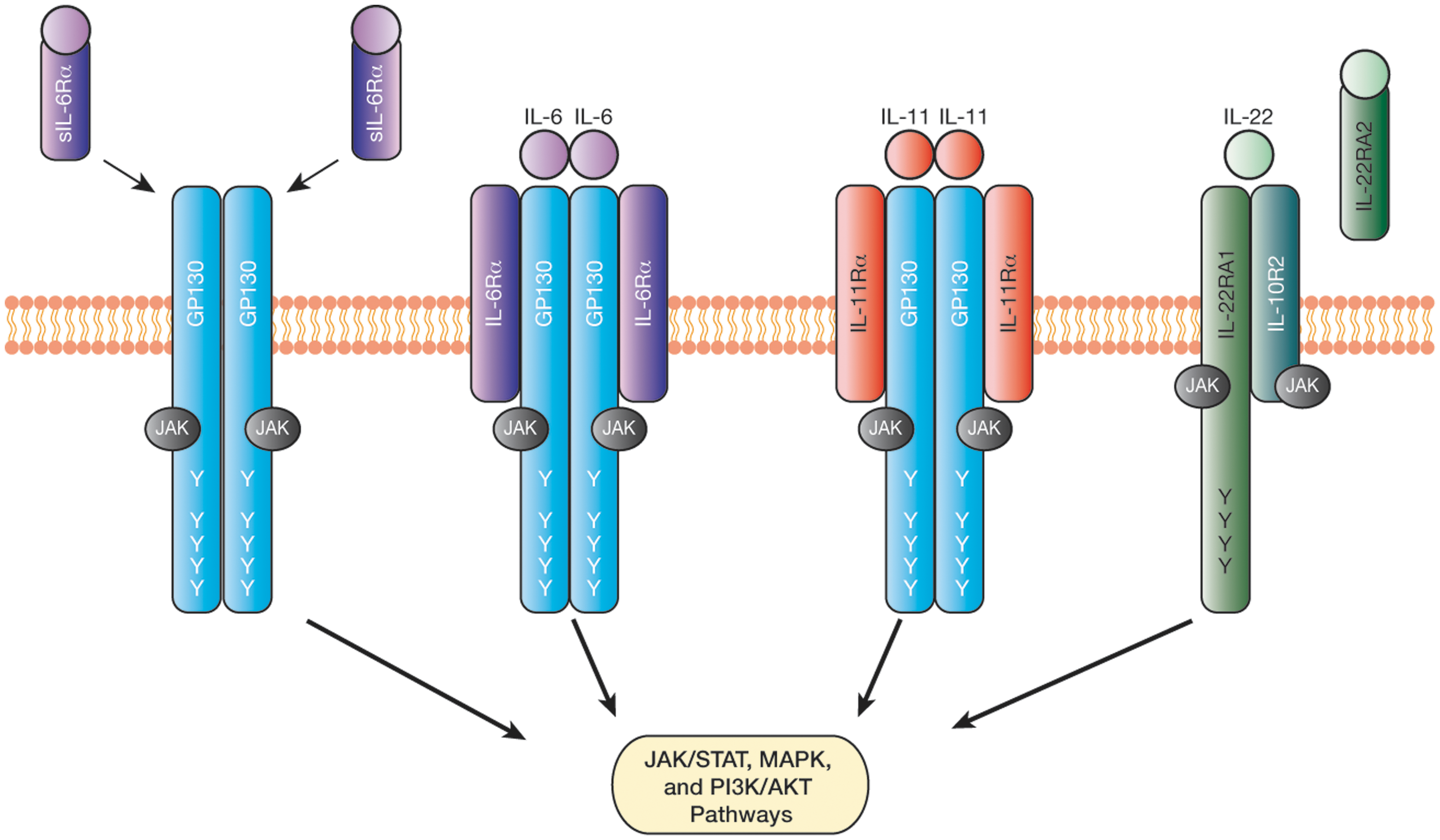
Activation of signaling pathways downstream of interleukin (IL)-6, IL-11, and IL-22. IL-6 and IL-11 signal through a hexameric receptor complex consisting of 2 ligands, 2 IL-6Rα or IL-11Rα receptors, and 2 gp130 receptors, while IL-22 signals through a heterodimeric complex comprising one IL-22RA1 and one IL-10R2 receptor. IL-6 can also signal through soluble IL-6Rα (sIL-6Rα) receptors in cells that do not express the transmembrane IL-6Rα receptor, a process referred to as trans-signaling. Meanwhile, IL-22 binding to its soluble receptor, IL-22RA2, prevents ligand-dependent formation of an activated receptor complex. Following receptor engagement, phosphorylation of tyrosine (Y) residues by JAKs allows for subsequent activation of the STAT, MAPK, and PI3K/AKT pathways. The membrane-proximal tyrosine (Y757 in mouse gp130) in gp130 acts as a docking site for the negative regulator SOCS3, and is also required for activation of MAPK signaling.
