
Abstract
The advent of genome editing has kindled the hope to cure previously uncurable, life-threatening genetic diseases. However, whether this promise can be ultimately fulfilled depends on how efficiently gene editing agents can be delivered to therapeutically relevant cells. Over time, viruses have evolved into sophisticated, versatile, and biocompatible nanomachines that can be engineered to shuttle payloads to specific cell types. Despite the advances in safety and selectivity, the long-term expression of gene editing agents sustained by viral vectors remains a cause for concern. Cell-derived vesicles (CDVs) are gaining traction as elegant alternatives. CDVs encompass extracellular vesicles (EVs), a diverse set of intrinsically biocompatible and low-immunogenic membranous nanoparticles, and virus-like particles (VLPs), bioparticles with virus-like scaffold and envelope structures, but devoid of genetic material. Both EVs and VLPs can efficiently deliver ribonucleoprotein cargo to the target cell cytoplasm, ensuring that the editing machinery is only transiently active in the cell and thereby increasing its safety. In this review, we explore the natural diversity of CDVs and their potential as delivery vectors for the clustered regularly interspaced short palindromic repeats (CRISPR) machinery. We illustrate different strategies for the optimization of CDV cargo loading and retargeting, highlighting the versatility and tunability of these vehicles. Nonetheless, the lack of robust and standardized protocols for CDV production, purification, and quality assessment still hinders their widespread adoption to further CRISPR-based therapies as advanced “living drugs.” We believe that a collective, multifaceted effort is urgently needed to address these critical issues and unlock the full potential of genome-editing technologies to yield safe, easy-to-manufacture, and pharmacologically well-defined therapies. Finally, we discuss the current clinical landscape of lung-directed gene therapies for cystic fibrosis and explore how CDVs could drive significant breakthroughs in in vivo gene editing for this disease.
INTRODUCTION
Gene editing in the clinic: Ribonucleoprotein delivery as the future of gene editing
The approval of the first ever clustered regularly interspaced short palindromic repeats (CRISPR)–Cas9 genome-editing therapy, Casgevy, by the U.S. Food and Drug Administration (FDA) in November 2023 for the genetic blood disorders sickle-cell disease and transfusion-dependent β-thalassemia has created great excitement for the future clinical development of CRISPR strategies to more effectively treat or even cure other genetic diseases. 1 Indeed, the CRISPR revolution has opened a new era for treating genetic diseases at their cause by inducing targeted and permanent genome modifications. A typical CRISPR strategy involves two key players: a Cas protein, most commonly Cas9 derived from Streptococcus pyogenes (SpCas9), that introduces a double-strand break in genomic DNA and a single-guide RNA (sgRNA) that directs the Cas protein to its genomic target. This machinery can be harnessed to alter genomic sequences or to facilitate the targeted integration of donor DNA sequences. Through site-directed mutagenesis and fusion of additional functional domains to the Cas protein, a diverse range of gene editing tools have been developed, including base editors capable of performing A-to-G and C-to-T conversions via nucleotide deamination and prime editors capable of introducing all types of base conversions, insertions, and deletions at precise genomic sites. 2
The possibility to correct mutations at their endogenous loci offers potential benefits compared with gene replacement strategies for monogenic disorders such as cystic fibrosis (CF), as restoring endogenous gene expression and regulation is likely favorable compared with gene addition/silencing for recovery of the biological function of the disease-causing target gene. 3,4 Central to the success of the gene editing product Casgevy is the combination of several factors as follows: (i) a well-understood genetic target, (ii) CRISPR technology to successfully knock out function of the target gene, (iii) the ability to selectively expand gene-corrected cells ex vivo, (iv) strong engraftment protocols to support the incorporation of the edited stem cells in the body, and (v) a clear understanding of the level of correction needed to improve clinical disease symptoms. 5,6 While other inherited blood diseases can directly build on these advances in ex vivo gene therapy, many genetic diseases require in vivo gene editing strategies, which will necessitate improvements in the molecular tools being used and a greater understanding of how these tools function in patients.
