
Abstract
Background:
Pressurized metered dose inhalers (pMDIs) currently contain propellants with relatively high global warming potential (GWP), such as hydrofluoroalkane-134a (HFA-134a). Hyrofluoroolefin-1234ze (HFO-1234ze) is a near-zero GWP propellant in development for use in future pMDIs.
Methods:
This Phase IIIb, multicenter, randomized, double-blind, single-dose crossover study aimed to assess post-dose lung function and clinical manifestations of bronchospasm following single doses of HFA-134a and HFO-1234ze via pMDI (four inhalations with no active drugs) in participants with well or partially controlled asthma. The primary endpoint was change from baseline in forced expiratory volume in one second area under the curve from 0 to 15 minutes (FEV1 AUC0–15 minutes) post-dose and was assessed using a linear mixed-effect model. Secondary endpoints included cumulative incidence of bronchospasm events (reduction in FEV1 of >15% from baseline at 5 or 15 minutes post-dose with associated wheezing, shortness of breath, or cough). Safety and tolerability were also assessed.
Results:
Among 52 participants randomized to treatment, noninferiority of HFO-1234ze pMDI versus HFA-134a was established for change from baseline in FEV1 AUC0–15 minutes post-dose between treatments (least squares mean [LSM]; 95% confidence intervals [CI] change: HFO-1234ze pMDI, –0.014 [–0.033, 0.006] L; HFA-134a, –0.004 [–0.024, 0.015] L; LSM [95% CI] difference: –0.009 [–0.037, 0.018; p = 0.492]). No bronchospasm events were reported. Two patients (3.8%) in each group experienced ≥1 adverse event (AE). No serious AEs, AEs of special interest, or AEs leading to discontinuation were reported.
Conclusions:
HFO-1234ze pMDI was well tolerated in participants with well or partially controlled asthma. As with HFA-134a pMDI, no significant effects on lung function or bronchospasm events were observed. As HFO-1234ze pMDI has a near-zero GWP, it represents a compelling, environmentally conscious alternative propellant for use in pMDI devices for treatment of chronic respiratory disease.
Trial Registration:
NCT05850494.
Introduction
Pressurized metered dose inhalers (pMDIs) are commonly used for the delivery of both maintenance and reliever therapy in persons living with obstructive lung diseases. 1 Asthma is characterized by chronic airway inflammation and bronchial hyperreactivity and is defined by a history of respiratory symptoms, 1 such as wheeze, shortness of breath, chest tightness, and cough, which can vary over time and in intensity, together with variable expiratory airflow obstruction.
Current propellants used in pMDIs, such as hydrofluoroalkane-134a (HFA-134a) and hydrofluoroalkane-227ea (HFA-227ea), were advances compared with chlorofluorocarbons, as the former has lower potential for ozone layer depletion; however, both are potent greenhouse gases with relatively high global warming potential (GWP).2–4 Transitioning patients to dry powder inhalers (DPIs) has been proposed as a potential solution to lower the carbon footprint of care for respiratory disease, but it is important to note that although DPIs have a lower GWP than current pMDIs, they may not always be used interchangeably,2,3,5 and patients may have a positive clinical response to medication delivered via pMDI for which there is no DPI alternative. Furthermore, nonclinical changes in inhalers can be detrimental to disease control.6,7 As such, a transition to pMDIs using propellants with low GWP is needed as a part of a comprehensive approach to reduce the carbon footprint of healthcare and thereby aid in addressing the climate emergency while safeguarding access to essential medicines.8,9
Hydrofluoroolefin-1234ze (HFO-1234ze) is a near-zero GWP propellant in development for use in pMDIs. 10 HFO-1234ze has undergone extensive preclinical and toxicology studies in the development as a pMDI propellant. 11 However, HFO-1234ze is a new excipient proposed for use in pMDIs, and, as such, there is a need to characterize its potential effects on the lungs including airway sensitivity reactions. HFO-1234ze showed no evidence of systemic or accumulated exposure in animals unless administered at extremely high doses; thus, it was considered to be safe for further human study. 11 Therefore, the propellant effects are primarily airway based, and the key tolerability consideration for this study was the potential for bronchospasm; this was assessed using comparative pre- and post-dose lung function measured over 30 minutes. Further studies are underway to assess the broader safety and tolerability considerations of the propellant.
