
Abstract
Abstract
Background:
People with cystic fibrosis (CF) suffer from chronic lung disease that is often treated with a bronchodilator. This trial evaluated the pharmacokinetics, safety, and tolerability of single and multiple doses of tiotropium inhaled via the Respimat® Soft Mist™ Inhaler in patients with CF.
Methods:
Patients received a single dose (placebo, 2.5 μg, 5 μg, or 10 μg) and/or multiple doses (placebo, 2.5 μg, or 5 μg) of tiotropium daily for 28 days.
Results:
Ninety-two patients, aged 5–57 years, were treated. All doses showed a satisfactory safety profile for adverse events, vital signs, laboratory evaluations, and physical examination. At steady-state, peak exposure to tiotropium was comparable between adult patients with CF and patients with chronic obstructive pulmonary disease.
Conclusions:
Tiotropium 2.5 μg or 5 μg inhaled via the Respimat® Soft Mist™ Inhaler once daily was well tolerated in patients with CF.
Introduction
T
Ipratropium bromide, a short-acting anticholinergic compound, has been shown to improve lung function in single-dose studies in patients with CF;(6–8) however, its long-term effects in CF have not been studied. Tiotropium is a long-acting, anticholinergic bronchodilator for the maintenance treatment of patients with chronic obstructive pulmonary disease (COPD). Because tiotropium has been shown to be more effective than ipratropium as a bronchodilator in COPD(9) and has a 24-h duration of action, it is expected to provide more effective and sustained bronchodilation than ipratropium in patients with CF. Boehringer Ingelheim initially developed tiotropium bromide in COPD as a powder for inhalation with the HandiHaler® and, more recently, as a solution for inhalation with the Respimat® Soft Mist™ Inhaler (SMI). The Respimat® SMI uses mechanical energy from a spring to generate a fine, low-velocity mist that is more easily inhaled than that from powder-based inhalers and metered-dose inhalers with propellants. The Respimat® SMI is currently under evaluation for asthma in adult and pediatric populations.(10) We present here the results of a phase 1 trial evaluating the safety, tolerability, and pharmacokinetics (PK) of single and multiple doses of tiotropium bromide administered via the Respimat® SMI in pediatric (age >5–≤11 years, “younger”) and adolescent/adult (age ≥12 years, “older”) patients with CF.
Materials and Methods
Trial design
This was a multicenter, randomized, double-blind, placebo-controlled, stepwise, dose-escalation trial in patients with CF, blinded to treatment allocation within each dose level. The trial was conducted at 12 sites in the United States between September 2006 and September 2008. At each study site, an institutional review board approved the protocol, and written informed consent was obtained from each patient or their legal representative prior to randomization. The trial was conducted in accordance with the International Conference on Harmonisation Good Clinical Practice guidelines and local regulations.
The study comprised a single-dose portion followed by a multiple-dose portion. In the single-dose arm, patients received one dose of placebo or tiotropium bromide 2.5 μg, 5 μg, or 10 μg from the Respimat® SMI as an add-on to standard therapy. In the multiple-dose arm, patients received a dose of placebo or tiotropium bromide 2.5 μg or 5 μg once daily for 28 days also as an add-on to standard therapy.
Dosing began with the lowest dose in the older age group (Fig. 1). The dose level was increased after the safety of the previous dose level had been evaluated and found acceptable by an external Data Monitoring Committee (DMC), convened by the Data Safety and Monitoring Board of the Cystic Fibrosis Foundation. The younger patients did not enter a dose arm until after the older patients completed the dose arm, and the safety and tolerability were reviewed and accepted by the DMC. The multiple-dose portion was initiated only after the single-dose portion was complete within any dose level. Patients could choose to participate in either the single-dose or multiple-dose arm, or both. For those patients who participated in both arms, a 30-day washout period was required between the single- and multiple-dose arms, and the same dose was administered in each arm. Open-label albuterol (salbutamol) was provided as rescue medication and could be administered, when needed, at any point during the study.