In vivo CRISPR approaches are necessary for target organs where stem cell isolation, expansion, and re-engraftment are challenging or not likely to produce therapeutic benefits, such as for the eye, liver, and lung. In humans, in vivo CRISPR studies have been done to target gene correction in the liver and the eye. For the inherited disease transthyretin amyloidosis, the faulty protein that circulates in the body is primarily produced in the liver, so CRISPR was delivered systemically using targeted lipid nanoparticles (LNPs) to transduce the liver as the target organ and edit the disease-causing gene in hepatocytes. 7 The treatment of Leber congenital amaurosis type 10, in contrast, was an adeno-associated viral vector (AAV) containing CRISPR cargo that was directly injected into the eye as the target organ. 8,9 These trials have reported successful treatment of patients, which underscores the great potential of in vivo gene editing. However, this approach encounters two major challenges: reaching the target cells at high enough levels to achieve sufficient editing efficiency, and editing in off-target cells throughout the body. The latter can be mitigated to a certain extent by opting for localized delivery, such as targeted injections into the subretinal space in the eye or local deposition of gene therapy agents into the lungs by aerosol delivery. Often, however, systemic delivery is the preferred strategy either because the target organ is easily reached—as is the case with the liver—or because of the multiorgan nature of the disease—such as for treating CF. A preclinical study on the systemic delivery of LNPs carrying Cas9 or adenine base editor (ABE) mRNA together with chemically synthesized sgRNA was reported by Sun et al., which was aimed at targeting the lungs to treat CF in mice. 10 The systemic LNP delivery transfected the endothelium of many organs through the circulatory system, as was expected, but remarkable uptake was achieved in the lung vasculature, which has high contact surface with the circulatory system, and in the airway epithelium, which is critical for treating CF. A key molecular tool to achieve selective targeting of airway cells was a selective organ targeting (SORT) nanoparticle. SORT nanoparticles have a chemical structure that interacts with specific proteins, which enhances transduction of cells that express the interacting protein’s receptor. 11 Sun et al. used a SORT molecule that interacts with vitronectin, which promoted their uptake in both vitronectin receptor-expressing lung endothelial and epithelial cells. 10 Intriguingly, the mechanisms through which these SORT-LNPs are capable of reaching the airway epithelium, which includes overcoming barriers such as traversing the endothelium and basement membranes, remain a matter of investigation. 4,12
One of the major advantages of LNPs is the ability to easily scale up their production to accommodate the large doses needed for systemic delivery. In most cases, LNPs are loaded with gene editing mRNAs because their inherently negative charge interacts well with the cationic or ionizable lipids used in LNPs, enhancing their formulation process. 13 Additional genetic tools such as plasmid DNA and ribonucleoprotein (RNP) complexes can be delivered as gene editing cargos. Plasmid DNA potentially offers prolonged expression compared with mRNA, which reduces the dose that needs to be delivered but increases the probability of off-target effects and immunogenic responses. 4 RNP delivery, however, provides ready-to-go, functional CRISPR complexes, which support the fastest initiation of editing and, importantly, have a short half-life—which lowers the chance of off-target effects. As RNPs consist of both nucleic acid and protein components, loading them into LNPs requires additional optimization of lipid composition and loading conditions to ensure proper encapsulation and stability, but proof-of-concept work has been described to begin standardizing this process. 14,15
Virus-like particles (VLPs), which are engineered from viral particles, have the ability to readily package RNPs during their assembly process in producer cells. VLPs have retained the intrinsic advantages of viruses to efficiently enter a target cell and prevent lysosomal degradation, allowing efficient cargo release (reviewed by Raguram, Banskota, et al. 16 ). Furthermore, VLPs have been engineered to exploit the unique properties of viral proteins, such as the structural protein Gag, to actively recruit gene editors, including Cas9 or zinc finger nucleases, into the budding particles within producer cells.
This review explores possible synergies between VLPs and extracellular vesicles (EVs), as similarities in their structure and production suggest that lessons learned from each can spur future developments of novel delivery vehicles for gene editing tools based on cell-derived vesicles (CDVs)—a category encompassing both VLPs and EVs. We focus on the technical aspects of CDV production, including cargo loading, functionalization, purification, and upscaling, and then discuss the possibilities of tailoring CDV therapies to correct genetic lung diseases—in particular, CF. For further details on current and future therapeutic applications of VLPs and other delivery vehicles such as LNPs and rAAVs, we refer readers to several extensive reviews. 15 –18
MOLECULAR MASQUERADE: FROM EVS TO VIRUS-INSPIRED PARTICLES FOR PROTEIN TRANSPORT