The aim of this study was to assess post-dose lung function and clinical manifestations of bronchospasm (e.g., wheezing), following single doses of two different propellants for pMDIs (and no active drugs) in adults with well or partially controlled asthma: HFO-1234ze versus reference HFA-134a.
Methods
Study design
This was a Phase IIIb, multicenter, randomized, double-blind, single-dose crossover study comparing the safety and tolerability of HFO-1234ze pMDI versus HFA-134a pMDI in adult participants with well-controlled or partially controlled asthma (NCT05850494).
The study design is summarized in Figure 1. The duration of participation was up to 37 days, with a screening period approximately 14 (±2) days prior to first dosing followed by Treatment Period 1 (1 day), a 3–12-day washout period, Treatment Period 2 (1 day), and a final safety follow-up visit via telephone contact 3–7 days after the final dose. Participants were randomized 1:1 to one of two treatment sequences (A
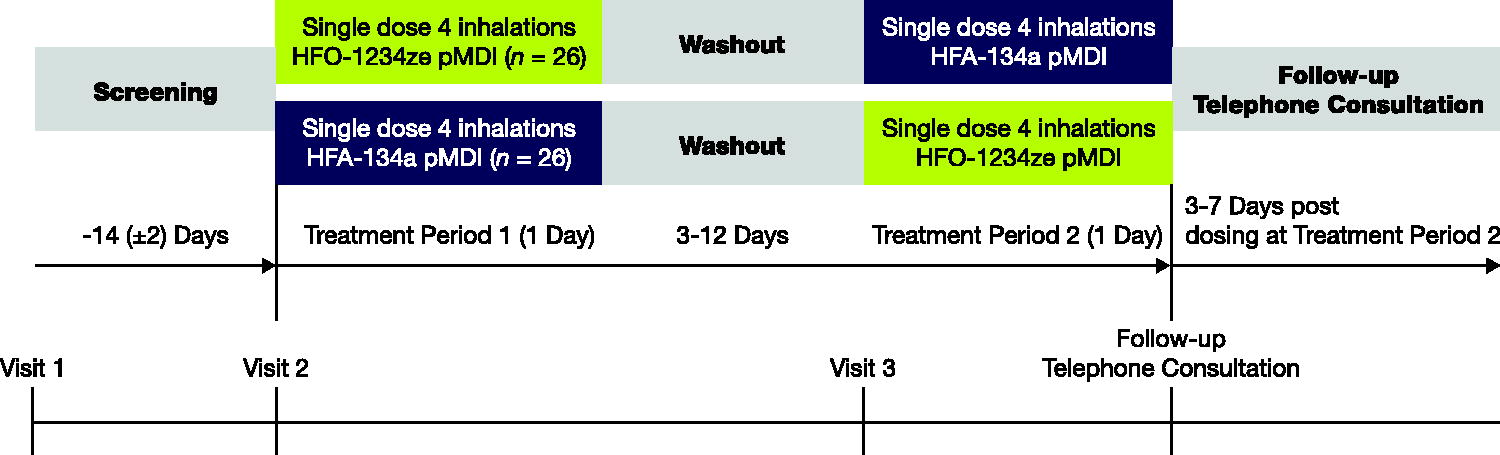
Study design. Study participants were randomized to two treatment sequences (n = 26 per sequence) with the following treatment schemes: AB—Treatment Period 1 (single dose—four inhalations HFO-1234ze pMDI) followed by Treatment Period 2 (single dose—four inhalations HFA-134a pMDI); BA—Treatment Period 1 (single dose—four inhalations HFA-134a pMDI) followed by Treatment Period 2 (single dose—four inhalations HFO-1234ze pMDI). HFA-134a, hydrofluoroalkane-134a; HFO-1234ze, hyrofluoroolefin-1234ze; pMDI, pressurized metered dose inhaler.
