
Abstract
Abstract
Background:
GFF MDI is a glycopyrrolate/formoterol fumarate fixed-dose combination metered dose inhaler formulated using co-suspension delivery technology. This open-label, single-arm multicenter study (NCT02268396) evaluated the accuracy, reliability, and functionality of the GFF MDI AeroCount® dose indicator when used by patients with chronic obstructive pulmonary disease (COPD).
Patients and Methods:
The study enrolled subjects (40–80 years of age) with an established clinical history (≥6 months) of COPD, who completed an electronic diary twice daily to record study-drug administration time, the number of actuations used, and pre- and post-dose dose indicator readings. The primary endpoint was the percentage of devices for which the number of subject-reported actuations was consistent (±20 actuations) with the dose indicator-based actuation count (equal to 130 minus the dose indicator reading) at the end of the treatment period (4 weeks). Safety was monitored throughout the study.
Results:
A total of 138 subjects with moderate-to-very severe COPD (50.7% male; mean [standard deviation (SD)] age 62.1 [8.3] years) were enrolled and treated. Subject-reported actuation count and dose indicator-based actuation counts were consistent for 96.4% (132/137) of devices at the end of the treatment period (4 weeks) in the intent-to-treat (ITT) population and for all devices in the per-protocol (PP) population. The mean (SD) dose indicator-based actuation and subject-reported actuation counts in the ITT population (n = 137) were 113.4 (18.9) and 117.0 (19.0), respectively, with a mean (SD) difference of 3.6 (7.9). The mean (SD) dose indicator-based actuation and subject-reported actuation counts in the PP population (n = 112) were 116.8 (8.7) and 119.7 (8.1), respectively. There were no unexpected safety findings.
Conclusions:
This study supported the accuracy, reliability, and utility of the dose indicator integrated into the GFF MDI device when used by patients with COPD.
Introduction
C
Inhaled bronchodilators are the principal treatment option for maintenance therapy in patients with COPD, including long-acting muscarinic antagonists (LAMAs) and long-acting β2-agonists (LABAs).(3) GFF MDI (Bevespi Aerosphere®), a LAMA/LABA fixed-dose combination of glycopyrrolate/formoterol fumarate 18/9.6 μg (equivalent to glycopyrronium/formoterol fumarate dihydrate 14.4/10 μg) delivered by metered dose inhaler using innovative co-suspension delivery technology, is approved in the United States for the long-term maintenance treatment of airflow obstruction in patients with COPD.(4)
Dose indicators in inhaler devices are intended to reduce patient error by allowing patients to track the number of actuations they have used.(5) In line with U.S. Food and Drug Administration (FDA) guidance, dose indicators should be designed to be as close to 100% reliable as possible and should specifically avoid undercounting. The FDA further recommends that the functionality, reliability, and accuracy of dose indicators should be evaluated in clinical trials within the target population.(5)
The GFF MDI device incorporates the second generation TMAI-200 series dose indicator (Trudell Medical International AeroCount® top mounted actuation indicator), a commercially available dose indicator that has been integrated into the GFF MDI container closure system without device modification, except for attaching the dose indicator on the base of the GFF MDI canister with a canister label. The indicator uses a force mechanism to count down the remaining actuations. The count display is advanced after every 10 actuations, and numbers are displayed in increments of 20.
For GFF MDI, the starting dose indicator position is set to 130, which accounts for 120 inhalations, 4 initial priming actuations, and 6 re-priming actuations. When the number of actuations remaining in the canister reaches 20, the indicator displays the number 20 with red backing to indicate to the patient that it is time to refill their prescription.
The aim of the present study was to evaluate the performance of the dose indicator of GFF MDI when used by subjects with COPD.
Patients and Methods
Study design
This was an open-label, single-arm multicenter study (NCT02268396) conducted at 10 sites in the United States, with a duration of ∼6 weeks per subject (a 1–2-week screening period, followed by a 4-week treatment period) (Fig. 1). This study was conducted in accordance with Good Clinical Practice, including International Conference on Harmonisation Guidelines, the Declaration of Helsinki, and the United States Code of Federal Regulations. An Institutional Review Board approved the study protocol and consent forms, and written informed consent was obtained from all subjects before screening and study entry.