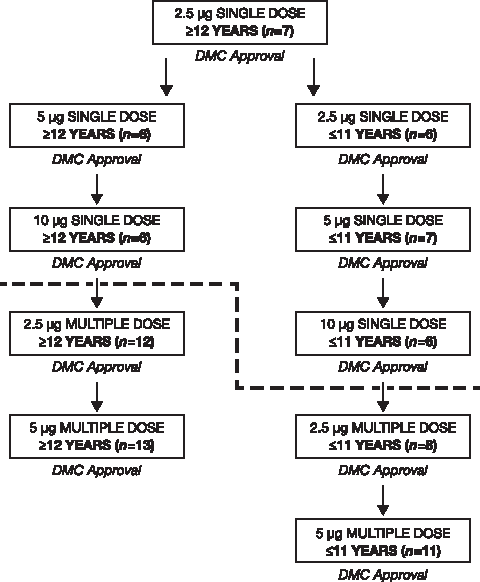
Dose escalation sequence and disposition of study patients by age group.
Patients
Male or female patients aged 5–≤11 years or ≥12 years, with clinically stable and verified CF (sweat chloride ≥60 mEq/L and/or a genotype with two identifiable mutations and symptomatology consistent with CF), with a forced expiratory volume in 1 second (FEV1)>25% of predicted value for healthy individuals of comparable age, sex, and height were eligible to participate.(11) Patients were also required to be able to inhale medication in a reproducible manner from the Respimat® SMI. Exclusion criteria included substance abuse, pregnancy or lactation, persistent colonization with Burkholderia cepacia, a new long-term medication for CF started within 4 weeks of screening, and clinically significant disease or medical conditions such as significant hematologic, hepatic, renal, cardiovascular, and neurologic disease, which, in the opinion of the study investigator, would compromise patient safety or data quality. Patients with diabetes could participate if their disease was under control prior to screening. Concomitant use of pulmonary medications, with the exception of other anticholinergics, long-acting β-adrenergics, or inhaled antibiotics, was allowed. When a patient qualified for entry into the randomized treatment period, treatment assignment was made by means of a third-party telephone interactive voice randomization system. For each dose group, two patients were to be randomized to placebo and four to active drug in the single-dose phase and four to placebo and eight to active drug in the multiple-dose phase.
Assessments
Safety and tolerability were reviewed on the basis of the following parameters: physical examination, vital signs, spirometry, laboratory evaluation, and adverse events (AEs). Because of the small number of subjects, no efficacy analyses were planned. Adverse events were reported using the Medical Dictionary for Regulatory Activities, version 11.0.(12)
For the purpose of providing additional support for dose optimization for subsequent phase 2 and 3 safety and efficacy studies, tiotropium PK was analyzed from blood samples taken after the first dose in all patients, and after 28 days of once-daily dosing (steady state) in those receiving multiple doses. Drug exposure was also compared with historical data from other non-CF patient populations. This was especially important for the pediatric patients, given that this was the first time that tiotropium had been administered to a pediatric patient population. The PK parameters reported in this study are the maximum plasma concentration of drug after a single dose (Cmax) and the time to reach Cmax. Blood was obtained predose, 5, 15, 30 min, and 1, 2, 4, and 6 h post drug administration. In the multiple-dose arm of the study in the patient group aged ≤11 years, blood sampling was only performed at the 2.5 μg dose.
Statistical analyses
A noncompartmental PK analysis of tiotropium plasma data was carried out using WinNonlin® Version 5.2 (Pharsight Corporation, Cary, NC). Descriptive statistics were presented. No formal testing of hypotheses was performed.
Results
Ninety-two patients were randomized to receive study drug, 30 participated in only the single-dose phase, 41 participated in only the multiple-dose phase, and 17 participated in both phases. The numbers of patients treated in each dose group are shown in Figure 1. Only one patient in the younger age group treated with tiotropium bromide 5 μg discontinued due to a nonserious AE of increased cough. Two patients in the older age group discontinued: a placebo-treated patient discontinued due to a serious AE of a pulmonary exacerbation, and a patient treated with tiotropium bromide 2.5 μg discontinued due to a nonserious AE of increased mucus thickness. Patient baseline characteristics are summarized in Table 1. Mean age and baseline pulmonary function were similar in all dose groups in both the younger and older patients.
BMI, body mass index; SD, standard deviation.
The frequencies of AEs by treatment (single- and multiple-dose phases) are summarized in Tables 2 and 3. Table 4 shows frequencies of moderate and severe AEs by treatment and preferred term in the multiple-dose phase. The frequencies of all AEs by treatment and preferred term in the multiple-dose phase are shown in Supplementary Table S1. In both the single-dose and multiple-dose phases, the frequency of AEs after inhalation of tiotropium was similar to that of placebo and was balanced among dose groups in both age groups, with the exception of cough in the multiple-dose phase. In the younger group in the multiple-dose phase, frequency of productive cough was slightly higher with tiotropium bromide 5 μg than with 2.5 μg, but in the older group the opposite was observed (Supplementary Table S1).