Although IL6 polymorphisms have been identified in the promoter region (c.-174C/G) as well as in an intron (g.4470G/A) of the human gene and they affect mRNA expression and splicing, these polymorphisms have not been associated with increased susceptibility to IBD (Koss and others 2000; Klein and others 2001; Cantor and others 2005). Likewise, an SNP identified in exon 4 and leading to an amino acid substitution (p.Arg112His) was not associated with increased IBD susceptibility (Klein and others 2002). However, a dinucleotide repeat in the IL-11 promoter region (IL11.A1 (GT)7(CT)8) was significantly more abundant in UC patients, although the functional relevance of this repeat was not explored (Klein and others 2002).
A significant association between elevated IL-6 expression levels and increased disease activity has been observed in CD and UC patients, with the production of IL-6 and sIL-6Rα by lamina propria mononuclear cells being associated with active disease (Hosokawa and others 1999; Mudter and Neurath 2007). Meanwhile, IL-11 levels have thus far not been investigated in IBD patients, although elevated IL-11 expression has been observed in a rat model of IBD (Torrence and others 2008). Due to its prominent capacity to promote IEC survival and epithelial regeneration, clinical trials with recombinant IL-11 have been undertaken, with a subset of CD patients showing reduced disease severity (Sands and others 2002; Herrlinger and others 2006).
The contribution of IL-6 and IL-11 to the pathogenesis of IBD has been studied extensively in various mouse models. Loss of IL-6 expression is associated with increased damage to the epithelium and inflammation in the DSS-induced colitis model (Tebbutt and others 2002; Grivennikov and others 2009). Interestingly, in Il6 KO mice with increased DSS-induced disease, the colonic inflammatory cell infiltrate was decreased and was accompanied by elevated IL-10 expression (Naito and others 2004). Similarly, treatment of mice with a neutralizing IL-6Rα antibody was effective in reducing the severity of inflammation in the CD4+CD45RBhi T cell transfer model of colitis, in the spontaneous colitis observed in IL-10-deficient mice, and in the trinitrobenzene sulphonic acid (TNBS) model of CD (Atreya and others 2000). A decrease in colitis severity was also observed in transgenic mice that express sgp130 to specifically bind to the IL-6/sIL-6Rα complex and abrogate trans-signaling (Becker and others 2004). Similarly, therapeutic administration of sgp130 as an Fc-fusion protein was also effective in reducing spontaneous intestinal inflammation observed in SAMP1/Yit mice and following DSS-induced colitis in wild-type mice (Atreya and others 2000; Mitsuyama and others 2006).
Compared with IL-6, the contributions of IL-11 to the pathogenesis of IBD are less well understood. However, treatment of mice or rats with recombinant IL-11 ameliorates intestinal inflammation and mucosal damage in various models of IBD (Qiu and others 1996; Peterson and others 2002; Gibson and others 2010). Likewise, in other models of intestinal injury, including radiation, ischemic bowel necrosis, and chemotherapy, administration of recombinant IL-11 reduced epithelial damage and promoted the regenerative response (Orazi 1996; Du and Williams 1997; Yang and others 2014).
