
Abstract
Cytokine (receptor) genes have traditionally attracted great interest as plausible genetic risk factors for autoimmune disease. Since 2007, the implementation of genome-wide association studies has facilitated the robust identification of allelic variants in more than 35 cytokine loci as susceptibility factors for a wide variety of over 15 autoimmune disorders. In this review, we catalog the gene loci of interleukin, chemokine, and tumor necrosis factor receptor superfamily and ligands that have emerged as autoimmune risk factors. We examine recent progress made in the clarification of the functional mechanisms by which polymorphisms in the genes coding for interleukin-2 receptor alpha (IL2RA), IL7R, and IL23R may alter risk for autoimmune disease, and discuss opposite autoimmune risk alleles found, among others, at the IL10 locus.
Introduction
A
Cytokine and cytokine receptor genes have traditionally attracted great interest as candidate genes for autoimmune diseases (see Vandenbroeck 2006). This interest was largely driven by the finding that it was possible to significantly alter disease outcome in animal models of autoimmune disease by means of either monoclonal antibodies against specific cytokines, administration of recombinant cytokines, or silencing of cytokine genes, and numerous examples of such successful interventions are available in the scientific literature. In 2007, the first GWAS in autoimmune diseases were published, and these revealed for the first time the real genetic size effects of non-HLA risk factors. What emerged was the finding that non-HLA risk factors have weak-to-modest effects, with odds ratios rarely exceeding 1.3. To robustly validate these genes with genome-wide significance (normally set at P=10−7), huge sample collections of carefully ascertained patients and controls are needed. With the benefit of hindsight, we now know that most pre-GWAS cytokine candidate gene studies did not have sufficient statistical power to reliably determine whether or not cytokine genes were autoimmune risk factors as the required thresholds for genome-wide significance were only very rarely met. Even if successful, the genomic context of multiple associated loci cannot be ascertained by candidate gene studies.
It should therefore not surprise that more than candidate gene studies, it is the GWAS approach that has facilitated the robust identification of those cytokine/receptor gene loci that act as clear-cut genetic risk factors for autoimmune disease. The aim of this review was to examine cytokine/receptor loci that have emerged as autoimmune risk factors mainly from GWAS but also, wherever relevant, from well-powered candidate gene studies performed after 2007 (Table 1, Figure 1). Cytokine/receptor loci emerging from GWAS were identified via the NHGRI GWAS catalog by means of the GWAS Integrator bioinformatics tool (Hindorff and others 2009; Yu and others 2011). A total of 17 autoimmune diseases were surveyed (Figure 1), that is, alopecia areata (AA), ankylosing spondylitis (AS), asthma, atopic dermatitis, Behçet's disease (BD), celiac disease (CeD), crohn's disease (CD), graves' disease (GD), multiple sclerosis (MS), primary biliary cirrosis (PBC), psoriasis, psoriatic arthritis, rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), type 1 diabetes (T1D), ulcerative colitis (UC), and vitiligo. Figure 1 provides a snapshot of a rapidly changing landscape of identified interleukin and chemokine (receptor) genes and members of the TNF receptor and ligand superfamily. Even if the functional implications of most of these associations are not yet clarified, we are beginning to understand the mechanisms by which associated polymorphisms in interleukin-2 receptor alpha (IL2RA), IL7R, cytokines belonging to the IL12 family, and IL10 may affect immune regulation in autoimmune disease. This review attempts to outline these emerging concepts.