The 10 μg dose was only included in the single-dose phase of the study; Data presented are n (%) unless otherwise specified.
Data presented are n (%) unless otherwise specified.
See Supplementary Table S1 for a full summary of AEs occurring by treatment and preferred term (multiple-dose phase).
CF exacerbation is coded to a preferred term of cystic fibrosis.
RTI, respiratory tract infection.
Two patients in the multiple-dose phase experienced a serious AE. A 23-year-old patient treated with placebo discontinued due to a CF exacerbation, and a 23-year-old patient treated with tiotropium bromide 2.5 μg discontinued due to an exacerbation; both reported an increase in sputum thickness, which is characteristic of an exacerbation. Neither event was considered by the investigator to be related to the study drug. There were no reports of symptoms associated with anticholinergics (e.g., dry mouth).
Laboratory results
Among the younger patients, one who received placebo, one who received tiotropium bromide 2.5 μg, and two who received tiotropium bromide 5 μg, had an increased plasma glucose concentration. This did not occur in the older patients. There were no significant changes in vital signs or findings on physical examination in these patients.
Vital signs and spirometry
There was a general decrease from baseline in systolic and diastolic blood pressure and pulse rate across all age and treatment groups. However, the extent of the change in each case was small, with a large amount of variability across the groups. In all patients, spirometry was performed 30 min before dose, as well as 30 min and 1 and 2 h post dose to evaluate for bronchoconstriction. No subjects met the criteria for bronchoconstriction, defined as a greater than 15% drop in FEV1 30 min post dosing (Table 5). The absolute change from baseline in FEV1 (L) is shown in Table 5. Meaningful improvements in spirometry were not observed at any time point post dosing.
The 10 μg dose was only included in the single-dose phase of the study.
SD, standard deviation.
Pharmacokinetics
Following inhalation of a single dose and following multiple dosing, tiotropium was rapidly absorbed, with Cmax achieved within 5 min post inhalation, corresponding to the first blood sample for all dose groups and treatment groups. Average and individual tiotropium plasma levels 5 min post dosing with a single dose, at steady state and Cmax were compared between the older and younger patients in this trial and with COPD patients and healthy volunteers from historical data (Fig. 2). The mean plasma concentrations were slightly higher in CF patients aged ≥12 years compared with those CF patients aged 5–≤11 years for all dose groups in the single-dose portion of the study. However, there was a substantial overlap of the individual values in the two age groups.The average plasma concentrations for healthy volunteers 5 min after a single dose (historical data—administered 5 μg) were comparable to those observed for all patients with CF (Fig. 2).
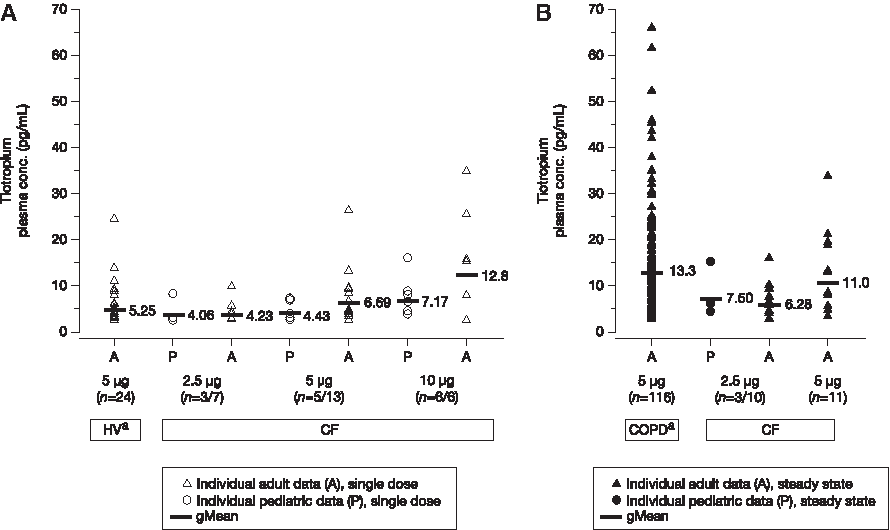
Comparison of average and individual tiotropium levels 5 min post dosing following the administration of a single dose
In the multiple-dosing arm, the Cmax at steady state values among CF patients aged ≥12 years compared with those aged 5–≤11 years administered the 2.5 μg dose were comparable (Fig 2). In the CF patient group aged ≥12 years, multiple dosing with 5 μg tiotropium bromide resulted in maximum plasma exposures that were similar to those observed in patients with COPD (Fig. 2). Finally, accumulation of tiotropium with multiple doses resulted in an approximately 1.5–2-fold increase in Cmax (in the older patients) compared with single doses.