Activation of STAT3 by the IL-10 Cytokine Family in IBD
The IL-10 family of cytokines comprises IL-10, IL-19, IL-20, IL-22, IL-24, IL-26, IL-28A, IL-28B, and IL-29 (Sabat 2010). These cytokines share a common 6 α-helical structure and signal through a heterodimeric receptor complex consisting of an R1 receptor (IL-10R1, IL-20R1, IL-22RA1, or IL-28R1) and an R2 receptor (IL-10R2 or IL-20R2) (Ernst and others 2014). While X-ray crystallography has demonstrated that IL-10 forms a dimer, IL-19, IL-20, IL-22, IL-24, and IL-28 are secreted as monomers (Ouyang and others 2011). Interestingly, dimeric and monomeric forms of secreted IL-26 have been reported (Knappe and others 2000; Corvaisier and others 2012). For the majority of these cytokines, signal transduction involves IL-10R2, with the exception of IL-19, IL-20, and IL-24, which utilize IL-20R2 (Dumoutier and others 2001a). While IL-10 signals through a tetrameric complex consisting of two IL-10R1 and IL-10R2 receptor chains each, IL-22 signals through a heterodimeric receptor complex consisting of one IL-22R1 and IL-10R2 chain (Fig. 1) (Kotenko and others 1997; Jones and others 2008).
While the IL-10R1 and IL-22R1 chains associate with Jak1 (Sabat 2010), the common IL-10R2 chain binds to the JAK family member, Tyk2. Phosphorylation of conserved intracellular tyrosine residues in these chains provides docking sites for Stat3 and, to a lesser extent, Stat1 and Stat5 (Lejeune and others 2002). Interestingly, Stat3-dependent gene transcription in response to IL-22 can also occur through interaction of the coiled-coil domain of Stat3 with the C-terminal region of IL-22RA1 (Dumoutier and others 2009). IL-22 has also been shown to activate the MAPK pathway through ERK, p38, and Jnk (Lejeune and others 2002). The presence of a soluble IL-22 receptor (referred to as IL-22RA2 or IL-22BP) with high binding affinity toward IL-22 serves a regulatory role by sequestering cytokines from binding to the transmembrane receptor complex (Fig. 1) (Dumoutier and others 2001b; Kotenko and others 2001; Jones and others 2008). While IL-10R2 expression is ubiquitous, IL-10R1 expression is limited to immune cells, including macrophages, T cells, B cells, and NK cells, as well as to cells in the bone marrow (Wolk and others 2002; Wolk and others 2004; Zenewicz and others 2008). By contrast, IL-22R1 expression is limited to nonimmune cell populations, including IECs, skin keratinocytes, subepithelial myofibroblasts, hepatocytes, and bronchial epithelial cells (Dumoutier and others 2000; Wolk and others 2004; Andoh and others 2005; Aujla and others 2008; Zenewicz and others 2008).
Gene polymorphisms have been identified in components of the IL-10 and IL-22 signaling complexes that correlate with IBD susceptibility. A polymorphism in IL10 (rs3024505) downstream the 3′ UTR region is associated with increased disease risk for UC and CD and has been suggested to affect regulation of IL-10, potentially through the transcription factor AP-1 (Franke and others 2008; Franke and others 2010). Loss-of-function mutations in IL10R1 (p.Gly141Arg, p.Thr84Ile) and IL10R2 (p.Trp159X) were identified in early onset IBD patients and were associated with impaired anti-inflammatory responses and increased secretion of proinflammatory cytokines, including TNFα, IL-1β, IL-6, and sIL-6Rα (Glocker and others 2009). In patients with active CD and UC, serum IL-10 concentrations were increased and found to correlate with disease severity and with the production of IL-10 by lamina propria T cells in UC patients with active disease (Kucharzik and others 1995; Melgar and others 2003). Similarly, IL-22 expression is elevated in patients with active IBD, which has been associated with increased cytokine production in CD4+ T cells (Andoh and others 2005; Brand and others 2006). In patients with CD, this was associated with an increase in Th17, while depletion of Th22 cells was observed in UC patients (Leung and others 2014). So far, few studies have investigated IL22 polymorphisms in IBD susceptibility; however, since polymorphisms (rs1558744, rs7134599, rs2870946, and c.-429C/T) that are associated with increased UC risk have been identified in the IL22 gene locus, it is likely that deregulated IL-22 is also involved in CD pathogenesis (Silverberg and others 2009; Chi and others 2014).
