
Abstract
Abstract
Background:
Loxapine, a first-generation antipsychotic, delivered with a novel inhalation delivery device developed for the acute treatment of agitation in patients with schizophrenia or bipolar disorder was evaluated in subjects with asthma or chronic obstructive pulmonary disease (COPD).
Methods:
Separate randomized, double-blind, parallel-arm, placebo-controlled trials compared two administrations of inhaled loxapine (10 mg) 10 hr apart with placebo in 52 subjects with asthma and in 53 subjects with COPD. A thermally-generated drug aerosol of loxapine was delivered to the deep lung for rapid systemic absorption. Controller medications were continued throughout the study, but quick-relief bronchodilators were withheld from 6–8 hr before through 34 hr after dose 1, unless indicated as rescue.
Results:
All airway adverse events (AEs) were of mild or moderate severity. Symptomatic bronchospasm occurred in 53.8% of subjects with asthma after inhaled loxapine and 11.5% after placebo; and in 19.2% of COPD subjects after inhaled loxapine and 11.1% after placebo. Subjects required inhaled albuterol as follows: asthma: 53.8% after inhaled loxapine and 11.5% after placebo; and COPD: 23.1% after inhaled loxapine and 14.8% after placebo. Respiratory signs/symptoms requiring treatment responded to rescue bronchodilator [forced expiratory volume in 1 sec (FEV1) return to within 10% of baseline] within 1 hr in 11 of 15 events in asthma subjects and four of seven events in COPD subjects, the remainder by the last spirometry.
Conclusions:
In subjects with either asthma or COPD, FEV1 decline and bronchospasm can occur following inhaled loxapine, but more frequently in asthmatic subjects. Most subjects with bronchospasm responded to rescue bronchodilator within 1 hr. No treatment-related serious AE occurred. Although inhaled loxapine is contraindicated in patients with active airways disease per the current approved US labeling, these studies demonstrated that rescue bronchodilator mitigated the symptomatic bronchospasms that may occur in case of inadvertent use.
Introduction
O
Oral loxapine is used for the treatment of schizophrenia in the United States. The intramuscular form of loxapine is no longer marketed in the United States, but is frequently used in France for the acute treatment of agitation.(8) Intramuscular antipsychotics have a Tmax of 90 min and can take up to 60 min to reduce agitation,(9–12) and symptoms can escalate during this period. Moreover, intramuscular administration is often resisted by patients. To address these concerns, an inhaled formulation of loxapine has been developed that has a Tmax of 2 min and has been shown in clinical trials to begin controlling agitation of patients with schizophrenia or bipolar I disorder within 10 min.(13,14) This formulation (ADASUVE®) uses a breath-actuated delivery system, the Staccato® system, which delivers loxapine with a pharmacokinetic profile comparable to that of intravenous administration, providing peak plasma levels in the systemic circulation within minutes after administration.(15) ADASUVE was approved by the US Food and Drug Administration in 2012 for a single dose and by the European Medicines Association in 2013 for two doses. In the United States, it is “indicated for the acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults.” It was commercially available in the European Union in 2013, and will be in the United States in 2014.
To address the possibility that inhalation of loxapine may have adverse pulmonary effects, we conducted two safety studies. Two 10-mg inhaled doses of loxapine or placebo were given 10 hr apart to subjects with asthma or chronic obstructive pulmonary disease (COPD) in separate studies. The objective of each study was to determine the time course and reversibility of potential pulmonary effects for inhaled loxapine compared with placebo.
Materials and Methods
Subjects and study design
Both studies were multicenter, randomized, double-blind, placebo-controlled, parallel-group studies conducted May 2009 to August 2009. There were four outpatient clinical research units for the asthma study and five for the COPD study. Before any protocol-related assessments or procedures, subjects signed an informed consent form. At Visit 1, subjects were screened for eligibility. At Visit 2 (≤28 days after Visit 1), randomization occurred, baseline measurements were obtained, treatment was administered (inhaled loxapine 10 mg or placebo; 2 doses, 10 hr apart), and post-treatment assessments, including serial spirometries, were performed at 0.25, 0.5, 1, 2, 4, 6, 10, 10.25, 10.5, 11, 12, 14, 16, 24, and 34 hr after dose 1. At Visit 3 (7±3 working days after Visit 2), end-of-study assessments were performed. The studies were approved by Western Institutional Review Board (3535 Seventh Avenue SW, Olympia, WA 98502) and conducted in accordance with the amended Declaration of Helsinki. The studies' design is presented schematically in Figure 1.

Study design schematic.
Principal eligibility criteria are shown in Table 1. In the asthma study, randomization was stratified based on screening pre-bronchodilator forced expiratory volume in 1 sec (FEV1: <80% or ≥80% of predicted) according to National Heart, Lung and Blood Institute criteria and was 1:1 within each stratum. In the COPD study, randomization was stratified based on screening post-bronchodilator FEV1 (<50% or ≥50% of predicted) and was 1:1 within each stratum.