Gene therapy delivery vehicles have been engineered from the natural mechanisms of how cells and viruses transport cargo. Eukaryotic cells produce a rich repertoire of EVs that mediate intercellular communication by delivering proteins, nucleic acids, metabolites, and lipids, which are capable of altering the homeostasis of the recipient cells. The main constituents of this repertoire, collectively termed CDVs, can be classified as ectosomes—which can be further subdivided in microvesicles (MVs) and apoptotic bodies—or exosomes based on their size, biosynthetic pathway, contents, and function. 19,20 Apoptotic cells undergo fragmentation, which produces apoptotic bodies. These are generally large (50–5000 nm) and contain stochastically determined cargo, which make them not suitable delivery vehicles for RNPs. MVs emerge directly from the outward budding of the host cell plasma membrane and are enriched with membrane-bound proteins such as tetraspanins. 20 MVs are highly heterogeneous due to the existence of distinct and not yet fully characterized budding pathways that give rise to functionally distinct subclasses of particle. 21 They form in a wide distribution of sizes, often between 50 and 500 nm, but can reach up to 10 μm. 22
Exosomes are generally smaller than MVs, ranging from 30 to 150 nm in size, 20 and originate as intraluminal vesicles (ILVs) through complex pathways that have been extensively characterized. 23,24 Briefly, the inward budding of the plasma membrane gives rise to endosomes. As endosomes mature into multivesicular bodies (MVBs), their limiting membrane invaginates, producing numerous smaller vesicles—exosomes—which can then be released into the extracellular space through fusion of the MVB with the plasma membrane. 20,21 This process can be driven by distinct and cell type-specific molecular pathways that influence the composition of the exosome cargo. 21,25 It is important to note that distinguishing between MVs and exosomes is often challenging due to a significant overlap in their size, cargo, and surface markers. 22
CDVs have a physiological role as specialized carriers of proteins, metabolites, and nucleic acids to orchestrate intercellular communication and exchange, which have inspired the design of nanovesicles for drug delivery known as “artificial exosomes.” These can be obtained either from the disruption of cells through physical and chemical methods (top-down approaches) or from in vitro synthesis (bottom-up approaches). These particles go beyond the scope of this review, and are discussed in detail elsewhere. 26
Owing to the many attributes that they share with EVs, enveloped viruses can be regarded as a type of CDV. They have, in fact, evolved to co-opt the cellular machinery that governs EV biogenesis and exploit similar cellular routes for their assembly, egress, target cell entry, and cell-to-cell transmission. Interestingly, viral infection is known to increase the production of EVs loaded with viral nucleic acids and proteins, thus mixing pathogen particles with host cellular components and shielding the virus from the host immune system to promote its survival and propagation. 27 –29
Similarly, active viral infections are known to alter the properties of secreted EVs to modulate host immune responses. Overall, the production and transport of viral particles using the host vesicle trafficking machinery make the distinction between CDVs and enveloped viral particles less clear than previously thought. 19,30,31
Two key features are necessary and sufficient to form a virus and make it infectious: a genome with the ability to replicate and a structured protein capsid that protects it. 32 Viral vectors are derived from naturally evolved viruses by replacing the protein-coding regions in their genome with alternative genetic sequences encoding a product of interest. Essential cis-acting viral sequences required for packaging, viral gene expression in the target cells, and—in the case of retroviral vectors—reverse transcription and integration into the target cell genome are retained, whereas the deleted coding sequences for the structural viral proteins are provided in trans. Viral vectors can leverage the same mechanisms used by their parental viruses to efficiently dock and transduce target cells. However, since they no longer package the genetic sequences that are required to replicate, viral vectors are not considered to be infectious and are referred to as single-round or dead-end vectors. Retrovirus-derived vectors retain the ability to integrate a transgene into the target cell genome, ensuring stable, long-term expression even after cell division. This integration can, however, result in the dysregulation of critical signaling pathways if the insertion site disrupts function or regulation of neighboring genes—an effect known as insertional mutagenesis. 33 In case the genetic material being delivered and integrated encodes a gene-editing agent, such as an active Cas9 with an sgRNA, the permanent expression significantly affects the likelihood of off-target effects and chromosomal rearrangements. 16
The boundary between viral vector particles and EVs became even more blurred with the development of VLPs. (Fig. 1) Here, we adopt the most common definition of VLPs as self-assembling nanoparticles that mimic the structural organization of native viruses but are generally devoid of viral genetic material. 34 However, there is a lack of consensus on the distinction between VLPs and viral vectors—a distinction further clouded by the fact that most Cas9-VLP architectures do not follow a standardization for naming, which is usually not informative of their origin, structure, or composition.
VLPs were first developed in the 1990s as a highly immunogenic and safer alternative to pathogen-based vaccines and continue to be used as vaccination strategy to elicit immune responses by presenting specific surface antigens to the host immune system. 34 In addition, VLPs have been steadily gaining traction in the last 15 years as novel and highly flexible vehicles for targeted delivery of small molecules, RNAs, and proteins. 35 The ability to shuttle cargo in the form of a protein—or, in the case of CRISPR-Cas cargos, of a Cas9-sgRNA RNP complex—is a unique feature of VLPs that sets them apart from viral vectors. However, certain architectures of Cas9-VLPs share more similarities with viral vectors, as they leverage cis-acting viral sequences to package sgRNA-expressing vector constructs 36,37 or to ensure the genomic integration of transgenes for concurrent gene editing and gene addition. 38 The entwined interplay between EVs and viruses has led to promising new approaches combining viruses and EVs as novel therapeutics (reviewed by Moulin et al. 29 ). Still, the effects of EVs and VLPs on target cells may differ substantially but interact with one another. For instance, Cocozza et al. recently showed that VLPs originating from the endogenous murine leukemia virus are a major component of the secretome of multiple murine cancer cell lines and are the primary mediators of some immune-regulatory effects typically ascribed to tumor-derived EVs. 39