Participants
The study population comprised male or females aged 18–45 years with a documented history of asthma ≥12 months prior to Visit 1 according to Global Initiative for Asthma (GINA) guidelines (GINA 2022 12 ) with an asthma control questionnaire (ACQ)-5 total score <1.5 at Visit 1, pre-bronchodilator forced expiratory volume in the first second (FEV1) >60% predicted normal value at Visit 1, acceptable pMDI administration technique, and well-controlled or partially controlled asthma 4 weeks prior to screening on their current treatment. Current asthma treatments included low-dose daily ICS (as defined by GINA 2022 12 ) low-dose ICS/formoterol as needed, SABA as needed, and low-dose ICS whenever SABA used as needed.
Key exclusion criteria included life-threatening asthma (defined as a history of significant asthma episodes requiring intubation that were associated with hypercapnia, respiratory arrest, hypoxic seizures, or asthma-related syncopal episodes), current smokers, former smokers with >10 pack-years or former smokers who stopped smoking <6 months prior to Visit 1, any respiratory infection or asthma exacerbation treated with systemic corticosteroids and/or additional ICS treatment in the 8 weeks prior to Visit 1 and throughout the screening period, hospitalization for asthma within 1 year prior to Visit 1, and current or prior history of disease, including cardiovascular, hepatic, renal, hematological, neurological, endocrine, gastrointestinal, or pulmonary (e.g., active tuberculosis, bronchiectasis, pulmonary eosinophilic syndromes, chronic obstructive pulmonary disease, and uncontrolled severe asthma), that, in the opinion of the investigator, could put the safety of the participant at risk or affect the safety/tolerability analysis.
Endpoints
The primary endpoint was change from baseline in FEV1 AUC0–15 minutes post-dose. The primary endpoint was selected since the focus of the study was tolerability of the propellant as measured by bronchospasm. This endpoint is aligned with similar inhaler-based studies assessing post-dose transient bronchospasm events.13–15 Secondary endpoints included cumulative incidence of bronchospasm events, defined as a reduction in FEV1 of >15% from baseline (i.e., the FEV1 value obtained within 30 minutes prior to study intervention administration) at 5 or 15 minutes post-dose with associated symptoms of wheezing, shortness of breath, or cough, and safety and tolerability, as assessed by adverse events (AEs). Repeated measurements of change from baseline in FEV1 at 5, 15, and 30 minutes post-dose during each treatment period were an exploratory endpoint. As post-dose lung function was also assessed at 30 minutes for safety, an assessment of the change in baseline FEV1 at 30 minutes post-dose was included as an exploratory safety endpoint.
Assessments
Site coordinators trained participants on appropriate inhaler use with the aid of training inhalers at the beginning of Treatment Periods 1 and 2. Site coordinators were provided education on inhaler technique by AstraZeneca staff and demonstrated appropriate usage at the study investigator meeting. At in-clinic Visit 2 (Treatment Period 1) and Visit 3 (Treatment Period 2), baseline pre-dose FEV1 values were obtained by spirometry within 30 minutes pre-dose, and post-dose FEV1 was assessed at 5, 15, and 30 minutes after dosing with four inhalations of randomized treatment (first or second single-dose treatment period). As repeated spirometric testing was conducted over short time periods, assessments between baseline, 5, 15, and 30 minutes were predefined and considered acceptable time intervals. Albuterol/salbutamol was withheld for at least 8 hours prior to FEV1 measurements. At each in-clinic Visit 2 or 3, spirometry at 5 and 15 minutes post-dose was performed to monitor participants for signs of bronchospasm. If FEV1 values were within 10% of baseline FEV1 performed that day, participants could be discharged.
AEs were assessed at each visit and were coded according to the Medical Dictionary for Regulatory Activities, version 26.1, by system organ class and preferred term. AEs of special interest (AESIs) included dysphonia, cough, dyspnea, wheezing, bronchospasm, and asthma exacerbations. Asthma exacerbations were defined as a worsening of asthma that required medical intervention and resulted in a deterioration in asthma symptoms, night-time awakening due to asthma, or physical examination findings consistent with deterioration of asthma. Moderate asthma exacerbations were defined as those that resulted in additional ICS treatment for ≥3 days and severe asthma exacerbations as those that resulted in a short course of systemic corticosteroids for ≥3 consecutive days, an emergency room or urgent care visit for <24 hours requiring systemic corticosteroids, in-patient hospitalization, or death related to asthma.