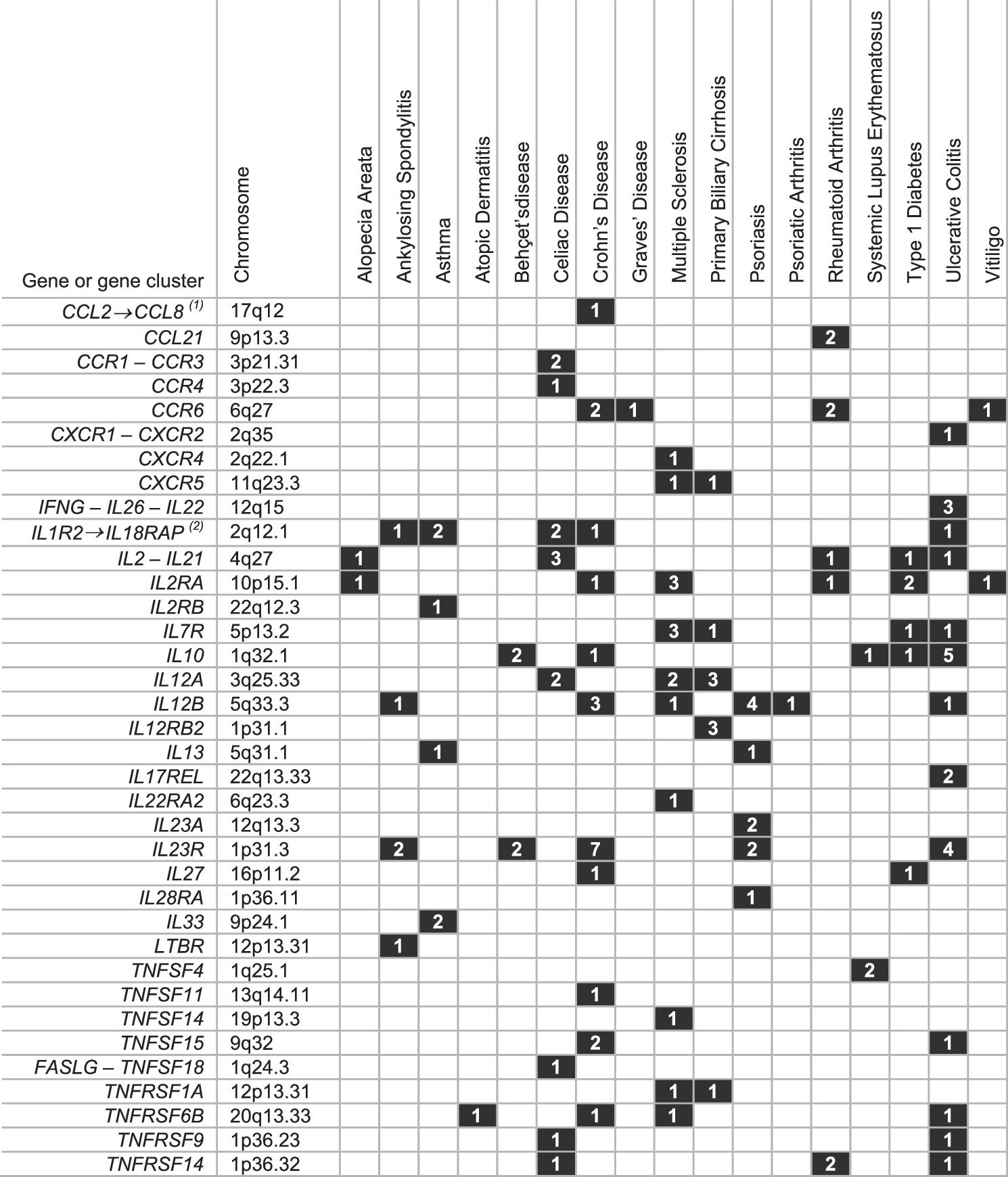
GWAS-based identification of cytokine gene loci as risk factors for 17 autoimmune/chronic inflammatory disorders. Included are chemokine and cytokine (receptor) and ligands/receptors of the TNF superfamily. Genes are considered to be associated with a disease, if they contain an associated SNP in the coding/intronic/3′ or 5′ regulatory sequences, or in the surrounding region within reach of linkage disequilibrium with the gene. Only GWAS and meta-analyses of GWAS that include validation of the associated variant through independent replication were examined. GWAS were accessed via the NHGRI Catalog of Published GWAS (
Included are only bona fide interleukin genes and their receptors.
Studies were selected from the NHGRI Catalog of Published GWAS, and from additional published GWAS. For loci that emerged more than once from independent GWAS on the same disease, only one representative study is included.
Pediatric onset form of the disease.
AA, alopecia areata; AS, ankylosing spondylitis; BD, Behçet's disease; CD, Crohn's disease; IBD, inflammatory bowel disease; MS, multiple sclerosis; PBC, primary biliary cirrhosis; Ps, psoriasis; PsA, psoriatic arthritis; RA, rheumatoid arthritis; T1D, type 1 diabetes; UC, ulcerative colitis; V, vitiligo; “-?,” risk allele was not reported; GWAS, genome-wide association studies; IL2RA, interleukin-2 receptor alpha.