The VLPs most commonly used for the delivery of protein cargo, and particularly of gene editing agents, originate from the Retroviridae family—specifically, the murine leukemia virus (MLV) from the Gammaretroviral genus 40 and human immunodeficiency virus 1 (HIV-1) from the Lentivirus genus. 41,42 For clarity, we hereafter refer to HIV-derived VLPs as VLPHIVs and MLV-derived ones as VLPMLVs. MLVs and HIVs are structurally alike retroviruses, and their genomes contain three essential open reading frames: Gag and Pol, which encode the polyprotein precursors of structural proteins and enzymes, respectively, and Env, which encodes the envelope glycoproteins. Gag is expressed either by itself, resulting in the Gag polyprotein, or together with Pol, giving rise to the larger Gag-Pol polyprotein. Gag consists of the matrix protein (MA), which forms a layer beneath the envelope, the capsid (CA), which forms the capsid core, and the nucleocapsid (NC), which associates with the viral genome RNA. Pol is constituted by the reverse transcriptase (RT), which converts the RNA genome into DNA during viral replication, the integrase (IN), which integrates the reverse-transcribed viral genome in the host DNA, and the protease (PR), which is responsible for the autoproteolytic processing of Gag and Gag-Pol that leads to virion maturation. This proteolytic maturation takes place during or shortly after virion release, which occurs through budding from the plasma membrane. During the budding process, the virion appropriates part of the cellular membrane, which then becomes the viral envelope. 43,44 Viral assembly in retroviruses and other viruses is a highly organized process that occurs with precise spatial and temporal coordination at the cellular plasma membrane. This process involves virus-encoded peptide motifs that hijack host factors responsible for vesicular trafficking—such as the endosomal sorting complexes required for transport (ESCRTs) and the ESCRT adaptor ALG-2-interacting protein X (ALIX) in the case of retroviruses (reviewed by Rose et al. 45 ). Although the exact mechanisms remain elusive and challenging to assess experimentally, the assembly and budding of retroviral particles are widely thought to occur at specific cholesterol- and glycolipid-enriched membrane domains known as lipid rafts. 46 –51
A notable difference between MLVs and HIV is that MLVs can only access the nucleus of actively dividing cells, whereas HIVs and other lentiviruses are capable of crossing the intact nuclear membrane. 52,53 Whether this property is maintained by VLPs delivering a protein cargo is still a matter of debate, with evidence both supporting 41 and refuting 54 the idea that proteins fused to GagHIV might be more readily imported into the cell nucleus. A thorough investigation of the mechanisms governing nuclear transport of viral and cargo proteins in dividing versus nondividing cells will be essential to further elucidate the differences between MLV- and HIV-based systems. Interestingly, Ngo et al. showed that virion maturation—a necessary process for productive retroviral infection—is dispensable for VLP-mediated delivery of functional Cas9-sgRNA RNPs, thus demonstrating that VLPs are biological entities fundamentally distinct from the corresponding native viruses. By showing that 78% of the protein content of a VLPHIV can be ablated without affecting its delivery efficiency, this work further obfuscates the distinction between EVs and VLPs. Notably, among the viral components that could be omitted was the entire Pol polyprotein. 54 It is not surprising that this deletion did not impair particle assembly and egress, as it has been known for more than 30 years that the Gag polyprotein is sufficient to support both processes. 55,56 More recently, VLPHIVs lacking the Pol polyprotein have been designed as modular platforms for ligand display and cargo delivery. 57 However, the Pol polyprotein is generally included in VLP architectures because it encodes the PR responsible for cleaving the Cas9 or other payloads fused to the Gag polyprotein. Such cleavage is typically thought to be essential for ensuring the correct intracellular trafficking and function of the payload—a belief that is challenged by the recent discoveries of Ngo et al. (Fig. 1).
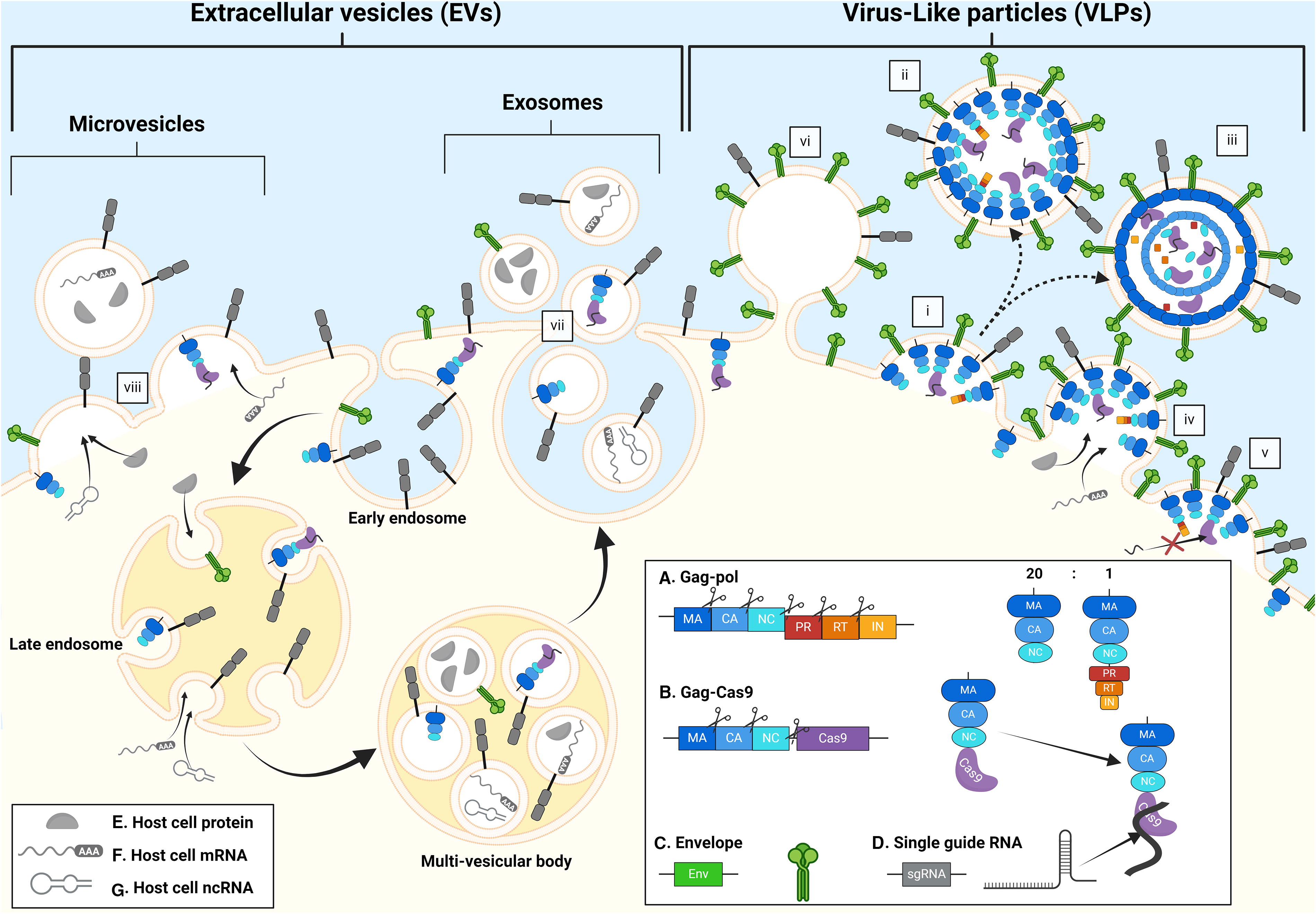
Interplay between the biosynthetic pathways of extracellular vesicles (EVs) and Cas9-virus-like particles (VLPs). The figure shows potential overlap between the assembly and budding pathways of Cas9-VLPs and the secretory pathways of EVs. A full Cas9-VLP particle
PACKAGING THE GOODS: ENCAPSULATION AND LOADING OF THE CRISPR-CAS TOOLBOX
Many therapeutic strategies seek to deliver mRNA cargos, but CDVs can be loaded with proteins as well. Protein cargo can be loaded into CDVs either during production or after isolation. Postisolation methods typically involve physical or chemical disruption of membranes, relying on their ability to reassemble afterward, but this limits their use to smaller cargos to preserve particle integrity, which makes them less suitable for delivering gene editing technologies. Production-stage approaches, which rely on genetic engineering of producer cells, are more commonly used for gene therapy strategies (Fig. 2; Table 1). 58,59 For conciseness, EVs, VLPs, and CDVs that shuttle an RNP cargo consisting of the Cas9 protein (or any derived editing tool, such as base and prime editors) and its cognate guide RNAs are hereafter referred to as Cas9-EVs, Cas9-VLPs, and Cas9-CDVs, respectively.