Although subjects were allowed to continue asthma controller medications, quick-relief agents (i.e., albuterol) were withheld from 6 hr (asthma) or 8 hr (COPD) before study drug administration through 24 hr after the second administration, unless required as rescue medication. The choice between metered-dose inhaler (MDI) and nebulizer rescue was determined by the responsible investigator's clinical judgment. There were no important changes to eligibility criteria, outcome measures, or other methods after trial commencement.
Study drug
The Staccato system is a single-use, hand-held, drug-device combination product (Fig. 2) that provides rapid systemic delivery by inhalation of a thermally generated aerosol of loxapine.(15–17) Oral inhalation through the product initiates the controlled rapid heating of a thin film of excipient-free loxapine to form a thermally generated, highly pure drug vapor. The study drug is fully vaporized within 0.5 sec and delivered in a single breath. The vapor condenses into aerosol particles with a particle size distribution appropriate for efficient delivery to the deep lung (mass median aerodynamic diameter of 2–3 μm). Placebo was a working device with no drug or excipients.
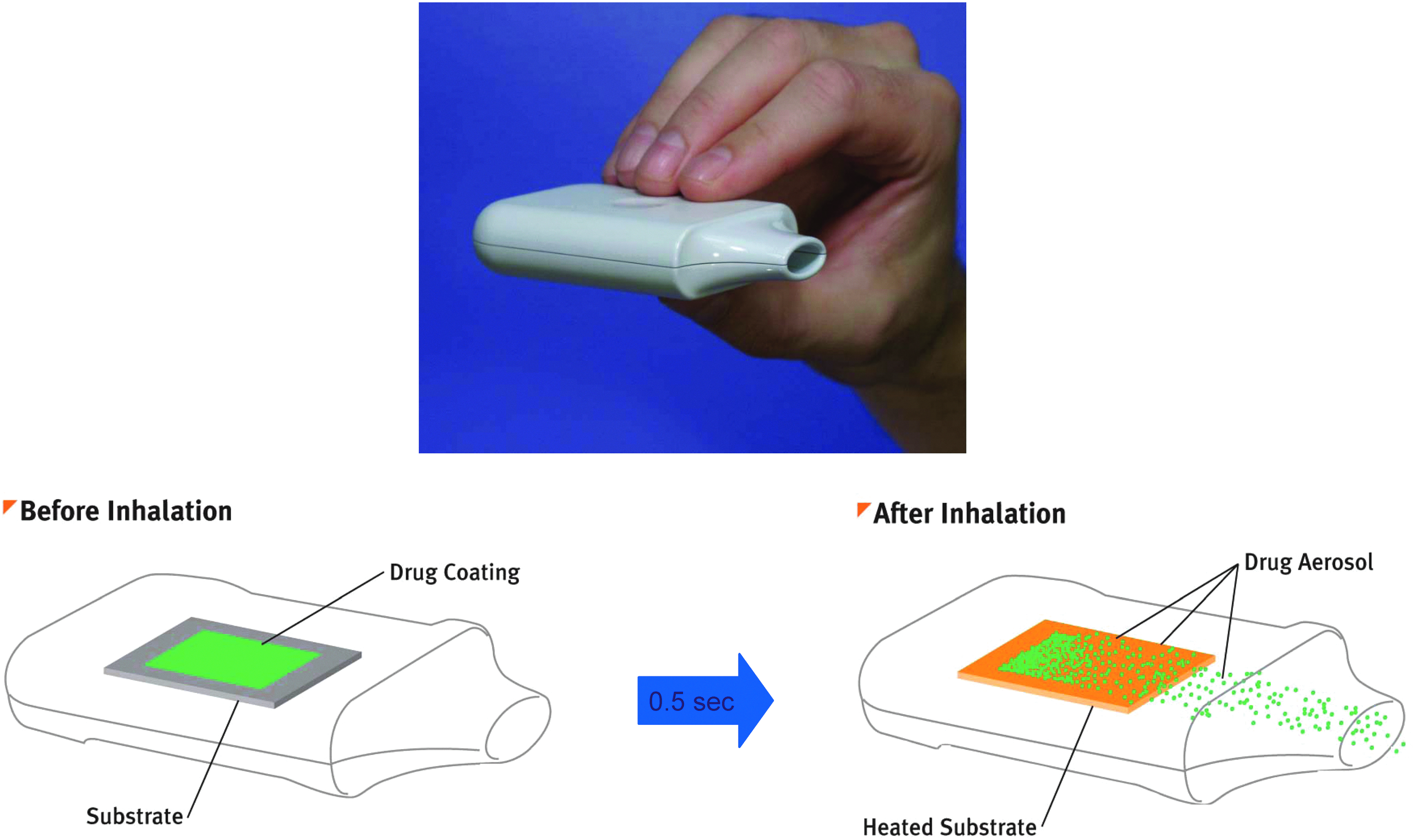
Staccato drug-device combination product.
The second dose of study drug was not given if a subject experienced an FEV1 decrease from baseline of ≥20% or an airway adverse event (AE), or required rescue medication.