Statistical analyses
The randomized population included all participants who were randomized to one of the two treatment sequences. The primary analysis set included all participants in the randomized population who received complete treatment in both periods. Participants who used any restricted medication (i.e., those that could potentially influence bronchodilator response testing) within the washout period prior to the clinical visit were excluded from the analysis. The safety analysis set included all participants in the randomized population who had received any amount of any study treatment (i.e., at least one inhalation). Since this was considered a safety study, participants were analyzed according to the actual treatment received. Demographic and baseline characteristics were analyzed in the randomized population and summarized by treatment sequence using counts and proportions for categorical variables and appropriate descriptive statistics for continuous variables.
The primary endpoint was analyzed in the primary analysis set using a linear mixed-effect model, which assumed that residuals and random effect coefficients were independent and identically distributed. This model included change from baseline in FEV1 AUC0–15 minutes post-dose as the dependent variable, actual treatment sequence, treatment period, average baseline FEV1, treatment received as independent fixed-effect variables, participant within treatment sequence as a random effect, and the corresponding two-sided 95% confidence interval (CI) for the difference between HFO-1234ze and HFA-134a were reported. Noninferiority was declared if the lower limit of the 95% CI for the difference between HFO-1234ze pMDI and HFA-134a pMDI was greater than –0.200 L. A noninferiority margin of 0.200 L was selected as it conservatively represents the minimum change in FEV1 required to classify the change as reversible and in keeping with a diagnosis of asthma. To demonstrate noninferiority, the lower bound of the 95% CI for the difference in FEV1 AUC0–15 minutes between HFO-1234ze and HFA-134a needed to exceed this threshold.
The secondary endpoint, the proportion of participants with a bronchospasm event by treatment received, was planned to be analyzed using McNemar’s test of marginal homogeneity.
For the exploratory endpoints, a linear mixed model was applied for each timepoint with change in FEV1 as the dependent variable and actual treatment sequence, treatment period, average baseline FEV1, treatment received, and random intercepts per participant as independent fixed-effect variables. The least squares mean (LSM) change from baseline estimates per treatment group, the LSM difference between treatment groups, and 95% CIs at 5, 15, and 30 minutes were analyzed. AEs were analyzed in the safety analysis set and were summarized by treatment group using counts and proportions.
The study was powered based on the ability to detect an event that occurred at a rate of 5% or greater. With 46 participants, there was a 90% probability of observing at least one event of bronchospasm in response to administration of HFO-1234ze pMDI if the true incidence was 5% or greater. Assuming a 10% dropout rate, 52 participants were to be randomized so that 46 participants completed the study. For the primary endpoint, based on a sample size of 46 participants and assuming a difference of 25 mL in terms of the change from baseline in FEV1 AUC0–15 minutes post-dose between HFO-1234ze pMDI and HFA-134a pMDI, and a within-participant standard deviation (SD) of 0.200 L, the total sample size for the 2 × 2 crossover design would have 98% power to detect the noninferiority of HFO-1234ze pMDI versus HFA-134a pMDI based on a noninferiority margin of –0.200 L at a one-sided significance level of 0.025.
All analyses were performed by Fortrea, using SAS version 9.4.
Results
The study was conducted at four sites in the United States from May 2023 to August 2023.