IL2RA
The IL2RA and beta (IL2RB) chains, in conjunction with the common gamma chain (IL2RG), form the heterotrimeric high-affinity cell surface receptor for IL-2, a cytokine with pronounced effects on proliferation of B and T lymphocytes. The IL2RA locus was one of the first to emerge from early GWAS as autoimmune risk factor. Thus far, this gene locus has been ascertained as risk factor for a diverse series of autoimmune diseases including MS (Hafler and others 2007; De Jager and others 2009), T1D (Barrett and others 2009), AA (Petukhova and others 2010), CD (Franke and others 2010), RA (Stahl and others 2010), and vitiligo (Jin and others 2010). Substantial progress has been made toward the characterization of IL2RA–associated susceptibility effects, and this has facilitated the elucidation of the discordant association patterns observed across different diseases. Lowe and others (2007) identified within the IL2RA-RBM17 region associated with T1D two independent groups of single nucleotide polymorphisms (SNPs), which were localized to a 40-kb region enclosing intron 1 and the intergenic region located upstream from IL2RA. None of the disease-associated SNPs appear to be located in known regulatory regions. Interestingly, T1D-associated allelic variants in both these regions were found to correlate with reduced levels of sIL-2RA, a soluble form of the IL-2RA chain (Lowe and others 2007). Building upon these observations, Maier and others (2009) discovered a third, novel group of SNPs at the IL2RA locus associated with T1D, and confirmed the extended degree of allelic heterogeneity at this autoimmune risk locus. Using a large collection of DNA samples from healthy controls, MS patients, and T1D patients, they found that Group I SNPs are associated with risk for T1D but not risk for MS, Group II SNPs confer susceptibility to T1D but protection from MS, and Group III SNP susceptibility alleles are shared between T1D and MS. The presence of two independent IL2RA allelic association signals for MS risk was also observed in an independent study (Cavanillas and others 2010). In addition, serum sIL-2RA levels appeared to be independently associated with SNPs belonging to each of the 3 groups (Maier and others 2009)—indicative for incongruence between sIL-2RA level and IL2RA–conferred susceptibility. Dendrou and others (2009) documented a series of phenotypic changes conferred by the different common haplotypes that account for most of the genetic diversity at the IL2RA locus. Essentially, IL2RA haplotype-specific expression patterns of the IL-2RA receptor showed pronounced differences between the different immune cell subsets analyzed. For rs2104286, a Group III SNP (Maier and others 2009), the protective genotype reduced the likelihood for CD4+-naïve T cells to express IL-2RA, thus limiting the extent of activation. On the other hand, the rs12722495 protective Group I genotype led to expression of higher IL-2RA levels on CD4+ memory T cells, which may increase their likelihood of activation by IL-2 (Dendrou and others 2009). Simultaneously, IL-2 secretion was observed by a higher proportion of stimulated CD69+ CD4+ memory T cells from homozygotes for the protective rs12722495 allele than from homozygotes for the susceptible allele. This is consistent with known defects in the production of IL-2 by individuals suffering from T1D (Zier and others 1984). Possibly, IL-2 produced by CD4+ memory T cells is required for FOXP3+ Treg homeostasis and competitive fitness, but alternatively, a higher expression level of IL-2RA on anti-self effector T cells may be beneficial and protect against MS and T1D through a reinforced sensitivity to activation-induced apoptosis (Willerford and others 1995).
Thus, multiple, complex phenotypic effects are conferred by autoimmune-associated allelic variants at the IL2RA locus, and some findings point to previously unrecognized differences and similarities between IL-2/IL-2RA–centered immune pathways in T1D and MS (Dendrou and others 2009). In fact, from a network-based analysis of shared susceptibility genes, IL2RA emerged as a powerful factor linking MS to T1D (Baranzini 2009). At any rate, full elucidation of the ultimate molecular basis of these effects is as yet largely unexplored. It is of interest to note that several autoimmune-associated IL2RA SNPs disrupt or create CpG dinucleotides, methylation of which may possibly affect transcriptional regulation (Lowe and others 2007). Lastly, epigenetic modifications such as acetylation of promoter nucleosomes are known to modify gene transcription activity, and this process maybe affected by SNPs that influence histone remodeling. Since transcription and chromatin regulatory elements in IL2RA are correlated with acetylation islands (Roh and others 2005), the extent to which SNPs may disrupt chromatin modifications and gene activity deserves further scrutiny.