Production-stage approaches for cargo loading
Nonspecific packaging
Expressing the desired cargo in producer cells is the most straightforward packaging strategy. Transfecting producer cells with plasmids encoding Cas9 and its cognate sgRNAs leads to the loading of functional Cas9-sgRNA RNPs into the EVs that are secreted either physiologically 60 –62 or due to the overexpression of exogenous factors that promote their budding and secretion, such as the viral fusogen vesicular stomatitis virus G glycoprotein (VSV-G). 63,64 The exact mechanism of cargo loading has not been fully elucidated. Chen et al. showed that sgRNAs can be recruited into EVs even in the absence of the Cas9 protein, and vice versa, at least in part, suggesting a stochastic encapsidation, 62 whereas Lainšček et al. found that the loading of Cas9 derivates into EVs (named GEDEX) was boosted by the presence of sgRNAs. In the same work, addition of a membrane-targeting motif to Cas9 did not improve encapsidation, indicating an intrinsic affinity of Cas9-sgRNA RNPs for packaging into EVs. 61 This is in sharp contrast with the strategy proposed by Whitley et al., who fused a myristoylation signal to Cas9 to promote its membrane anchoring, and thus its packaging. 65 Montagna et al. identified colocalization of the Cas9 protein and the sgRNA at the cell periphery as a limiting factor for Cas9-sgRNA RNP packaging. By placing the sgRNA transcription under the control of the cytosolic RNA polymerase of the bacteriophage T7—which is stably expressed by the producer cell line 66 —instead of the more commonly used nuclear polymerase III, the investigators generated vesicles (named VEsiCas) that mediated editing levels comparable with direct transfection of Cas9- and sgRNA-encoding plasmids. 63
Fusion of cargo proteins to structural viral proteins
In the first example of Cas9-VLPHIV design, Cas9 was fused to the N-terminus of GagHIV through a HIV-1 PR-cleavable linker to ensure its release inside the capsid during particle maturation. In the same construct, a membrane-targeting protein domain was added to the Cas9-Gag fusion protein to promote its encapsulation. The sgRNA, however, was recruited inefficiently, and was therefore encoded in the lentiviral genome. 36 Hamilton et al. reported that the fusion of Cas9 to the C-terminus of GagHIV allowed sufficient incorporation of Cas9-sgRNA RNPs to support robust editing, even in the absence of an sgRNA-expressing genome—possibly due to the C-terminal fusion allowing more efficient Cas9-sgRNA interactions compared with the N-terminal one (system named Cas9-EDVs). 38 The same group has now shown that fusing Cas9 to a truncated GagHIV, containing only the MA domain and the C-terminus of CA, and expressing this fusion protein in producer cells that do not additionally express the native Gag-PolHIV yields particles that are 25% smaller and less complex than the original architecture (named mini-EDVs) but are equally or more efficient. 54 Recently, Haldrup et al. designed novel Cas9-VLPHIVs—termed lentivirus-derived nanoparticles (LVNPs)—in which Cas9 is fused to the C-terminus of a truncated Gag-PolHIV, and where sgRNAs are equipped with optimized scaffolds that increase their stability and promote their interaction with Cas9 to facilitate corecruitment. This architecture was also adapted to shuttle base editor and prime editor RNPs. 67
The first all-in-one Cas9-VLP was the friend MLV-based design developed by Mangeot et al. (named Nanoblades) in which Cas9 was fused to the C-terminus of GagMLV and efficiently complexed with sgRNAs. 68 Building upon this architecture, Banskota et al. developed a platform (named eVLPs) capable of delivering Cas9, the base editor ABE8e, 69 and, in a more recent adaptation, the prime editor RNP as well—the latter a significantly larger cargo (∼250 kDa for the prime editor, ∼160 kDa for Cas9) that required the systematic identification of specific packaging bottlenecks. 70 A considerable improvement came from switching to a Moloney MLV-based architecture. The better understanding of substrate specificity of the Moloney MLV PR enabled the inclusion of more efficient PR-cleavable sites in GagMLV-Cas9 linkers that enhanced Cas9 release upon particle maturation. 69 Interestingly, this is in sharp contrast with the previously reported findings of Ngo et al. that most Cas9-VLPs do not complete maturation, and that uncleaved GagHIV-Cas9 is fully functional and localizes to the cell nucleus. This strongly suggests that, at least in this design, GagHIV-Cas9 is associated with the VLP membrane rather than the capsid core, supporting a model in which PR-mediated maturation is responsible for forming a highly structured capsid core in retroviruses and retroviral vectors, but is not required to generate VLPs. 54