Participants
Of 60 participants screened, 52 were randomized to treatment. All randomized participants completed both treatment periods and the study, with no participants discontinuing treatment or withdrawing from the study. Participant demographics and clinical characteristics among Sequences HFO-1234ze
Compared with Sequence HFA-134a
Key respiratory characteristics were typical for this population. Both Sequences HFO-1234ze
Lung function
Baseline FEV1% predicted (SD) was similar for each treatment sequence, 87.9% (14.4) for Sequence HFO-1234ze
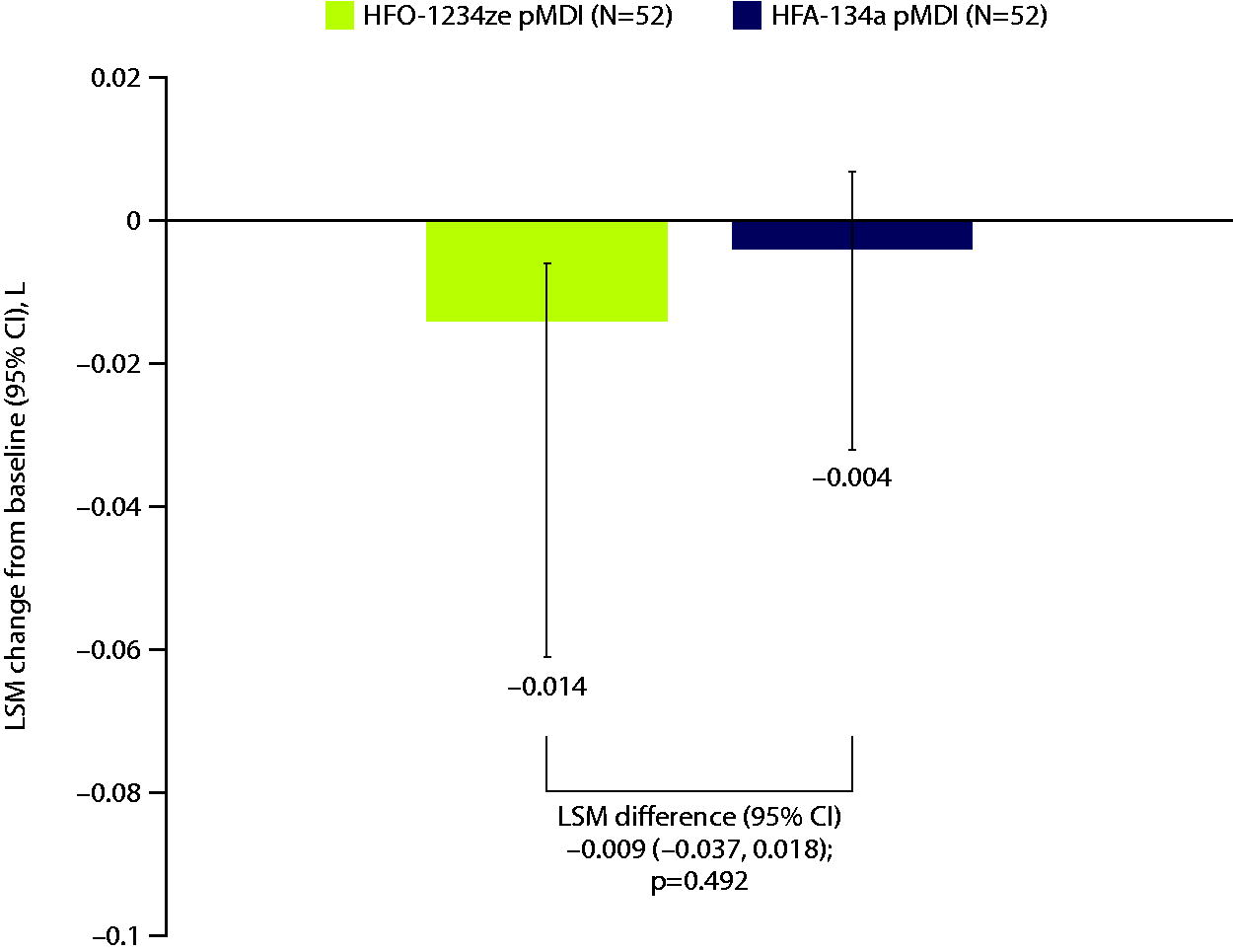
FEV1 AUC0–15 minutes post-dose change from baseline for HFO-1234ze and HFA-134a pMDIs. Data shown are the primary analysis set; this includes all participants randomized to treatment who completed both treatment periods and did not use any restricted treatment within the washout period prior to the clinical visit. LSM change from baseline in FEV1 AUC0–15 minutes post-dose was estimated using a linear mixed-effects model with change from baseline in FEV1 AUC0–15 minutes post-dose as a dependent variable and independent fixed-effects variables of actual treatment sequence, treatment period, average baseline FEV1, treatment received, and a participant within treatment sequence random effect. p-Value reflects two-sided t-test. CI, confidence interval; FEV1 AUC0–15 minutes, forced expiratory volume in the first second area under the cover from 0 to 15 minutes; LSM, least squares mean; pMDI, pressurized metered dose inhaler.