IL7R
The IL-7 receptor alpha-chain is instrumental in V(D)J recombination during lymphocyte development, and regulates access of the TCRγ locus by STAT5. Furthermore, evidence from knockout studies in mice indicates that IL7R blocks apoptosis during differentiation of T lymphocytes. These, and additional, biological activities position IL-7Rα as a crucial mediator of naïve and memory T cell homeostasis (Surh and Sprent 2008). A nonsynonymous missense SNP, rs6897932, located in the 6th exon of the IL7R gene that changes T244 in I has emerged from GWAS as risk factor for MS (Hafler and others 2007; De Jager and others 2009) and T1D (Todd and others 2007). Haplotype analysis of IL7R has shown that rs6897932, or haplotypes including this marker, constitutes the most strongly associated determinant for susceptibility to MS at this locus (Lundmark and others 2007; O'Doherty and others 2008; Vandenbroeck and others 2011), suggesting that in contrast to IL2RA, susceptibility to MS conferred by IL7R is less likely to be driven by complex patterns of allelic heterogeneity. Gregory and others (2007) demonstrated that the presence of the C risk allele of rs6897932 resulted in a 2-fold increase in the skipping of exon 6 in IL7R transcripts when compared with transcripts containing the alternate T allele. Exon-6–free transcripts code for a soluble form of the protein (Goodwin and others 1990). The ratio of membrane-bound to soluble IL-7Ra is likely to affect T cell responses to myelin proteins in MS via neutralization of IL-7 (Traggiai and others 2001). Analysis of the effect of IL7R haplotypes on transcript isoform ratios in different immune cell subsets showed that maximal differences were found in dendritic cells, in which the MS-protective haplotype produced a marked decline in the ratio of soluble to membrane-bound receptor (Hoe and others 2010). Possibly, this altered balance may make dendritic cells more responsive to thymic stromal lymphopoietin (TSLP), a cytokine employing the IL-7Rα receptor chain. TSLP-activated dendritic cells regulate the positive selection of regulatory T cells (Ziegler and Liu 2006). TSLP activation of either myeloid or plasmacytoid dendritic cell subsets via IL-7Rα is associated with the generation, as well as altered IL-10 and TGF-β cytokine production potential, of FOXP3+ regulatory T cells (Hanabuchi and others 2010). Haas and others (2011) found evidence for reduced IL-7Rα expression on conventional T cells, increased plasma IL-7 levels, and reduced myeloid dendritic cell TSLPR expression in MS, which may contribute to inhibition of Treg neogenesis in MS, even though these effects seemed not to be associated with rs6897932.
Neither TSLP nor TSLPR has emerged from GWAS as genetic risk factors for MS. However, a large-scale analysis of genes functionally related to the IL-7Rα pathway identified IL7 and SOCS1 as novel MS risk genes (Zuvich and others 2010). IL7 was also identified in a recent GWAS in MS (IMSGC and WTCCC2 2011). While IL7 did not emerge from another independent large-scale study in a Spanish-Basque cohort, SOCS1 appeared as the single most strongly MS-associated gene out of 44 cytokine/receptor genes scrutinized by means of a haplotype-tagging approach (Vandenbroeck and others 2011). IL-7 is crucial in the regulation of expansion of effector Th17 cells in experimental autoimmune encephalomyelitis as well as of Th17 cells in subjects with MS, but has no effect on Th17 differentiation (Liu and others 2010b). Moreover, IL-7R antagonism induced apoptosis of differentiated Th17 cells, but less so of Th1 and regulatory T cells, and these effects were correlated with low levels of IL-7Rα presentation on Treg cells and high level of SOCS1 in Th1 cells (Liu and others 2010b). Importantly, SOCS1 was recently found to be required for Treg cell integrity by upholding FOXP3 expression, and this process was associated with suppression of both STAT1-driven IFN-γ expression and STAT3-driven IL-17 production (Takahashi and others 2011). Thus, from these studies has appeared a functional link between IL-7, IL-7Rα, and SOCS1 in the regulation and homeostasis of Th17/Th1/Treg cells of relevance to autoimmunity, which seems to be vindicated by the identification of “hard-wired” polymorphisms in the 3 corresponding gene loci that act as susceptibility factors for MS (Zuvich and others 2010; Vandenbroeck and others 2011). The decipherment of how these polymorphisms potentially intersect to change the phenotypic behavior of helper and regulatory T cell function in MS remains for now an important and exciting challenge.