Outcome measures
Spirometry results included FEV1, forced vital capacity (FVC), and FEV1/FVC, with FEV1 serving as the primary outcome measure. Spirometry tests were performed in the hour before the first dose of study treatment and at 0.25, 0.5, 1, 2, 4, 6, 10, 10.25, 10.5, 11, 12, 14, 16, 24, and 34 hr after that dose. The 10-hr assessments were performed just before the second dose was administered. Other quantitative (secondary) outcome measures, i.e., respiratory rate, heart rate, and oxygen saturation by pulse oximetry (SpO2), were measured, and sedation was assessed using a visual analogue scale (VAS). Blood pressure was measured at all spirometry assessment times in the first 4 hr after each dose and at 10 and 34 hr. AEs that occurred
All spirometry tests used the same controlled clinical standards and were conducted according to American Thoracic Society/European Respiratory Society standards(18) using NHANES III predicted values.(19) Tests were assessed for adequacy by an external, blinded, pulmonologist rater and were judged adequate if there were a minimum of three acceptable FVC maneuvers (i.e., flow-volume loops). Baseline spirometry measurements were documented in the hour before the first dose of each treatment period (i.e., same-period baseline) and at 15 post-treatment time points after dose 1.
Other safety assessments
Symptoms of bronchospasm such as wheezing, dyspnea, and cough were scored as AEs. Sedation, measured by VAS, vital signs, and pulse oximetry were also assessed.
Statistics
Approximately 50 subjects were expected to complete each study (approximately 25 randomized to inhaled loxapine and 25 to inhaled placebo). This sample size was intended to gauge the typical effects, if any, of inhaled loxapine on expiratory flow rates.
Masked treatment was assigned via sequentially numbered packages provided to each center from a random number list for each stratum with a block size of 2 for each study prepared by the contract research organization. All participants, care providers, and those assessing outcomes were masked as to treatment.