A crucial step in the architectural optimization of Cas9-VLPs is the titration of the Gag-cargo: Gag-Pol plasmid ratios in producer cells to ensure the ideal stoichiometry that maximizes cargo loading while efficiently supporting particle assembly. Indeed, Gag-Pol is widely believed to ensure the production of sufficient native structural polyprotein for efficient particle formation and egress while also supplying the PR, which is still considered necessary for the maturation of VLPs into functional particles according to the current consensus. 67,69 Indikova et al. circumvented this issue by fusing Cas9 to the accessory HIV protein Vpr, and by equipping the fusion transcript, which is expressed in packaging cells, with the same regulatory elements that govern the spatiotemporal distribution of the Gag-PolHIV mRNA. 37
Tethering cargo proteins to vesicle-enriched transmembrane proteins via dimerization modules
In contrast to VLPs, EVs lack a structured protein core, so their cargo is often anchored to vesicle-enriched transmembrane proteins. However, due to the large size of RNPs, direct fusion would likely hinder the function and folding of both proteins. Anchoring is therefore typically mediated by dimerization modules—which additionally ensure the reversibility of the assembly.
Wang et al. first applied this principle to arrestin domain-containing protein 1 (ARRDC1)-mediated MVs, or ARMMs—a subtype of small MVs (∼50 nm) whose budding from the plasma membrane is mediated by ARRDC1. They exploited the specific interaction of AARDC1 with the WW protein domain—a 40-amino acid module characterized by two conserved tryptophans (W)—by fusing WW to Cas9, which they demonstrated did not impair its enzymatic activity. 71 This delivery vector has now been refined into a patented platform for tissue-specific in vivo delivery of gene editor RNPs. 72
Zhang et al. devised a similar strategy to encase protein cargo in a broader spectrum of MVs by exploiting two self-associating fragments of a split-GFP system 73 to tether Cas9 to the cytoplasmic end of VSV-G and thus promote its active loading in VSV-G-containing MVs (named gectosomes). 74 Similarly, Ye at al. leveraged the affinity between GFP and a GFP-targeting nanobody to tether Cas9 to the tetraspanin CD63—a well-known exosomal marker 19 —to produce exosomes enriched in functional Cas9-sgRNA RNPs (named M-CRISPR-Cas9 exosomes). 75 An equivalent approach based on the P3/P4 coiled coil peptides—a pair of α-helical peptides that selectively and strongly bind to each other—was proposed by An et al. to tether prime editor RNPs to Gag in VLPMLVs. 70
A tighter control on particle production can be introduced by governing cargo loading with an inducible dimerization system. Campbell et al. first demonstrated this by anchoring the FK506-binding protein 12 (FKBP12, also known as DmrA) to the cell membrane through myristoylation, and by fusing the T82L mutant FKBP12-rapamycin binding domain (FRB, also known as DmrC) to Cas9. Addition of a rapamycin analogue triggers DmrA and DmrC to dimerize in producer cells, thus promoting loading of the Cas9-sgRNA RNPs in secreted EVs (named gesicles). Once inside the target cells, rapamycin becomes diluted, prompting cargo release. 76 This is in line with strategies proposed by Ilahibaks and Ardisasmita et al. 77 (named Cas9-TOP-EVs) as well as by Gee et al., 78 (named NanoMEDIC), which were developed based on this principle. Gee et al. fused FKBP12 to GagHIV to promote cell membrane localization of the viral protein and equipped the sgRNA with the HIV Ψ packaging signal to enable its Gag-mediated recruitment into the vesicle. The sgRNA is transcribed in the producer cells from the HIV long-terminal repeat promoter and subsequently released from the viral RNA through the self-cleavage of two flanking ribozymes. This combined approach was shown to maximize Cas9 and sgRNA recruitment into the VLP. 78