Autoimmune Risk Factors Belonging to the IL-12 Family of Heterodimeric Cytokines: IL12A, IL12B, IL12RB2, IL23A, IL23R, and IL27
IL-12, a heterodimeric cytokine composed of disulfide-bonded p35 (coded for by IL12A) and p40 subunits (coded for by IL12B), is of crucial relevance to cell-mediated immunity and Th1 differentiation (Collison and Vignali 2008). IL-12 has been implicated in the pathogenesis of a multitude of diverse autoimmune diseases (reviewed by Kastelein and others 2007; and by Paunovic and others 2008) and exerts its biological effects via binding to a heterodimeric receptor consisting of IL-12Rβ2 and IL-12Rβ1 subunits. Three of the genes coding for proteins involved in the initial stages of the IL-12 binding and signaling cascade, that is, IL12A, IL12B and IL12RB2, have emerged as risk factors from GWAS on a variety of autoimmune disorders (Figure 1, Table 1), though the precise patterns of diseases associated with each of the 3 genes overlap only partially. For IL12A, the MS-associated risk allele was reported to be protective in CeD (De Jager and others 2009; IMSGC 2010). The MS- and CeD-associated IL12A risk SNPs are located 5′ from the gene in the same haplotype block, and show only limited correlation with the PBC-associated risk SNPs located 3′ from the gene in a distinct haplotype block (Hunt and others 2008; IMSGC 2010; Liu and others 2010a). To date, IL12RB2 has emerged only as risk factor for PBC, with the reported top associated SNPs mainly located in intronic sequences (Hirschfield and others 2009; Liu and others 2010a; Mells and others 2011). IL12B has been identified in GWAS on 6 autoimmune conditions, and the majority of reported risk SNPs are located 5′ from the IL12B gene at varying positions (Table 1). Whereas alternative splice variants for both IL12RB2 and IL12A are documented in the Ensembl database (
Like IL-12, IL-23 is a heterodimeric cytokine that shares the former's p40 subunit, this time covalently linked to a p19 subunit (coded for by IL23A). It is mainly produced by dendritic cells and macrophages, and, in conjunction with IL-6 and TGF-β1, plays a pivotal role in the induction of Th17 cells and secretion of lineage-specific IL-17A (Betelli and others 2008). These effects are initiated via the engagement of L-23 with a heterodimeric receptor consisting of IL-23R and IL-12Rβ1 subunits, which leads to phosphorylation of STAT3 and further downstream signaling events (Paunovic and others 2008). Silencing of the il23a gene or selective targeting of IL-23 inhibits inflammation in experimental animal models of MS (Cua and others 2003), RA (Murphy and others 2003), IBD (Hue and others 2006), as well as psoriasis development in a clinically relevant xenotransplant mouse psoriasis model (Tonel and others 2010). Set against this background of immunological evidence, the identification of the IL23R gene locus as risk factor for 5 autoimmune conditions based on 17 GWAS is, perhaps, not entirely unexpected (Table 1, Figure 1). The most frequently encountered peak SNP at this locus is rs11209026, which is associated with 4 autoimmune diseases. The rs11209026 is a nonsynonymous G>A SNP that changes R381, located in the cytoplasmic domain of IL-23R, into Q. The Q allele is much less common than the R allele and has an allele frequency <9% in each of the 10 HapMap reference populations analyzed to date. The R and Q alleles function, respectively, as risk and protective factors for AS, UC, CD, and psoriasis (Table 1). Recent attempts at elucidating the functional repercussions of this amino acid substitution have revealed that the 381Q variant does not affect Th17 cell differentiation, but reduces Th17 effector response by limiting IL-23–induced IL-17A production and STAT3 phosphorylation (Di Meglio and others 2011). Sarin and others (2011) found that the 381Q variant impaired IL-23–dependent CD8+ T cell expansion and STAT3 activation, and reduced IL-17 and IL-22 levels in CD4+ and CD8+ T cells. In general, these observations are reconcilable with a role for IL23R R381Q in IL-17A–induced peripheral tissue pathology rather than systemic inflammation (Di Meglio and others 2011).