FEV1 data were examined using mean change statistics and change categories (≥10%, ≥15%, ≥20%) for changes from same-day baseline. Descriptive statistics were calculated by treatment group at each time point. For percent change from baseline in FEV1 at each post-baseline time point, means and standard errors of the mean were calculated. Least square means (LSmeans) and 90% confidence intervals (CIs) for the differences between treatments were calculated for the change from baseline for each secondary outcome measure for each post-baseline time point. All CIs were based on two-factor ANOVA models including terms for stratum and treatment.
Results
Subjects
Fifty-two subjects with asthma and 53 subjects with COPD were randomized in the studies between May through August 2009 (Fig. 3). In general, subjects assigned to the two treatment groups were well matched for demographic and baseline characteristics in both studies (Table 2).
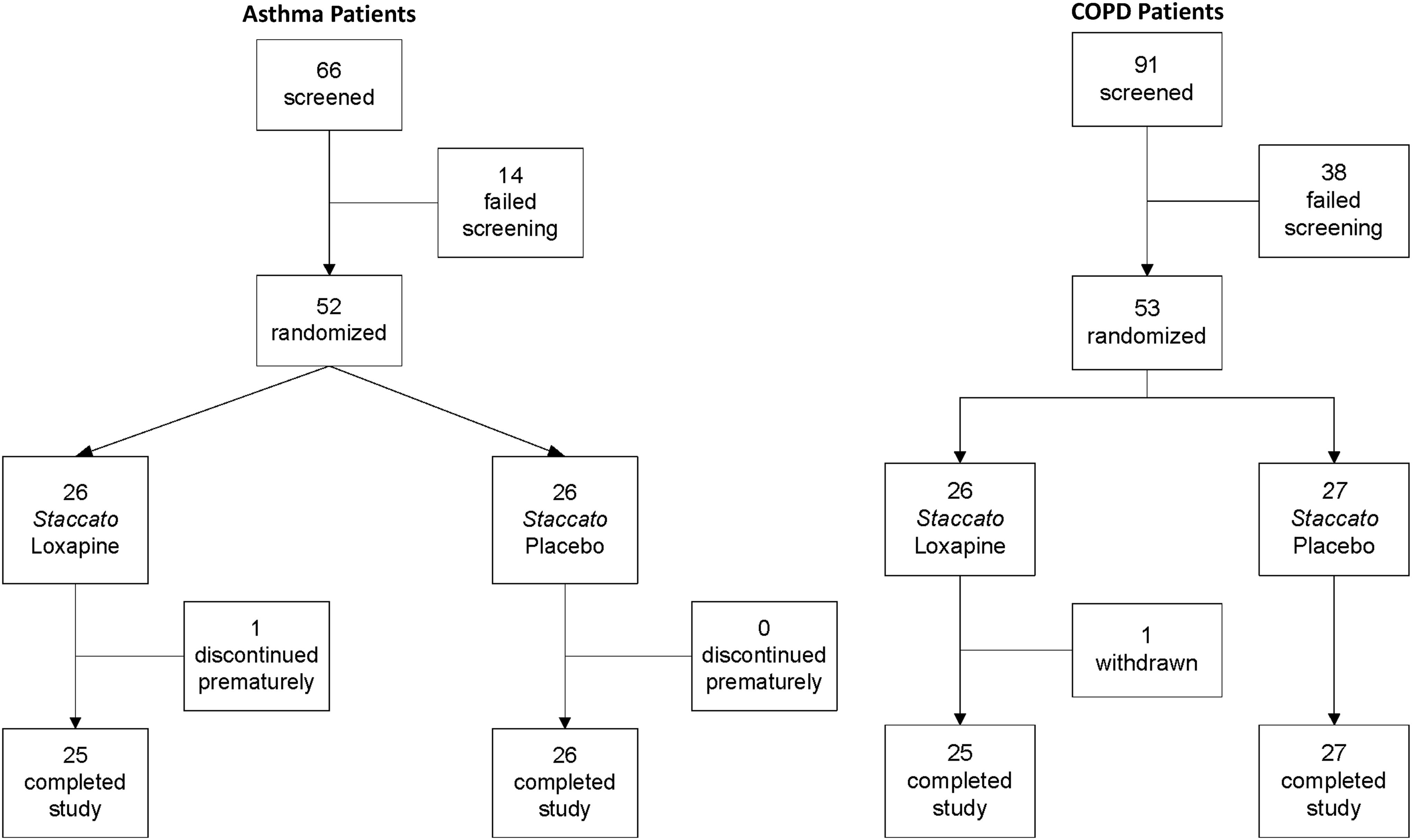
Disposition of study participants (asthma and COPD studies).
GOLD, Global Initiative for Chronic Obstructive Lung Disease; na, not applicable; NHLBI, National Heart, Lung, and Blood Institute.
Adverse events
In the asthma study, a greater percentage of loxapine-treated subjects had at least one AE compared with placebo-treated subjects; the converse was the case in the COPD study (Table 3). Airway AEs (e.g., wheezing, dyspnea, and cough) occurred in a greater percentage of asthma and COPD subjects after receipt of inhaled loxapine than after placebo, with the treatment difference greater in asthma subjects (Table 3). Among inhaled loxapine subjects who experienced an airway AE, the event occurred within 25 min of dosing for 12 of 14 asthma subjects and four of five COPD subjects. Airway AEs were mild or moderate in severity and not associated with clinically significant changes in respiratory rate or oxygen saturation. All respiratory symptoms developing after treatment in inhaled loxapine subjects were either self-limiting (one asthma subject, three COPD subjects) or treated (13 asthma subjects, two COPD subjects) with an inhaled bronchodilator (albuterol). Three placebo asthma subjects and four placebo COPD subjects received albuterol for AEs.
All AEs presented were treatment emergent. Subjects with more than one occurrence of a specific AE are counted only once.
For one subject, the investigator reported a “greater than 20% drop in FEV1 from baseline” as an AE; there were no other airway AEs for this subject.
Sedation