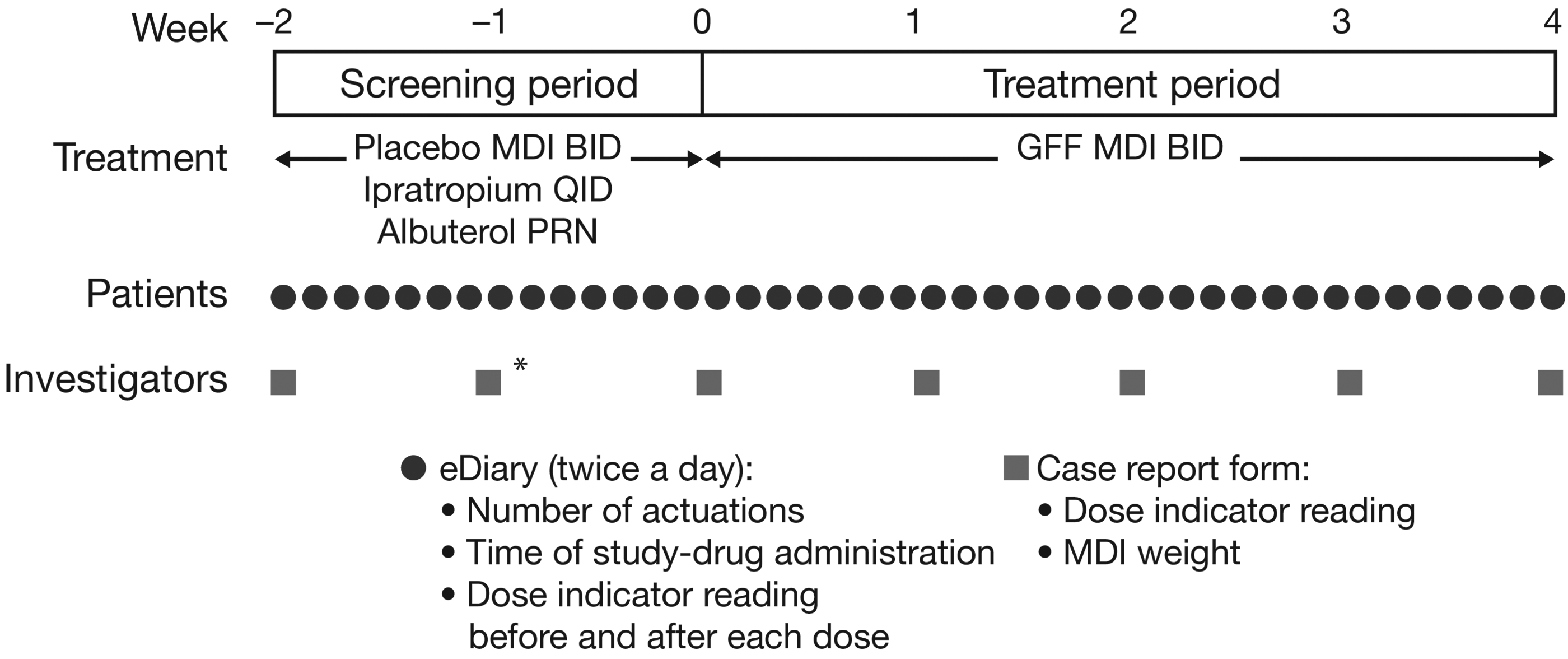
Study design. BID, twice daily; GFF, glycopyrrolate/formoterol fumarate; MDI, metered dose inhaler; PRN, as required; QID, four times daily. *Compliance reassessment visit.
Subject population
Eligible subjects were 40 − 80 years of age with COPD and current or former smokers with a smoking history of ≥10 pack-years and a minimum of 6 months' established clinical history of COPD (as defined by American Thoracic Society/European Respiratory Society guidelines).(6) Subjects were required to have a forced expiratory volume in 1 second (FEV1)/forced vital capacity ratio of <0.70, and a post-bronchodilator FEV1 value ≥30% and <80% of predicted normal (calculated using the Third National Health and Nutrition Examination Survey reference equations(7,8)) that was also ≥750 mL. Exclusion criteria included a clinically abnormal chest X-ray or computed tomography scan (if abnormalities were not due to COPD) and significant diseases other than COPD.
Subjects unable to comply with study procedures, including noncompliance with electronic diary (eDiary) completion, and those who required the use of a spacer device to compensate for poor hand-to-breath MDI coordination were also excluded.