The IL23R gene has also been implicated in BD based on both Japanese and Turkish GWAS (Mizuki and others 2010; Remmers and others 2010). The R381Q SNP, rs11209026, appeared monomorphic in the Japanese population, and did not emerge from the Turkish GWAS. However, both studies coincided in locating the peak association signal at around 30 kb 3′ from the IL23R gene on the IL23R side of a recombination hotspot separating the IL23R and IL12RB2 haplotype blocks. Of relevance to this finding is the observation that noncoding IL23R SNPs that are not or only weakly correlated with rs11209026 have been reported to constitute residual, independent risk variants for other autoimmune disorders including IBD/UC (Duerr and others 2006; Silverberg and others 2009; McGovern and others 2010), psoriasis (Nair and others 2009), and AS (Evans and others 2011). Possibly, the functional implications of these secondary signals maybe related to the complex splicing strategies utilized by the IL23R gene. At least six alternatively spliced naturally occurring mRNA isoforms of IL23R have been reported, that can result in premature termination producing a different receptor ectodomain or a frame shift that modifies the lengths of the endodomain (Zhang and others 2006). Kan and others (2008) reported up to 24 different IL23R transcripts in activated human leukocytes, including 4 prematurely terminated isoforms. Exon-9 deletion transcripts have been identified that code for a truncated receptor containing the complete ectodomain. This soluble variant is secreted and functions as IL-23 antagonist by intercepting the cytokine and inhibiting IL-23–mediated STAT3 phosphorylation and production of the Th17 cytokines IL-17A and IL-17F (Yu and Gallagher 2010).
Notwithstanding functional indications for a role of the IL-23/Th17 axis in neuroinflammation (Becher and others, 2011), IL23R polymorphisms appear not to be associated with overall risk for MS (Begovich and others 2007; Roos and others 2008; Vandenbroeck and others 2011), even though two studies have suggested a modest, although differential, impact on clinical course of the disease (Illes and others 2008; Núñez and others 2008). Taken in conjunction with the lack of efficacy in a phase II trial of RR MS of ustekinumab (Segal and others 2008), an antibody neutralizing the p40 subunit shared by IL-23 and IL-12 that was shown to be highly effective in psoriasis (Leonardi and others 2008) and promising in CD (Sandborn and others 2008), this data sheds doubt on the pathogenetic role of IL-12 and IL-23 in MS (Martin 2008). Though the final verdict will depend on further mechanistic studies, the validated association of the IL12A and IL12B genes with risk for MS (De Jager and others 2009; IMSGC and WTCCC2 2011) seems to affirm the IL-12 pathway in MS. However, as the IL12A–encoded p35 subunit can associate with EBI3 (coded for by IL27B) to form the T cell regulatory cytokine IL-35 (Collison and others 2007), the currently undocumented role of IL-35 in neuroinflammation and MS will have to be resolved so as to factor it in into the overall phenotypic manifestation of IL12A–conferred genetic risk. In addition, the IL12B–encoded p40 subunit can homodimerize to form a cytokine with both antagonistic and agonistic activities mediated by binding to the IL-12Rβ1 chain only (reviewed by Brombacher and others 2003). While only limited data are available on the agonist role of (p40)2 in neuroinflammation, the observation that (p40)2 induces expression of iNOS and lymphotoxin-α in microglia is suggestive for a causative role in the pathogenesis of neuroinflammatory processes (Jana and others 2009; Jana and Pahan 2009). There are no records as to whether ustekinumab binds and neutralizes the (p40)2 homodimer. Consequently, the ascertainment of IL12A and IL12B as genetic risk factors for MS could well point to IL-35– and (p40)2-associated effector processes that work beyond the classical IL-12 (Th1)/IL-23 (Th17) paradigms and that are reconcilable with both the observed lack of therapeutic benefit of ustekinumab in MS and of evidence for a role of IL23R in genetic risk.
An SNP located in an intronic sequence of the STAT2 gene located 3.7 kb downstream from IL23A was found to be associated with risk for psoriasis (Nair and others 2009). Though its functional effects are as yet unknown, highly significant differences in IL23A expression levels between involved and uninvolved skin biopsies, and between normal skins from controls and uninvolved skin from cases were observed (Nair and others 2009). Taken in conjunction with the identification of IL23R and IL12B as additional risk genes for this disease, psoriasis appears to come forth as the single autoimmune disease with the clearest genetic impact of the IL-23 pathway on its pathogenesis. Finally, SNPs located in the region of the IL27 gene that codes for the p28 subunit known to heterodimerize with the EBI3 subunit to form the highly pleiotropic regulatory cytokine IL-27 (Collison and Vignali 2008) were found to be associated with risk for T1D (Barrett and others 2009) and CD (Franke and others 2010). Since this region is very gene dense, it remains to be seen whether these SNPs impact de facto upon IL-27 regulation or biology.