Discussion
This is the first study to describe the safety and tolerability of inhaling single and multiple doses of the once-daily, long-acting anticholinergic agent tiotropium bromide in children and adults with CF. Tiotropium was administered via the Respimat® SMI instead of the HandiHaler dry powder inhaler because of its ease of use, and also because it can be used with a spacer, particularly among children, as demonstrated in handling studies conducted in children aged <5 years and those aged 4–12 years.(13,14) Overall, relatively few AEs were reported and most were mild to moderate in severity. Only two patients experienced a serious AE, neither of which was considered related to study drug. In the multiple-dose phase, both age groups had a slight increase in cough and productive cough compared with placebo (Table 4 and Supplementary Table S1). There was no evidence of bronchoconstriction when spirometry was assessed during and shortly after inhalation of tiotropium.
Ipratropium bromide, a short-acting anticholinergic compound, has previously been shown to improve lung function in patients with CF.(6–8) In patients with COPD, tiotropium delivered using the Respimat® SMI at doses of 5 and 10 μg has consistently shown superior efficacy to ipratropium bromide.(15) Given the differences between the diseases, it is unknown whether tiotropium will have a similar effect in patients with CF. This warrants further investigation based on the safety and tolerability results of this phase 1 trial.
In younger and older patients with CF, multiple dosing resulted in a 1.5–2-fold increase in Cmax compared with single dosing. This accumulation in tiotropium peak plasma concentration is similar to that seen in healthy volunteers and in patients with COPD.(16) In both children and adults with CF, tiotropium was rapidly absorbed following inhalation. Cmax, values for tiotropium were similar among children aged ≥12 years and those between the ages of 5 and 11 years, as well as to the historical data from healthy volunteers. Likewise, Cmax steady-state values were comparable between the older age group (≥12 years) administered the 5 μg dose and historical data from patients with COPD. All doses showed a satisfactory safety and tolerability profile with respect to AEs, vital signs, laboratory evaluations, and physical examination. The results of this study support the safety and tolerability of 4 weeks of tiotropium bromide in patients with CF. There were no safety or tolerability concerns that would preclude further clinical evaluation of tiotropium in patients with CF using doses up to 5 μg once daily. Therefore, the 2.5 and 5 μg doses were chosen to move forward into a phase 2 study in patients with CF.
Footnotes
Acknowledgments
The study site principal investigators were: R. Ahrens (Iowa City, IA), H. Carveth (Salt Lake City, UT), T. Ferkol (St. Louis, MO), J. Finder (Pittsburgh, PA), D. Geller (Orlando, FL), M. Konstan (Cleveland, OH), C. Merlo (Baltimore, MD), S. Nasr (Ann Arbor, MI), M. Pian (San Diego, CA), C. Ren (Rochester, NY), A. Uluer (Boston, MA), J. Wooldridge (Cincinnati, OH), and J. Nick (Denver, CO). The authors acknowledge the Cystic Fibrosis Foundation Data Safety Monitoring Board and the Cystic Fibrosis Therapeutics Development Network for their assistance with this study. The study was sponsored and supported by Boehringer Ingelheim Pharmaceuticals, Inc.
The authors were fully responsible for all content and editorial decisions, were involved at all stages of manuscript development, and have approved the final version. Medical writing assistance, supported financially by Boehringer Ingelheim, was provided by Godfrey Lisk, of PAREXEL International, during the preparation of this manuscript.
Clinical trial number: 205.338 / U09-3457-02.
Author Disclosure Statement
Ashish Sharma, Petra Moroni-Zentgraf, Fei Wang, and Paul Koker are employees of Boehringer Ingelheim. Michael Konstan has served as a paid consultant to Boehringer Ingelheim Pharmaceuticals, Inc.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.