IL-10 dampens the inflammatory response following intestinal injury. This is best demonstrated in il10 KO mice, which develop CD4+ T cell-mediated spontaneous enterocolitis at 3 months of age (Kühn and others 1993; Berg and others 1996; Davidson and others 1996). In contrast, IL-22 expression is important for maintenance of epithelial barrier integrity with loss of IL-22 resulting in more severe colitis as a result of impaired Stat3 signaling (Pickert and others 2009). The production of IL-22 by T cells and ILCs, as well as neutrophils mediates protection from colitis (Zenewicz and others 2008; Zindl and others 2013). Importantly, the production of IL-22 during disease is dependent on IL-23, which reinforces Th17 cell identity, as well as on Lyn tyrosine kinase, which mediates signaling from pattern recognition receptors as part of the innate immune responses (Eken and others 2013; Zindl and others 2013; Bishop and others 2014). However, some studies suggest that IL-22 may also have a pathogenic role in colitis, because IL-22 neutralization significantly reduced colon pathology and inflammation in an acute colitis model that depends on innate immune responses (Eken and others 2013). These conflicting results suggest both protective and pathogenic roles for IL-22-mediated Stat3 activation, which may be dependent on the severity of the disease.
Cytokine-Mediated STAT3 Activation Maintains Epithelial Homeostasis
Animal models of colitis have demonstrated both protective and pathogenic roles for IL-6, IL-11, and IL-22 in IBD through regulation of the innate immune response, epithelial cell proliferation and apoptosis, wound repair, and antimicrobial defense. Below we compare the pathogenic and protective attributes of these cytokine-signaling pathways in the context of epithelial Stat3 activation.
During the initial onset of injury in inflammatory intestinal diseases, IL-6 can protect from epithelial damage through complementing responses in IECs (Kuhn and others 2014). In chemically induced models of colitis, for instance, IL-6 promotes Stat3-dependent induction of proliferative, antiapoptotic, and cytoprotective gene programs, including proliferating cellular nuclear antigen, Bcl-xL, and heat shock protein (Hsp)-72 (Grivennikov and others 2009) (Fig. 2). However, in Muc2 KO mice, which spontaneously develop colitis associated with epithelial barrier defects, IECs show elevated expression of the proinflammatory cytokines, IL-6, TNFα, and IL-1β, which may perpetuate chronic inflammation in this model (Bao and others 2014). Macrophage-derived IL-6 on the other hand can upregulate expression of Muc1 and downregulate that of Muc2 in colon epithelial cell lines (Li and others 2009). In immune cells, Stat3 activation through IL-6 trans-signaling can also promote the survival of lamina propria T cells by inducing expression of antiapoptotic Bcl-2 and Bcl-xL proteins to enable T cell survival and perpetuation of sustained inflammation in CD (Atreya and others 2000). Collectively, these results demonstrate that during intestinal inflammation, IL-6 can confer both IEC-specific protective and immune cell-mediated pathogenic effects.
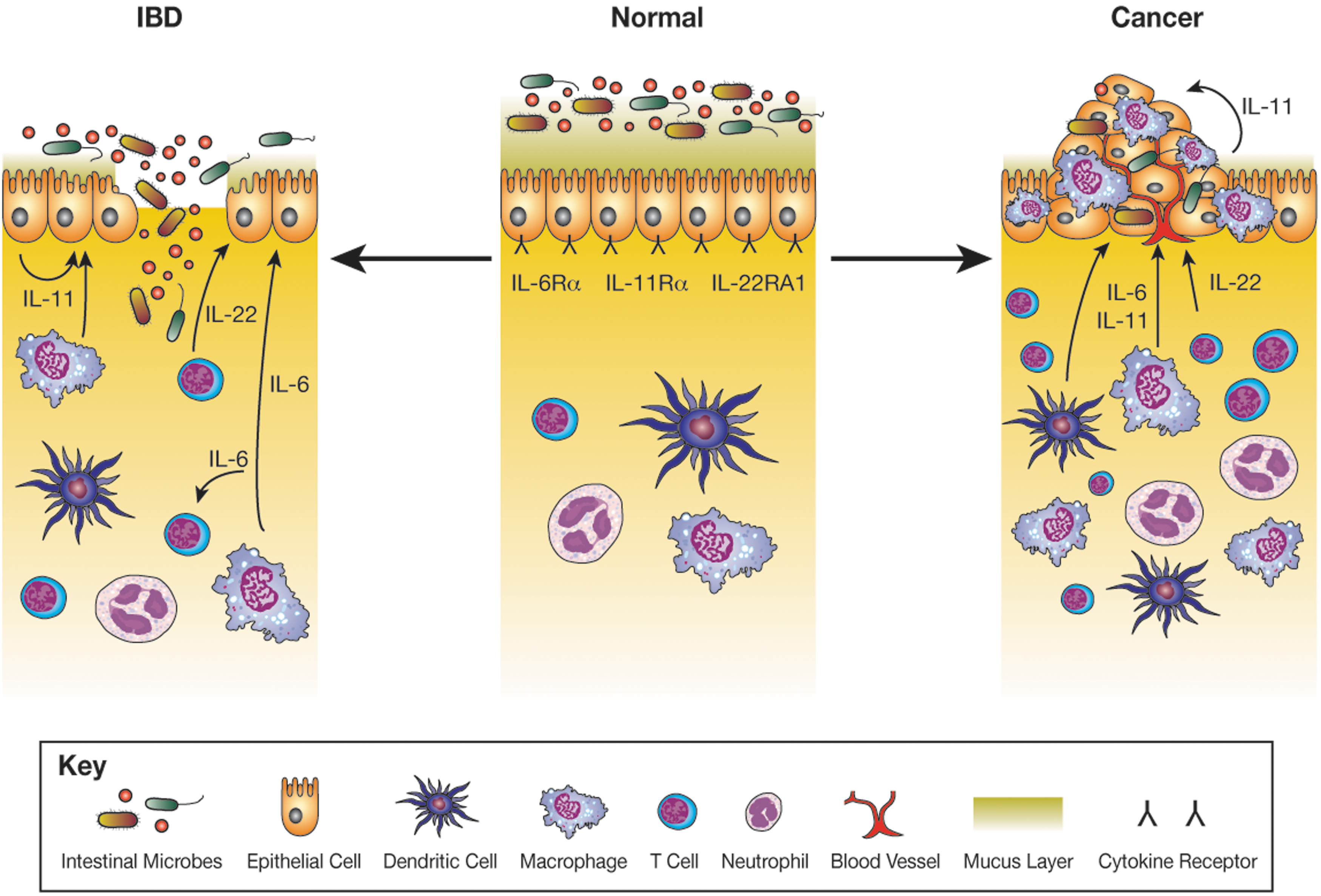
Production of IL-6, IL-11, and IL-22 in inflammatory bowel disease (IBD) and Cancer. The receptors for IL-6, IL-11, and IL-22 are expressed on the surface of intestinal epithelial cells (IECs), and during normal tissue homeostasis, low levels of IL-6, IL-11, and IL-22 are produced by IECs and immune cells. In IBD, epithelial barrier defects result in aberrant activation of the immune system with infiltration of inflammatory cells and increased production of inflammatory cytokines. Among these, IL-11 and IL-22 induce Stat3-dependent gene expression in IECs that confer a proliferative, antiapoptotic, and antimicrobial response. In addition to these protective effects, IL-6 can also sustain survival of pathogenic T cells. IBD patients have an increased risk of colon cancer development with IL-6, IL-11, and IL-22 also associated with the survival and growth of tumorigenic cells, highlighting a delicate balance between beneficial and pathogenic IEC Stat3 activation.
Similar to chemical models of colitis, expression of IL-6 by colonic IECs and macrophages or of IL-22 by DCs, ILC3, and γδ T cells helps protect against infection by pathogenic bacteria, including Citrobacter rodentium (Dann and others 2008; Zheng and others 2008; Mielke and others 2013). As a consequence of Stat3 activation, these cytokines mediate cellular protection through expression of antiapoptotic genes in IECs, including Bcl-x L , Mcl-1, cIAP-2, and Bcl-3, and these effects were no longer observed in Il6 KO, Il22 KO, or Il22ra1 KO mice or after administration of the corresponding neutralizing antibodies, resulting in increased epithelial damage and inflammation and associated mortality (Dann and others 2008; Zheng and others 2008; Pham and others 2014). Consistent with these findings, Stat3-depedent expression of IL-22 is required for the production of S100A8, S100A9, Reg3β, and Reg3γ by IECs to confer antimicrobial protection. Accordingly, decreased expression of these cytokines attenuates epithelial cell restitution and the general recovery response to infection and chemically induced colitis (Zheng and others 2008; Sokol and others 2013; Backert and others 2014; Guo and others 2014).