IL10, and Opposite Autoimmune Risk Alleles
Polymorphisms within or near many other cytokine and chemokine gene loci have been implicated in a wide array of different autoimmune diseases through GWAS (Table 1). Of these, a few stand out with reproducible patterns of association. A potent antiinflammatory cytokine, IL-10, is known to be an essential immunoregulator of inflammatory processes in the intestinal tract (Steidler and others 2000) and inhibits the costimulatory activity of macrophages on T cell activation. In two GWAS on BD, promoter and intronic variants in IL10 emerged with genome-wide significance (Remmers and others 2010; Mizuki and others 2010). The associated rs1518111 variant was associated with altered mRNA transcription and protein expression (Remmers and others 2010). BD was not associated with rs3024505, an SNP that has been linked to various autoimmune diseases (see next paragraph), indicating a different IL-10–related causative pathway (Remmers and others 2010) substantiated by the absence of correlation between the BD-associated SNPs and rs3024505 (r2 ≅0.07).
The T allele of the rs3024505 SNP, which is located close to the 3′ untranslated region of IL10, has emerged as a risk factor for UC (Franke and others 2008), CD (Franke and others 2010), and SLE (Gateva and others 2009). Interestingly, the T allele of rs3024505 protects against T1D (Wang and others 2010). This sharing of opposite risk alleles across a spectrum of autoimmune diseases is not restricted to IL10 and extends to other well-documented autoimmune risk loci such as the MHC region, the R602W variant in PTPN22, as well as other cytokine loci (Wang and others 2010). For instance, rs4788084 located 5′ from IL27 and rs917997 located 3′ from IL18RAP show opposite patterns of association with IBD versus T1D (Wang and others 2010), and similarly, opposite association patterns have been recorded for rs11594656 located 5′ from IL2RA with MS versus T1D (discussed in the section on IL2RA; Maier and others 2009), and for an intronic SNP in IL23R, rs2201841, in susceptibility to psoriasis versus UC (Nair and others 2009; McGovern and others 2010). This data raises the question as to the evolutionary forces and selection pressures that have driven maintenance of these alleles in the human population. Indeed, as either of both alleles at these risk loci is unfavorable by predisposing to a different disease, the question arises under which circumstances they do confer evolutionary advantages. Since gene variants associated with complex traits contribute only little to overall disease risk, autoimmune disease itself is unlikely to have acted as selective pressure (Barreiro and Quintana-Murci 2010). Susceptibility loci for autoimmune diseases have been suggested to occur under a balancing selection, by which multiple alleles are actively perpetuated in the gene pool at high frequencies through interactions with heterogeneous environmental factors (Wang and others 2010). This does not necessarily reflect heterozygosity advantage but could be the result of competition between diverse immune pathways to control distinct infectious pathogens. A signature for balancing selection was identified at the IL10 promoter locus (Wilson and others 2006). A seminal population genetics study analyzing 91 cytokine/receptor genes demonstrated that five IL genes, including the established autoimmune risk genes IL7R and IL18RAP, have been possible targets of pathogen-driven balancing selection (Fumagalli and others 2009). Allelic variation at these loci may have been maintained by balancing selection achieved through antagonistic pleiotropic effects. However, analysis of 11 autoimmune risk loci with opposite risk alleles confined balancing selection largely to the extended MHC region, and suggested that the observed level of nucleotide diversity at most of these loci, including IL10, is consistent with evolutionary neutrality (Cagliani and others 2011). According to this theory, evolutionary changes are caused by random drift of neutral allelic variants that impact little or not upon fitness. In some cases, opposite patterns of allelic association maybe brought about by underlying complex haplotype structures in gene areas with pronounced genetic diversity (Cagliani and others 2011).