Based on reports that rapamycin-induced heterodimerization may not be fully reversible, 79 Stranford et al. developed a similar system based on the plant hormone abscisic acid-induced dimerization between a truncated form of the abscisic acid insensitive protein 1, which was fused to Cas9, and a pyrabactin resistance-like protein, which was tethered to an antibody single-chain variable fragment (scFv) protein embedded in the EV membrane. 80 Cas9-sgRNA RNPs have been encapsulated in EVs also by exploiting the blue light-dependent heterodimerization of the cryptochrome 2 (CRY2) and its ligand protein CIBN, which can be enriched on the plasma membrane of secreted vesicles by fusing it either to an EV marker such as CD9 or to a membrane-anchoring peptide. 81,82
sgRNA-mediated recruitment
Most Cas9-sgRNA RNPs loading strategies focus on active recruitment of Cas9 while relying on the intrinsic affinity of Cas9 for sgRNAs to encase functional RNPs into CDVs. Conversely, Lyu et al. designed an sgRNA-mediated approach to load Cas9-sgRNA RNPs in VLPHIVs by replacing the sgRNA tetraloop with the com RNA aptamer—a phage-derived sequence characterized by a secondary hairpin structure—and fusing GagHIV to the aptamer-binding protein COM, which selectively and specifically binds the com hairpin. 83,84 This system (named LVLPs) was originally developed to deliver SaCas9, but was later adapted to shuttle both SpCas9 85 and ABE 86 RNPs. The same group was also able to enrich such cargos in EVs by fusing COM to CD63. This was shown to increase the average vesicle size without a significant impact on morphology or yield. 87
Postisolation approaches for cargo loading
Chemical/physical methods
Wan et al. were the first to electroporate a cargo as large as the Cas9-sgRNA RNP into exosomes purified from cultured cells, which resulted in a highly transient (half-life <3 h) and nonimmunogenic delivery vehicle (named ExosomeRNP). 88 More recently, Mun et al. applied the same technique to EVs isolated from human serum (named EV@RNP). 89 Both studies reported that electroporation did not alter the morphology and size distribution of the particles. 88,89 Consistent with this, Majeau et al. reported comparable results using protein transfection with the Lipofectamine CRISPRMAX reagent. 90 In Busatto et al., Cas9-sgRNA RNPs were incubated with cationic lipids to form synthetic LNPs. These could then be hybridized with exosomes resulting in only slight changes in the mean size and the zeta potential of the exosomes 91 —the latter being an indicator of surface charge and colloidal stability. 92 A similar technique was leveraged by Zhou et al. to convert the RNA-targeting nuclease Cas13a into a detection system for cancer-associated miRNAs in circulating exosomes. 93
As final note, it is crucial to emphasize that the CDV architectures discussed in this section were designed to target distinct genomic loci in various in vitro or in vivo models. Therefore, it is difficult to compare efficacy outcomes or direct performances between delivery systems (Fig. 2; Table 1).
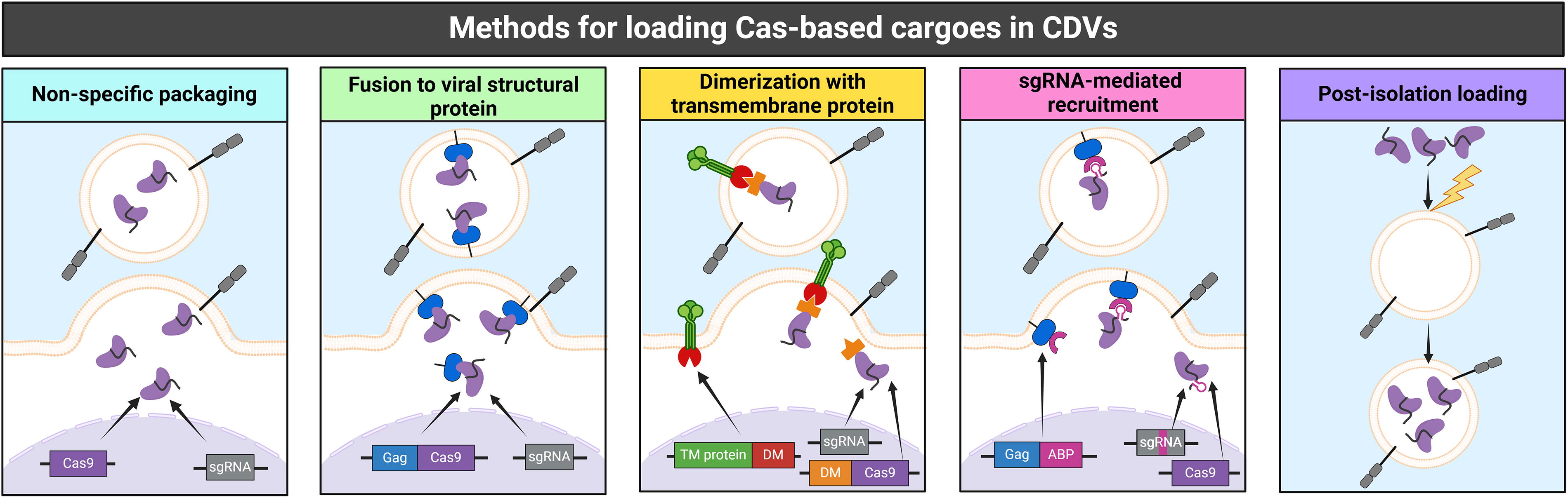
Methods for loading clustered regularly interspaced short palindromic repeats (CRISPR)-Cas cargoes into cell-derived vesicles (CDVs). The figure illustrates the methods for cargo loading used in the Cas9-CDV systems that are discussed in Chapter 3 and referenced in Table 1 using the same color scheme. Nonspecific packaging (light blue): The Cas9 protein and sgRNAs are coexpressed in the producer cell line, usually via plasmid transfection. They assemble into Cas9-sgRNA RNPs due to their intrinsic affinity and are packaged into budding CDVs via stochastic, nonspecific interactions. Fusion to viral structural protein (green): Cas9 is fused to the precursor of a viral structural protein—most commonly Gag. Coexpression of the Gag-Cas9 fusion protein together with the sgRNA and the native Gag-Pol polyprotein allows the assembly of VLPs that encapsulate Cas9-sgRNA RNPs. Dimerization with transmembrane protein (yellow): Cas9-sgRNA RNPs are tethered to plasma-membrane proteins enriched on the EVs via dimerization modules. One of the dimerizing proteins (DM) is fused to the cytosolic domain of the EV-enriched transmembrane protein, whereas its binding partner is fused to Cas9. The dimerization may be constitutive or triggered by small molecules or photons. sgRNA-mediated recruitment
Overview of existing Cas9-CDV systems
The table summarizes the various Cas9-CDV systems discussed in Chapter 3, organized by method of cargo loading: nonspecific packaging (light blue), fusion to viral structural proteins (green), constitutive (yellow), or inducible (orange) dimerization with vesicle-enriched transmembrane proteins, sgRNA-mediated recruitment of Cas9 (pink), and postisolation cargo loading through physical or chemical membrane disruption (indigo).