In both studies, sedation was apparent after each dose of inhaled loxapine (Fig. 4), with the maximum change in VAS score occurring 30 min to 1 hr after each dose of loxapine (1-hr and 10.5-hr post-dose time points). Notable changes from baseline in the sedation VAS were evident in both treatment groups at the 14- and/or 16-hr assessments.
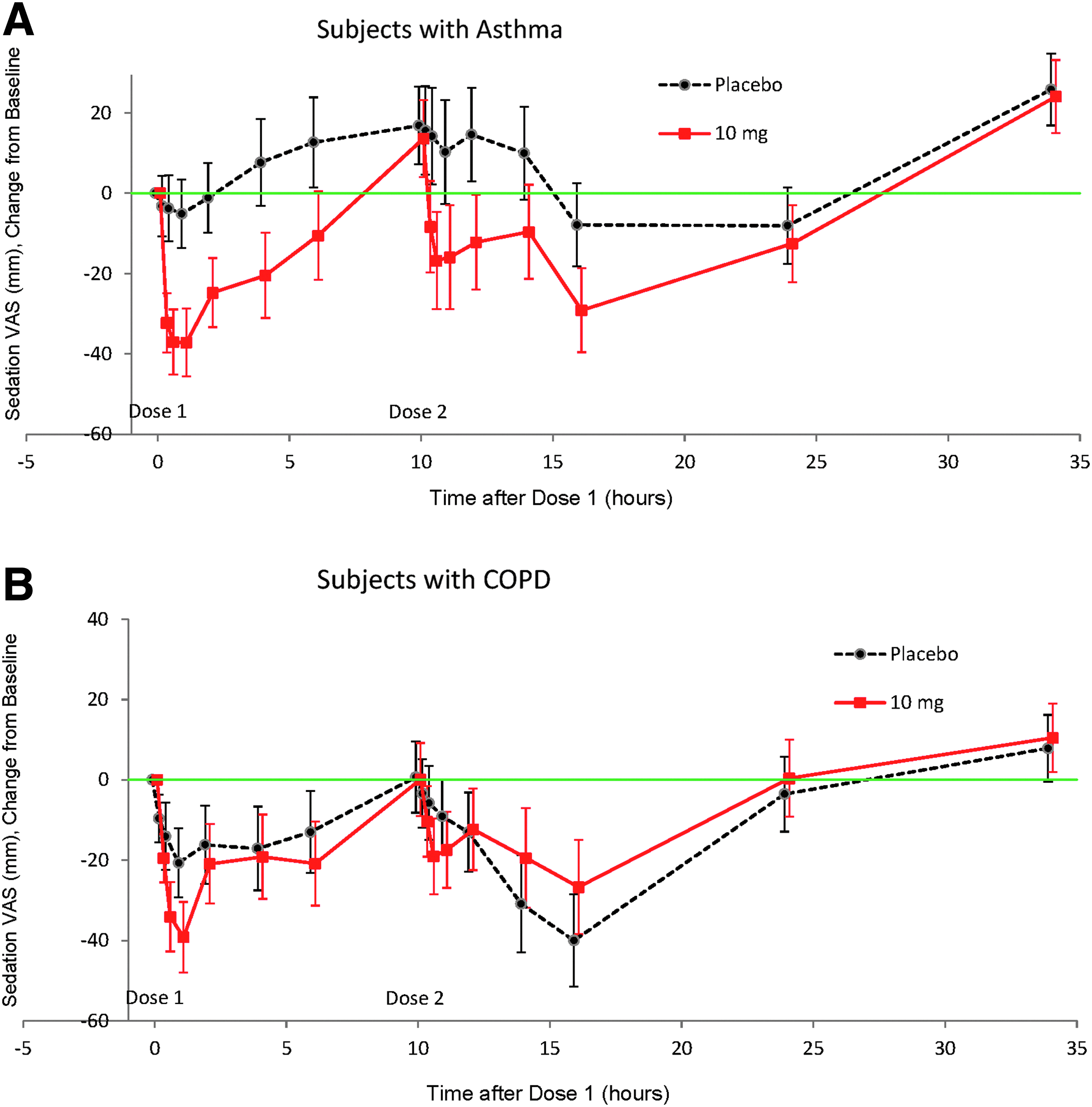
Sedation VAS change from baseline (LSMean, 90% CI):
Spirometry findings
In asthma subjects, baseline FEV1 was similar in the two treatment groups before dose 1 [3.33±0.74 L before placebo and 2.92±0.69 L before inhaled loxapine (means±SD)]. In order to avoid bias in the data display toward a return to baseline, subjects who used rescue medication and/or did not receive dose 2 at hour 10 were excluded from data analysis at all subsequent time points (i.e., after rescue for those who received rescue; after hour 10 for those who did not receive dose 2); consequently, the population size represented on the figures decreases over time. After inhaled loxapine treatment, there were notable decreases in mean FEV1, especially at the 0.25- and 10.25-hr time points (i.e., 15 min after the administration of dose 1 and dose 2, respectively) (Fig. 5A; Table 5). These decreases in FEV1 returned to within 10% of baseline with a median time of 29 min. Beyond the 10.25-hr time point, there was a small (4.8%) mean difference between the treatment groups for those subjects remaining in the analysis. However, in 24 of the 26 inhaled loxapine-treated subjects, FEV1 was within 10% of baseline at 24 hr, and in the remaining two subjects it was within 10% of baseline at 34 hr, the final spirometry.
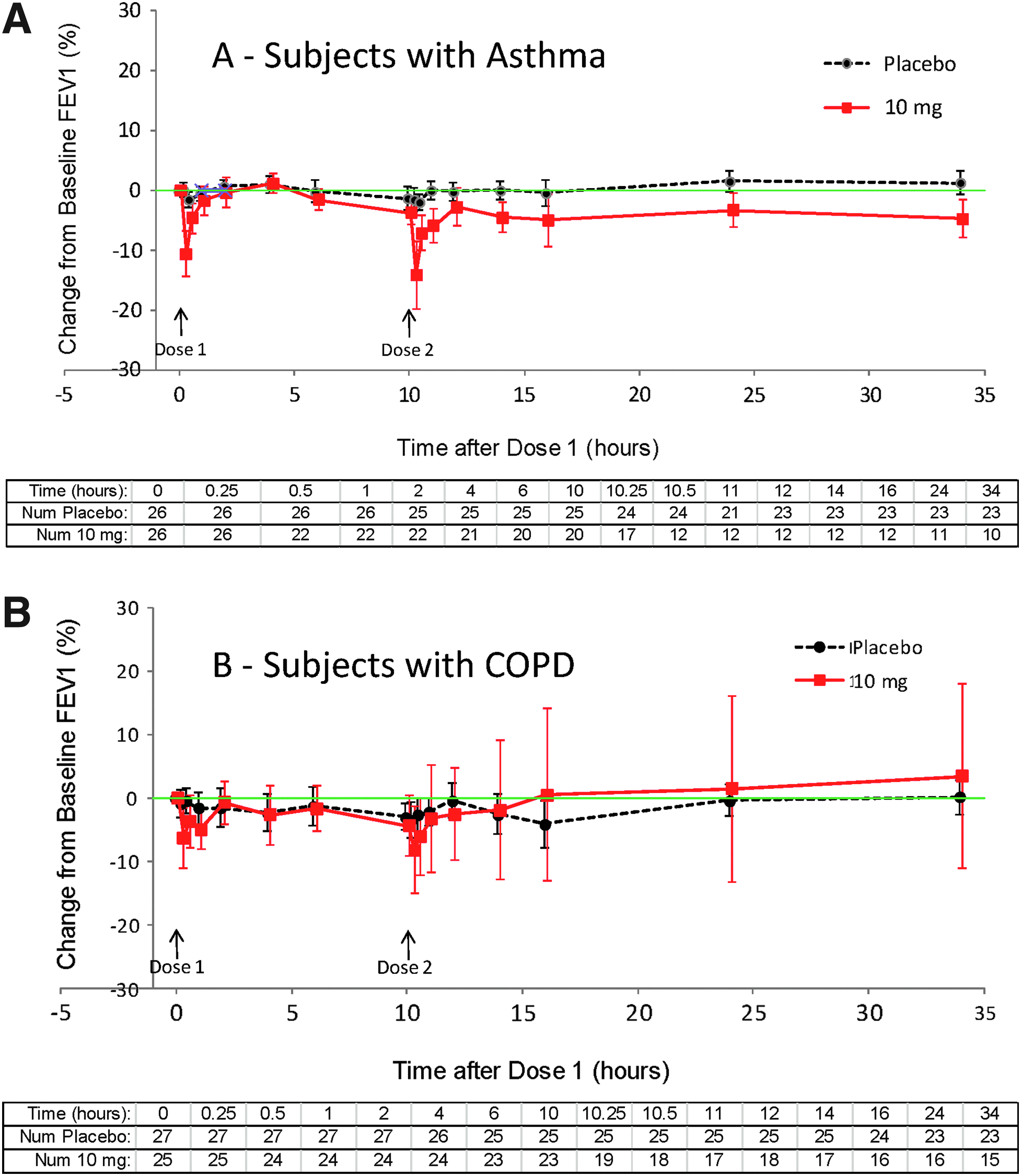
FEV1 % change from baseline, dropouts censored (means±1 SEM).
Data are the LSmean difference in change from baseline (loxapine – placebo) and 90% CIs (safety or spirometry population).
Spirometry population.
90% CI excludes 0 (difference in change statistically significant).
In COPD subjects, baseline FEV1 was similar in the two treatment groups before dose 1 [1.603±0.588 L before placebo and 1.551±0.411 L before inhaled loxapine (means±SD)]. There were very small decreases from baseline in mean FEV1 at most assessment times after placebo or inhaled loxapine treatment, with a slightly larger decrease after inhaled loxapine treatment (Fig. 5B). The difference, although small, was most noticeable in the hour after each dose.