IL-22 is critical for disease resolution in DSS-induced colitis through Stat3-dependent signaling, resulting in induction of mucus components, Muc1, Muc3, Muc10, and Muc13, as well as antimicrobial peptide secretion from neutrophils (Sugimoto and others 2008; Zindl and others 2013). Accordingly, loss of IL-22 resulted in severe colitis and this was reverted in response to IL-22 gene delivery (Sugimoto and others 2008). By contrast, this protective effect is abolished upon IL-22BP gene delivery. Mucins and, in particular, Muc2 are essential for protection of the epithelium, with Muc2 deficiency resulting in spontaneous disease that is associated with increased IL-22 expression and Jak/Stat signaling, presumably as a compensatory protective measure (Bergstrom and others 2010; Lu and others 2011). As discussed above for other Stat3-activating cytokines, IL-22 also mediates a tissue repair response in IECs through Stat3-dependent expression of Mcl1, Survivin, Myc, Reg3β, Pla2g5, and Smo (Pickert and others 2009). In addition to Stat3, the transcription factor, aryl hydrocarbon receptor (AhR), can synergistically promote the expression of IL-22 expression in ILCs and T cells to mediate protection during intestinal damage and infection (Yeste and others 2014). Accordingly, AhR-dependent induction of IL-22 was effective in ameliorating inflammation in TNBS-induced colitis in mice (Monteleone and others 2011).
The mechanism by which IL-11 reduces susceptibility to, and extent of, colitis is less well characterized; however, given that recombinant IL-11 can ameliorate symptoms of colitis in numerous animal models and induce Stat3 signaling through engagement of the Gp130 receptor complex, IL-11 is also likely to regulate the expression of genes that are important in the repair response (Gibson and others 2010). These involve Reg3β and Reg3γ, vascular endothelial growth factor, and others (Putoczki and others 2013). Thus, IL-11 has a dual effect by inducing proliferation of IEC progenitor and stem cells in response to, for instance, 5-fluorouracil-induced intestinal injury, while prophylactic IL-11 administration protects from cytotoxic injury (Potten 1995). Additionally, IL-11 was effective in reducing symptoms of mucositis during chemotherapy and mediated protection during oxidative stress through induction of Hsp25 and other cytoprotective proteins in IECs (Gibson and others 2002; Ropeleski and others 2003). Collectively, these results suggest that IL-11 affords amelioration of intestinal inflammation through induction of proliferative, antimicrobial, and cytoprotective genes.
IL-6, IL-11, and IL-22: New Therapeutic Opportunities for IBD?
Given the potential for IL-6, IL-11, and IL-22 to mediate protective effects on IECs through epithelial Stat3 expression and the induction of proliferative, antiapoptotic, antimicrobial, and wound healing genes to maintain epithelial barrier integrity, these cytokines may represent therapeutic opportunities for IBD. However, the relative clinical benefit of each of these cytokines needs to be considered in two important areas of what could be considered as on-target side effects. The first limitation arises from the capacity of some of these cytokines to also confer activities on inflammation-perpetuating immune cells, and the second limitation is in the context of their capacity to promote survival and proliferation of neoplastic epithelium at sites of primary and secondary tumors.
Given the widespread activity of IL-6 on immune cells, its therapeutic use may be limited, since IL-6 can perpetuate colitis by promoting the survival of pathogenic T cells during chronic inflammation (Atreya and others 2000; Becker and others 2004; Mitsuyama and others 2006), although administration of recombinant IL-6 was sufficient to protect IECs during acute intestinal injury by induction of cytoprotective genes, Hsp70 and Bcl-xL (Spehlmann and others 2013). Moreover, continuous IL-6 production is observed in mouse models of colitis-associated cancer (CAC) and facilitates Stat3-dependent survival of tumorigenic epithelial cells and T cells (Becker and others 2004). This most likely occurs through IL-6 trans-signaling, since recombinant Hyper-IL6, a designer peptide where IL-6 is fused to the sIL-6Rα, but not recombinant IL-6, increased CAC-associated tumor burden (Becker and others 2004; Grivennikov and others 2009).