Other Cytokine Risk Loci
Of the large family of chemokine and their receptors, the CCR6 gene region in particular leaps out through multiple associations with risk for RA (Stahl and others 2010), CD (Barrett and others 2008), vitiligo (Quan and others 2010), and GD (Chu and others 2011). In a Japanese population, Kochi and others (2010) identified a triallelic dinucleotide polymorphism in strong LD with the RA-associated CCR6 promoter risk variant rs3093024 that affects transcription regulation and is correlated with CCR6 expression levels and IL-17+ status of RA cases. The rs3093024 is in virtually perfect LD with the nearby SNP rs3093023 (r2=1), that is, the CCR6 RA risk variant identified in a GWAS based on patients and controls of European ancestry (Stahl and others 2010). CCR6 is a cell surface marker for Th17 cells (Annunziato and others 2007). Synoviocytes from arthritic joints of mice and humans secrete large amounts of the chemokine CCL20, the ligand of CCR6, which controls the migration of arthritogenic CCR6+ Th17 cells to the inflamed tissues (Hirota and others 2007). Though other additional mechanisms may pertain to CCR6-associated risk for autoimmune disease, CCR6-driven Th17 cell recruitment to inflammatory lesions constitutes an attractive working model that is worthy of further scrutiny in CD (Brand 2009) and vitiligo (Wang and others 2011).
Finally, a multitude of SNPs in regions encompassing more than 8 genes belonging to the tumor necrosis factor receptor (TNFR) superfamily and its ligands has emerged from GWAS in 7 autoimmune diseases (Figure 1). However, so far little mechanistic information is available explaining the functional consequences of the genetic basis of these associations. Interactions between TNF superfamily receptors expressed on T cells and TNF-like ligands have wide-ranging implications on inflammatory processes by affecting T cell stimulation, communication between T cell subsets, and amplification of inflammatory responses via non–T cells (Croft 2009).
Concluding Remarks
Since 2007, a rapidly expanding series of GWAS diseases has identified and validated numerous cytokine/receptor gene loci as autoimmune risk factors. It is important to note that the majority of the associated DNA variants are unlikely to constitute the ultimate causative risk factors of disease, but have probably emerged as a consequence of linkage disequilibrium with the actual causative variant. One of the main challenges for the near future therefore will be the fine mapping of each associated locus with dense panels of markers. Through linkage disequilibrium, some autoimmune-associated variants located within or close to cytokine gene loci may, as well, impact upon the functional genomics of neighboring, noncytokine genes located in the same haplotype block. In addition, uncommon variants with large effect size could explain part of the hidden heritability of autoimmune diseases. Rare variants tend to be underrepresented in current databases as well as in commercially available genome-wide SNP arrays (Oksenberg and Baranzini 2010) and their identification may require deep sequencing of DNA from patients, an option that is rapidly becoming a cost-effective perspective. Further understanding of causality will be dependent on functional studies that link allelic variants with biological processes, which will help to explain disease-specific pathophysiological manifestations.
For the purpose of this review, literature survey was confined to cytokine/receptor genes. The phenotypic effects of polymorphisms in these genes cannot be uncoupled from those of other autoimmune risk loci if we wish to gain comprehensive insights. Epistatic interactions will need to be addressed and may refine and expand current concepts of cytokine-centered pathways. One of the most important findings from current GWAS, which is particularly true with regard to cytokine/receptor genes, is the sharing of cytokine genes and allelic variants across a wide spectrum of autoimmune diseases. Whereas this clearly indicates that common underlying pathogenetic mechanisms exist, the reality might be more complex. For many of the gene loci covered in this review including, but not limited to, IL2RA, IL10, IL23R, and IL7R, association signals for one autoimmune disease appear not to be correlated with those found for other such conditions at the same locus. For some autoimmune diseases, secondary association signals are found within a cytokine gene locus that are uncorrelated with the primary signal at the same locus. Opposite risk alleles are documented to occur at the IL10, IL23R, IL2RA and IL18RAP loci. This global picture may reflect the complex genetic architecture and modularity of human autoimmune diseases, and several strategies have already been developed to define the unique and shared gene assortments for autoimmune diseases (Baranzini 2009; Sirota and others 2009).
Traditional pre-GWAS candidate gene approaches have highlighted numerous genetic associations between cytokine genes and risk for, or clinical parameters of, autoimmune diseases, and many of these associations may remain valid even if their effects are modest (for review see Goris and Vandenbroeck 2003; Vandenbroeck 2006). However, GWAS rather than candidate gene approaches have provided the power and resolution to unequivocally pin down those cytokine/receptor genes that affect susceptibility with genome-wide significance. For the cytokine biologist, a treasure trove of new information is now available that may act as a powerful guide to further research into the functional and immunological ramifications of these associations.
Footnotes
Author Disclosure Statement
No competing financial interests exist.