In both asthma and COPD subjects, decreases in FEV1 occurred more frequently following loxapine than following placebo (Table 4). The maximum decrease in FEV1 nearly always occurred within the first hour after dosing (Fig. 5).
Table presents number (%) of subjects with indicated percentage decrease at 15 min through 24 hr after dose 1; data obtained 34 hr after dose 1 were not included.
FEV1 categories are cumulative (e.g., a subject with a maximum decrease of 21% would be included in the ≥10%, ≥15%, and ≥20% categories).
Relative risk 95% confidence interval excludes 1.
We examined the relationship of airway response (FEV1 % decrease from baseline and need for rescue) to disease severity (baseline FEV1 % of predicted). There was no statistically significant relationship between baseline FEV1 and the decrease in FEV1 following drug administration.
Response to albuterol treatment in subjects with notable respiratory signs and symptoms
Notable respiratory signs or symptoms were defined as any FEV1 decrease from baseline of ≥20%, any airway AE, or use of rescue medication. Of the inhaled loxapine subjects, 18 (69.2%) with asthma and 15 (57.7%) with COPD had notable respiratory signs and/or symptoms. Relief of post-treatment respiratory symptoms in both asthma and COPD subjects required treatment with albuterol via MDI or nebulizer.
In the asthma study, 14 of the inhaled loxapine subjects were treated with albuterol: 13 of them for airway-related AEs, and one subject used her own albuterol for a cough judged by the investigator not to be an AE because it was a symptom of the subject's asthma. Of the 14 inhaled loxapine asthma subjects who received rescue medication, 13 received only one treatment with albuterol via MDI or nebulizer (one of these subjects had two events), and one received two treatments for a single event. Recovery to an FEV1 within 10% of baseline occurred within 1 hr for the majority of subjects (Fig. 6A); 10 of the 14 subjects (11 of 15 events) who received albuterol had an FEV1 within 10% of baseline in the subsequent 1 hr. The other four had recovery to within 10% of baseline at later scheduled spirometry time points (range: 1 hr 20 min to 7 hr 57 min).