Participant Demographics and Baseline Characteristics, by Treatment Sequence a
Data are shown for the randomized set; this includes all participants randomized to study treatment.
ACQ, asthma control questionnaire; BMI, body mass index; HFA-134, hydrofluoralkane-134a; HFO-1234ze, hydrofluoroolefin-1234ze; ICS, inhaled corticosteroid; LABA, long-acting β2-agonist; LTRA, leukotriene receptor antagonists; SD, standard deviation.
Change from Baseline in FEV1 AUC0–15 Minutes, L Mean (SD), by Treatment Period and Overall a
The primary analysis set includes all participants randomized to treatment who completed both treatment periods and did not use any restricted treatment within the washout period prior to the clinical visit.
AUC0–15 minutes, area under the curve from 0 to 15 minutes; FEV1, forced expiratory volume in the first second; pMDI, pressurized metered dose inhaler.
Fig. 3 reports the overall LSM change from baseline in FEV1 for each treatment group over time. There was an LSM difference between treatment groups of –0.0012 at 5 minutes (p = 0.0543), –0.0019 (p = 0.400) at 15 minutes, and 0.0013 (p = 0.577) at 30 minutes, showing there was no significant change in FEV1 between HFA-134a pMDI and HFO-1234ze pMDI.
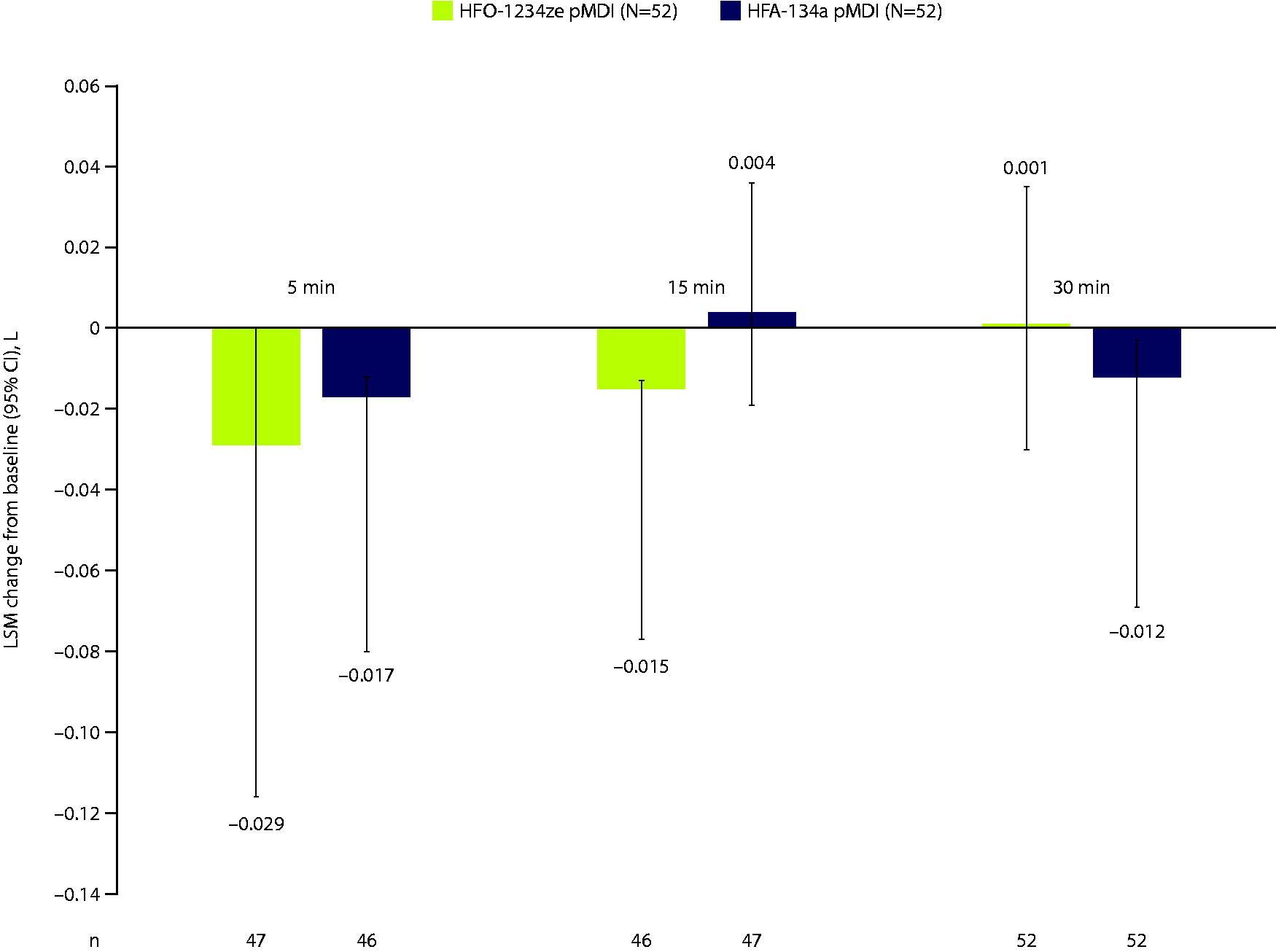
FEV1 change from baseline for HFO-1234ze and HFA-134a pMDIs. Data shown are the primary analysis set, which includes all participants randomized to treatment who completed both treatment periods and did not use any restricted treatment within the washout period prior to the clinical visit. LSM change from baseline in FEV1 was estimated for each timepoint using a linear mixed-effects model with change from baseline FEV1 as the dependent variable and independent fixed variables of actual treatment sequence, treatment period, average baseline FEV1, treatment received, and a random intercept per participant.
Safety
Overall, the incidence of AEs was similar between treatments, with two AEs reported in two participants treated with HFO-1234ze pMDI and four AEs reported in two participants treated with HFA-134a pMDI (Table 3). With HFO-123ze, one participant had an AE of dysgeusia that was considered possibly related to the study treatment and one participant had an AE of pyrexia. With HFA-134a, one participant had an