Any eligible subjects who were using prohibited COPD medications (such as oral β2-agonists and LABAs), cromoglycate or nedocromil inhalers, aclidinium, tiotropium, or fixed-dose combination treatment with an inhaled corticosteroid discontinued these medications for the duration of the trial and were switched to ipratropium bromide MDI four times daily and albuterol sulfate MDI as needed.
Endpoints and assessments
The primary endpoint was the percentage of devices that showed consistency between the subject-reported actuation count and the dose indicator-based actuation count at Week 4 (end of study). Owing to the dose indicator only advancing in increments of 10 actuations and displaying numbers in increments of 20 actuations,(4) consistency was met when recorded within ±20 actuations. The subject-reported actuation count was the total number of actuations recorded by the subject in an eDiary, including priming shots before initial use and after cleaning. The dose indicator-based actuation count was defined as the total number of actuations of the MDI obtained by subtracting the dose indicator reading (recorded by the investigator) from 130.
Safety assessments included physical examinations, electrocardiograms, vital sign measurements, clinical laboratory tests, and monitoring of adverse events (AEs). AEs that occurred on or after the date of the first dose of GFF MDI were considered treatment emergent. AEs that occurred between the time of signing the informed consent form and receiving the first dose of GFF MDI were recorded as medical history, unless they were serious treatment-emergent AEs (TEAEs).
Screening period
Subject compliance to treatment and eDiary use was assessed in the screening period through use of a placebo MDI. At the start of the screening period, subjects were issued with a handheld eDiary device (PROLogic nSpire Health) and placebo MDI, trained on their use, and instructed how to read and record information from the dose indicator. The information that subjects entered in the eDiary included information relating to dosing, i.e., the time of study-drug administration; the number of actuations used; and both pre-and post-dose dose indicator readings. Subjects used their MDI twice daily and entered information into the eDiary after each use. In addition, the eDiary also provided a weekly reminder to wash the actuator, which was confirmed by the subject.
MDI weight and dose indicator readings were recorded weekly at each study visit by the site staff. Figure 2 shows the position of the dose indicator in GFF MDI and the display window showing the number of actuations left in the device. MDI weight and dose indicator readings were recorded weekly at each study visit by the site staff. Study medication compliance (time of dosing) was checked at all study visits.

Schematic of a GFF MDI device with the AeroCount® dose indicator. Reprinted by permission from AstraZeneca Pharmaceuticals LP.(4)
Subjects were reassessed for continued eligibility after 1–2 weeks. Subjects whose reported actuation count differed by >6 versus the weight-based actuation count during screening were issued a new placebo MDI and required to return to the clinic in 1 week for reassessment. Subjects who failed to complete >80% of eDiary assessments, or whose subject-reported actuation count differed by >6 versus the weight-based actuation count after the second attempt, were considered screening failures.
Treatment period
Subjects compliant in the screening period continued into the treatment period and received GFF MDI, two inhalations twice daily, over a 4-week period, and recorded use in the eDiary as during the screening period.
Statistical analyses
The primary endpoint was analyzed in the intent-to-treat (ITT) population (defined as all subjects enrolled into the treatment period and who used ≥10 actuations of GFF MDI, as recorded in the eDiary) and in the per-protocol (PP) population (subjects who completed all visits with ≥80% eDiary compliance and who had no major protocol deviations that could affect dose indicator assessment).
Data for the primary endpoint were summarized descriptively and displayed using a scatter plot. AE data were summarized descriptively in the safety population, defined as all subjects who were enrolled into the treatment period and who received any amount of GFF MDI.
Results
In total, 138 subjects were enrolled and treated with GFF MDI, and 10 (7.2%) withdrew before study completion due to: AEs (n = 3); administrative reasons (n = 2); withdrawal of consent (n = 1); loss to follow-up (n = 2); and discontinuation due to protocol-specific criteria (n = 2; included lack of compliance with study visits [n = 1] and COPD exacerbation [n = 1]). The ITT population was predominantly White (94.9%) and around half (50.7%) were male, with a mean (standard deviation [SD]) age of 62.1 (8.3) years. The mean (SD) disease duration of COPD was 7.7 (5.9) years. COPD severity in subjects was mostly moderate (65.2%) or severe (33.3%), and a small proportion of subjects had very severe COPD (1.4%).