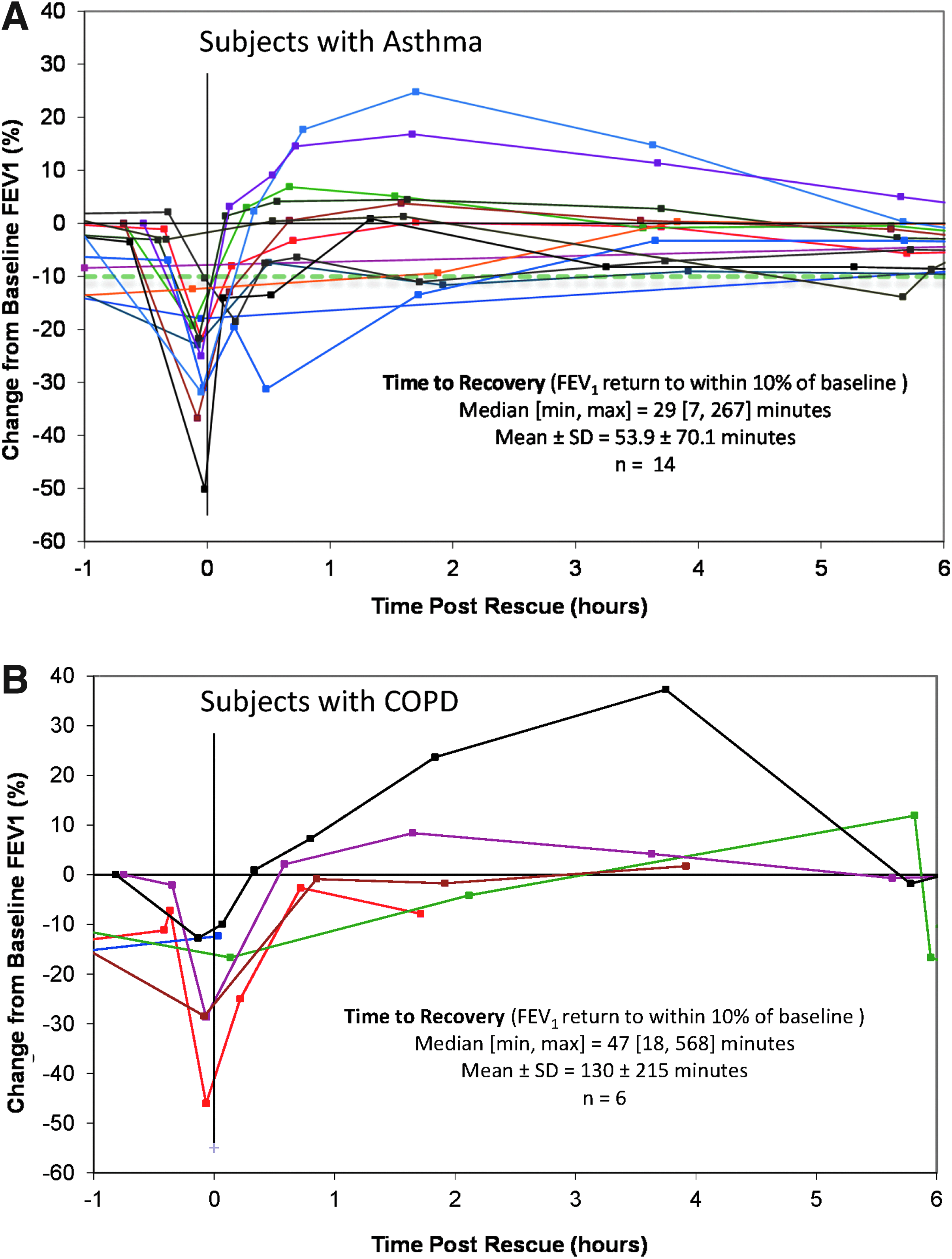
FEV1 response to albuterol in the individual subjects
In the COPD study, six inhaled loxapine subjects were treated with albuterol: three of them for airway-related AEs, two for a decrease in FEV1, and one for shortness of breath that was not recorded as an AE. Of the six subjects treated with albuterol, all received only one treatment with albuterol via MDI or nebulizer. Four of the six inhaled loxapine subjects in which albuterol was administered had an FEV1 within 10% of baseline documented in the subsequent 1 hr (Fig. 6B). For an additional event in one of these four subjects and for the remaining subjects, recovery of FEV1 to within 10% of baseline or higher was documented at later scheduled spirometry time points (range: 2 hr 7 min to 17 hr 58 min).
Discussion
Limitations of these studies reflect the methodology of carrying out parallel studies in healthy volunteers. Patients regularly taking antipsychotic medications tolerate loxapine in higher doses than healthy volunteers. In the clinical trials, agitated patients received the second dose at 2 hr, whereas in these studies in order to reduce the impact of sedation on pulmonary function testing, the second dose was given at 10 hr. Thus, although these effects were largely reversible in these studies, the degree to which these results are directly applicable to the patients for whom ADASUVE is indicated is unknown. Severe asthmatics and very severe COPD patients were excluded from these studies, and the results may not reflect those in patients with more severe airway diseases. The degree to which asthma or COPD could alter aerosol deposition and the fraction of loxapine dose absorbed is unknown. However, the significant and comparable level of sedation in drug-treated subjects, indicated by VAS, suggests efficient loxapine drug absorption in both asthma and COPD subjects.