Compared with IL-6, IL-11 may have a more favorable range of target cells in particular since megakaryocytes, the main hematopoietic cell target for IL-11, are less likely to play a major role during colitis. In addition, the IL-11-responsive IECs may include stem cells that do respond to IL-6 trans-signaling, but not to IL-6 (Putoczki and others 2013). Thus, Stat3-dependent gene transcription in IECs in response to administration of recombinant IL-11 is effective in reducing colitis severity (Gibson and others 2010). Additionally, recombinant IL-11 is also able to protect the epithelium during other forms of intestinal injury, including chemotherapy, radiation, and ischemic necrosis (Orazi 1996; Du and Williams 1997; Yang and others 2014). However, we have demonstrated that IL-11 is also the prominent cytokine that drives Stat3-dependent gastric tumor formation (Ernst and others 2008) and colon tumorigenesis in the CAC model by acting on radioresistant mutant IECs (Putoczki and others 2013). The latter resulted in suppression of the Bcl-2 survival antagonist, Bim, and induction of the cell cycle regulators, cyclin D1, D2, and D3, alongside a range of genes that promote tumor cell invasion (Putoczki and others 2013). In the context of CAC tumorigenesis, the latter activities could be reversed following administration of the IL-11 mutein signaling antagonist (Putoczki and others 2013).
The directed activity of IL-22 as a cytokine produced by ILCs and other immune cells, but acting primarily on epithelial cells, is an attractive mechanism to therapeutically exploit, notwithstanding some contextual discrepancies outlined above with respect to the capacity of IL-22 to affect progression of colitis. Additionally, IL-22 expression by Th17 cells mediates protection in a mouse model of hepatitis (Zenewicz and others 2007). IL-22 also exerts a proinflammatory effect in keratinocytes with transgenic IL-22 mice displaying epidermal thickening and infiltration of macrophages, resembling psoriasis-like inflammation (Wolk and others 2009). These changes may be due to IL-22-induced expression of hyperproliferative genes (keratin 14) and inflammatory chemokines (CCL3, CXCL3) (Van Belle and others 2012). Interestingly, Il22 KO mice in the CAC model develop increased tumor burden, suggesting that the exacerbated colitis observed in these mice arising from IL-22 deficiency in the early stage of this model may trigger accelerated tumorigenesis at later stages (Huber and others 2012). Meanwhile, IL-22-dependent induction of the antimicrobial peptides, Reg3γ, Reg3β, and S100A8, may trigger dysbiosis and colonization of the intestine with colitogenic microflora such as Helicobacter hepaticus (Kirchberger and others 2013; Behnsen and others 2014). The latter, associated with epithelial barrier defects, which arise in response to oncogenic mutations in IECs, may result in production of proinflammatory cytokines (Grivennikov and others 2012). Aberrant IL-22 production by ILCs can also promote tumorigenesis through Stat3-dependent transcription in malignant IECs of cell cycle regulators, including cyclin D1. Likewise, Il22bp KO mice had increased CAC-induced tumor burden, while Il22 gene deficiency reduced tumor burden in the Apc min model of intestinal tumorigenesis, demonstrating that dysregulated IL-22 signaling can promote tumorigenesis (Huber and others 2012).
Conclusions
Maintenance of epithelial barrier integrity is critical for intestinal homeostasis. Perturbation of this leads to aberrant immune activation in response to the intestinal microbiota and, as a consequence, results in autoimmune conditions, including IBD. Stat3 induction of proliferative, antimicrobial, and cytoprotective genes by cytokines, such as IL-6, IL-11, and IL-22, is important for maintenance of intestinal homeostasis. Therefore, and notwithstanding the potential of these cytokines to support tumor formation, there is a strong mechanistic rationale to consider the possibility of exploiting these cytokines therapeutically to ameliorate colitis and reestablish epithelial barrier integrity.
Footnotes
Acknowledgments
P.M.N. is supported by an Australian Postgraduate Award. The research in the laboratory of M.E. is, in part, funded by Ludwig Cancer Research. The work in the laboratories of M.E. and T.L.P is supported by the Victorian State Government Operational Infrastructure Support and NHMRC grants 1008614 and 1080498 (to T.L.P.) and 487822, 603122, and 1069024 (to M.E.) and WCRF, formally AICR (to T.L.P.).
Author Disclosure Statement
M.E. and T.L.P. are inventors of patents on the use of IL-11 antagonists for the treatment of cancer.