At the last available visit, dose indicator-based actuation count and subject-reported actuation count in the ITT population were consistent for 96.4% of devices (132 of 137 devices) (Fig. 3A). The mean (SD) dose indicator-based actuation count (n = 137) was 113.4 (18.9), and mean (SD) subject-reported actuation count (n = 137) was 117.0 (19.0) (mean [SD] difference = 3.6 [7.9]); note that due to the design of the dose indicator, each individual dose indicator-based actuation count was a multiple of 10, but the average of these individual counts did not have to be a multiple of 10. The median value for both counts was 120.0.

Subject-recorded actuation count versus dose indicator-based actuation count at the last available visit for
In the PP population (n = 112), dose indicator-based actuation count and subject-reported actuation count at the last available visit were consistent for 100% of devices (Fig. 3B). The mean (SD) dose indicator-based actuation count was 116.8 (8.7), and the mean (SD) subject-reported actuation count was 119.7 (8.1). The median value for both counts was 120.0.
Thirty-eight subjects (27.5%) reported a total of 70 TEAEs during the study, including six subjects (4.3%) who experienced TEAEs that the investigator considered to be treatment related. Three subjects (2.2%) experienced serious TEAEs (COPD exacerbation [n = 2]; metastatic neoplasm and metastatic pain [both in the same subject; n = 1]). Four subjects withdrew due to TEAEs (COPD exacerbation [n = 3]; atrial fibrillation [n = 1]) (Table 1). No important trends were observed in changes from baseline in clinical laboratory results, vital signs, and electrocardiograms.
Data shown are n (%).
COPD, chronic obstructive pulmonary disease; TEAE, treatment-emergent adverse event.
Discussion
Subject-reported actuation count and dose indicator-based actuation counts were highly consistent in both the ITT population (96.4%) and PP population (100%), demonstrating accuracy, functionality, and reliability of the dose indicator over the lifetime of the inhaler. There was very little undercounting in the ITT population, and no undercounting was observed in the PP population. Avoiding undercounting is important(5) as it prevents patients from mistakenly assuming that they have medication left in their MDI when they do not.
Our study shows that the high reliability of this series of AeroCount dose indicators that was previously demonstrated in a simulated-use study(9) is also achieved with GFF MDI in clinical trials in the intended target population of patients with COPD. The study duration was consistent with the anticipated lifetime of GFF MDI (i.e., 130 actuations to last 4 weeks of treatment, including priming actuations).
The safety and tolerability profile of GFF MDI in patients with COPD has previously been investigated in large, long-term,(10,11) and short-term(12) Phase III studies with a duration of up to 52 weeks. Consistent with these studies, no unexpected safety findings were identified in the present study.
In conclusion, this study supports the accuracy, reliability, and functionality of the dose indicator integrated into the GFF MDI device when used by patients with COPD.
Footnotes
Acknowledgments
This study was supported by Pearl—a member of the AstraZeneca Group. The authors thank all the subjects and their families, and the team of investigators, research nurses, and operations staff, involved in these studies. The authors thank Amber Doty for her support with the site training and coordination of the laboratory-based testing and Mervin Taylor for his technical insight into the dose indicator function. Medical writing support, under the direction of the authors, was provided by Joanna Wilson, PhD, of CMC CONNECT, a division of Complete Medical Communications Ltd., Glasgow, United Kingdom, which was funded by AstraZeneca, Cambridge, UK in accordance with Good Publication Practice (GPP3) guidelines.(13)
Authors' Contributions
A.M., C.R., E.S.R., P.M., and S.S. contributed to the conception or design of the study. K.P., F.F., and G.F. participated in the acquisition of the data. A.M., C.R., E.S.R., P.M., and S.S. participated in the analysis of the data. All authors contributed to the interpretation of the data, critically reviewed the article, and approved the final version for submission.
Authors' Disclosure Statement
K.P. and G.F. have no potential conflicts of interest to disclose. F.F. is an employee of Florida Premier Research Institute, LLC, and has performed clinical trials for AstraZeneca and Pearl—a member of the AstraZeneca Group. E.S.R., P.M., and A.M. are employees of Pearl—a member of the AstraZeneca Group. C.R. is an employee of Pearl—a member of the AstraZeneca Group and an employee of AstraZeneca. S.S. is an employee of AstraZeneca and former employee of Pearl—a member of the AstraZeneca Group.
Reviewed by:
Gur Jai Pal Singh
Kurt Nikander