AE of fatigue, and one participant had AEs of urinary tract infection, nephrolithiasis, and ovarian cyst. All AEs were either mild or moderate in intensity, and no AESIs (dysphonia, cough, dyspnea, wheezing, bronchospasm, or asthma exacerbation) were reported during the study. No AEs led to discontinuation of the study treatment, and no serious AEs, serious AEs with outcome of death, or possibly treatment-related serious AEs were reported during the study.
Summary of Adverse Events a
AEs are listed by the preferred term per the Medical Dictionary for Regulatory Activities version 26.1.
AEs with start date on or after the date of treatment during Treatment Period 1 up to (and including) 7 days after the last dose date are reported.
Data shown are the safety analysis set, which included all participants randomized to study treatment who received at least one dose of any study drug.
Possibly related is defined as reasonable possibility that the AE was caused by the investigational product, as assessed by the investigator.
AE, adverse event; SAE, serious adverse event.
Discussion
This Phase IIIb, single-dose, crossover, lung function monitoring study is the first published study to assess lung function reduction and tolerability associated with HFO-1234ze in people diagnosed with asthma. For the primary endpoint of FEV1 AUC0–15 minutes, noninferiority was demonstrated between HFO-1234ze pMDI and the reference propellant, HFA-134a pMDI, as changes from baseline were minimal with both propellants. Overall, there were no clinically relevant differences in FEV1 AUC0–15 minutes between treatment groups.
Exploratory analyses found that changes in FEV1 from baseline at 5, 15, and 30 minutes post-dose were minimal, with no notable differences between HFO-1234ze pMDI and HFA-134a pMDI, supportive of the primary analysis. Additionally, the potential for bronchospasm induced by HFO-1234ze pMDI did not differ from HFA-134a pMDI, as no bronchospasm events were reported in either treatment group at any time during the study. The safety profile of HFO-1234ze pMDI was similar to HFA-134a pMDI, as both treatments were well tolerated in participants with asthma, with AEs reported in fewer than 5% of participants in either treatment sequence and no new or unexpected safety findings.