Oral loxapine is indicated for the treatment of schizophrenia. An intramuscular formulation is not currently marketed in the United States. Time to effect after administration of oral loxapine is relatively slow, and intramuscular injection of other anti-agitation agents (antipsychotics and/or benzodiazepines) can be poorly accepted by a disturbed, agitated patient. These drawbacks can be avoided by administering loxapine via inhalation, for which a delivery system, Staccato Loxapine, has been developed. Delivery of loxapine by the Staccato device provides for very rapid systemic absorption and effective relief of agitation.(13–15)
In these two studies of inhaled loxapine and inhaled placebo in subjects with asthma and COPD, the most common AEs were known effects of loxapine (e.g., sedation) or minor oral effects common with inhaled medications (e.g., dysgeusia). However, inhaled loxapine resulted in more airway AEs and changes in measures of pulmonary function than placebo. Although subjects with either condition could develop signs and spirometric evidence of bronchospasm following loxapine inhalation, these effects were more common in asthmatics than COPD patients.
Airway AEs occurring after inhaled loxapine were consistent with an irritant effect of the inhaled aerosol and were observed almost immediately after dosing. Any notable respiratory signs and symptoms were not accompanied by clinically significant changes in respiratory rate or oxygen saturation, and those that were treated generally responded promptly when albuterol was administered (a single treatment via MDI in most cases).
Of note, the results from the current studies in subjects with known airway disorders contrast with those from previous studies of inhaled loxapine in subjects with schizophrenia. The incidence of airway AEs following loxapine inhalation in subjects with schizophrenia or bipolar disorder I was low, although subjects with known airways disease were excluded from those studies.(13,14) In all 209 subjects with bipolar I disorder that were studied, there were no reports of coughing, wheezing, bronchospasm, or other AEs following loxapine inhalation.(13) In subjects with schizophrenia, four of 229 subjects experienced wheezing, bronchospasm, or cough after inhaled loxapine.(14) Only one of these subjects required albuterol; the events in the other three subjects resolved without intervention.
The results suggest that, in subjects with asthma and, to a lesser degree, in subjects with COPD, inhaled loxapine can produce an airway effect that is generally reversible when managed with albuterol. A pulmonary assessment (i.e., history and screening physical examination) can allow for successful selection of appropriate patients for the treatment of agitation with inhaled loxapine. Bronchospasm in patients with asthma and/or COPD may be effectively treated with a short-acting β-agonist bronchodilator, which should be readily available in real-world medical or psychiatric emergency settings. Thus, although inhaled loxapine is contraindicated in patients with active airways disease per the approved US labeling, the risks of bronchospasms may be reduced for such patients in case of inadvertent use.
Footnotes
Acknowledgments
The authors thank Pamela Lindroos, PhD (WebbWrites, LLC), for medical writing support. These studies were sponsored by Alexza Pharmaceuticals, Mountain View, CA. Clinical trials registration is as follows: ClinicalTrials.gov identifiers: Asthma NCT00890175, and COPD NCT00889837
Author Disclosure Statement
Dr. Gross has served as an advisor/consultant to Alexza, Almirall, AstraZeneca, Boehringer Ingelheim, Dey Specialty, Elevation, Forest, Pfizer, and Sunovion; has received research grants from Boehringer Ingelheim, Forest, Mylan Specialty, and Nycomed; and is a speaker for AstraZeneca, Boehringer-Ingelheim, Forest, and Sunovion.
Dr. Greos was an investigator for the asthma study; has served as an advisor/consultant to Alexza; has received research grants from Amgen, Amphastar, Array, Boehringer Ingelheim, Forest, GlaxoSmithKline, Merck, Novartis, Sunovion, and Teva, and Vectura; and is a speaker for Sunovion.
Dr. Meltzer was an investigator for the asthma study; has served as an advisor/consultant to Alexza and to Alcon, Apotex, AstraZeneca, Bausch & Lomb, Boehringer Ingelheim, Dey, Forest, ISTA, Johnson & Johnson, Kalypsys, Meda, Merck, Mylan, ONO, OptiNose, Procter & Gamble, Rigel, SanofiAventis, Stallergenes, Sunovion, and Teva; has received research grants from Alcon, Amgen, Apotex, AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, HRA, MedImmune, Merck, Novartis, Procter & Gamble, Sunovion, and Teva; and is a speaker for Alcon, Ista, Merck, Mylan, Sunovion, and Teva.
Dr. Spangenthal was an investigator for the COPD study; and is a speaker for Pfizer, Novartis, Forrest Labs, and Boehringer Ingelheim.
Drs. Fishman, Spyker, and Cassella were full-time employees of Alexza during the design, conduct, and reporting of these studies and have Alexza stock and stock options.