The climate emergency has brought scrutiny to the use of pMDIs due to the GWP of current propellants, prompting the need for alternative propellants with reduced climate impact. It is important to note that DPIs and pMDIs are not always interchangeable for each individual, so careful consideration must be given when choosing the correct device for each patient and in ensuring correct usage.2,5,16 Potential advantages of pMDIs over DPIs include flexibility in dosing, ability to employ a holding chamber to decrease oropharyngeal side effects, a substantially longer inhaler user-life, and a larger selection of medications—permitting personalization of inhaled therapies in patients with obstructive lung diseases. 17
The lung can be sensitive to foreign materials, and paradoxical respiratory effects can occur with inhaled medicines, often due to excipients (e.g., soya bean lecithin) and preservatives in the formulation18–23 that are sometimes related to direct pharmacological effects.20,24 Prior pMDIs have contained sulfites, 25 while multidose nebulized solutions previously contained benzalkonium chloride—both known to cause bronchospasm in asthma.18,21,23,25 It is essential to determine that any excipient used in inhaled medications is assessed for effects on lung function. 26
Limitations may include the usage of four inhalations of HFO-1234ze to assess for bronchospasm events. The selected dose should suffice for study endpoints since propellant dosing is not anticipated to exceed two inhalations when used with combination inhalers using active ingredients. Although this study is limited by its small sample size, it is based upon an a priori power calculation and our findings further support HFO-1234ze as a viable alternative propellant with near-zero GWP for use in pMDIs, even among people with reactive airways, such as those with well or partially controlled asthma included in the present study. Additional tolerability will ultimately be assessed in clinical studies using this new propellant with active medications. While this study was conducted exclusively in adults, there is no evidence to suggest that comparative paradoxical bronchospasm events would be any different in younger-aged populations. 27
Conclusions
In conclusion, similarly to the reference propellant HFA-134a pMDI, HFO-1234ze pMDI is well tolerated in participants with well or partially controlled asthma, with no bronchospasm events reported and no significant effect on lung function. Therefore, as HFO-1234ze pMDI has near-zero GWP, it may represent a useful alternative propellant for use in pMDI devices, which could help reduce the current contribution of greenhouse gases generated by the treatment of chronic respiratory diseases.
Footnotes
Acknowledgments
The authors would like to thank the study participants. Medical writing support, under the direction of the authors, was provided by Rebecca Douglas, PhD, of CMC Connect, a division of IPG Health Medical Communications, and was funded by AstraZeneca, in accordance with Good Publication Practice (2022) guidelines. 28
Authors’ Contributions
All authors read and approved the final article. R.A.P. contributed to methodology and writing—review and editing. A.S.B. contributed to conceptualization, investigation, methodology, project administration, supervision, visualization, and writing—review and editing. M.J. contributed to formal analysis, investigation, methodology, and writing—review and editing. J.X. contributed to data curation, formal analysis, methodology, supervision, and writing—review and editing. D.P. contributed to conceptualization, formal analysis, and writing—review and editing. I.R. contributed to conceptualization, investigation, methodology, project administration, supervision, visualization, and writing—review and editing. M.A. contributed to conceptualization, data curation, formal analysis, investigation, methodology, project administration, supervision, and writing—review and editing. P.B. contributed to conceptualization, funding acquisition, project administration, resources, supervision, and writing—review and editing. M.P. contributed to formal analysis, methodology, project administration, visualization, and writing—review and editing.
Ethics Approval and Consent to Participate
The clinical study protocol and participant informed consent documents were submitted for review to the Institutional Review Board. This study was performed in accordance with the ethical principles that have their origin in the Declaration of Helsinki and that are consistent with ICH/GCP, applicable regulatory requirements and the AstraZeneca policy on Bioethics. Each participant provided written informed consent.
Data Availability
Data underlying the findings described in this article may be obtained in accordance with AstraZeneca’s data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure. Data for studies directly listed on Vivli can be requested through Vivli at http://www.vivli.org/. Data for studies not listed on Vivli could be requested through Vivli at https://vivli.org/members/enquiries-about-studies-not-listed-on-the-vivli-platform/. The AstraZeneca Vivli member page is also available outlining further details: ![]() .
.
Author Disclosure Statement
R.A.P. reports research grants and consulting fees from
Funding Information
This study was funded by AstraZeneca.