For each system, the purification and quality control techniques mentioned in the corresponding publication are reported. These methods were used as part of the same protocol or applied separately in distinct procedures.
Purification techniques: AEX, anion exchange chromatography; B, benzonase treatment; C, centrifugation; gUC, gradient ultracentrifugation; MAB, magnetic affinity beads; MF, microfiltration; P, precipitation; SEC, size exclusion chromatography; TFF, tangential flow filtration; UC, ultracentrifugation.
Quality control techniques: AC, affinity chromatography; BCA, protein quantification by Pierce bicinchoninic acid assay; ddPCR, droplet digital polymerase chain reaction; DLS, dynamic light scattering; ELISA, enzyme-linked immunosorbent assay; FC, flow cytometry; FT, functional titration; HTS, high-throughput sequencing; IB, immunoblot; MS, mass spectrometry; NB, northern blot; NTA, nanoparticle tracking analysis; RT-qPCR, reverse transcription quantitative polymerase chain reaction; TEM, transmission electron microscopy.
SEALING THE DEAL: DOWNSTREAM PROCESSING AND QUALITY CONTROL OF CDVS
Purification and concentration of CDVs
Producing pure and homogeneous CDVs with defined architectures and functional payloads is a complex task, and the properties of the product are heavily influenced by the methods adopted for downstream processing steps, such as purification and concentration. Due to the novelty of CDVs harboring gene editor RNPs, there is no consensus on the best practices for their purification, and their application so far has been limited to academic, preclinical research. CDVs, however, have a long-established history in molecular biology: lentiviral vector (LV)-based gene therapies are in the clinic, 94 VLPs are well-recognized vaccine platforms, 34,95 and EVs are being investigated as disease biomarkers. 20,96 We posit that the accumulated insights and knowledge from these fields can fuel the development of scalable pipelines for the production, purification, and concentration of CDV-based delivery vehicles for gene editor RNPs without losing sight of the distinct goals of each field: High immunogenicity is crucial for vaccines but not desirable for gene editing, resulting in different purity requirements; vaccine manufacturing and diagnostics prioritize the preservation of morphology and surface antigens, but not of cargo functionality, which is necessary for gene editing.
A well-known challenge to producing clinical-grade CDVs is contamination by several sources of impurities, which may reduce transduction efficiency and increase immunogenicity or toxicity. 97 Contaminants are depleted through the following three phases: clarification, intermediate purification, and polishing (Fig. 3). 98
Clarification
Clarification involves the removal of coarse contaminants, such as cell debris or intact cells—the latter being particularly relevant for producer cell lines grown in suspension, which is becoming the standard practice in the industry. Clarification is usually achieved through low-speed centrifugation, followed by microfiltration 97,99,100 (pore size 0.1–1 μm). 101
Intermediate purification
Intermediate purification eliminates impurities such as salts, serum components, and macromolecules produced by the packaging cells. It is usually coupled with a concentration step to reduce the particles’ suspension volume—which is crucial to achieve sufficient in vivo potency. 100,102
Proteins and nucleic acids are particularly challenging contaminants. Host cell proteins (HCPs) derived from the packaging cells or from serum 97,100 can form aggregates with similar size and density as the desired products. 101 DNA originating from the genome of packaging cells or from the plasmids used to transfect them tends to copurify with retroviral particles due to its high molecular weight and negative charge—which are similar to those of proteoglycans. While digestive enzymes can effectively eliminate these contaminants, their removal requires a supplementary purification step. 97,100,103 Alternatively, contaminants can be depleted upstream by implementing earlier harvest times, which minimizes dead cells, 99,100 and by using serum-free culture medium, 97,103 which also reduces costs, interbatch variability, and microbial contamination risks. 102 All of these strategies, however, can hinder particle yield. 99,104 The removal of cosecreted vesicles and defective particles—that is, particles carrying incomplete cargos—is more demanding 98 and further complicated by the fact that CDV-producer cells often exhibit a general increase in the secretion of vesicles with altered lipid composition and size distribution profile after being transfected. 105 –107 Techniques to enrich the yield of full capsids over empty and partially full ones have been developed for AAVs, but not for enveloped viral vectors. 98 Protocols for efficient production of VLPs lag even further behind, partly due to their internal configuration being less well understood to date—as recently highlighted by Hamilton et al. 108
Ultracentrifugation
At the laboratory scale, intermediate purification is usually achieved through ultracentrifugation (UCF). Scalability, however, has been limited by small rotor capacities, which is being overcome by using continuous-flow systems. The high hydrodynamic pressure and shear stress in UCF enhance aggregation, damage, and possibly particle inactivation, posing another challenge to achieving high yields of therapeutic particles. This is partially mitigable by underlaying sucrose or iodixanol cushions. 97,98,109 Equilibrium density UCF leverages density gradients to separate particles based on their buoyant density. 97,110 Although reports of separation between HIV-1 virions and exosomes exist, 111 the density parameter generally overlaps for most CDVs, HCP aggregates, and organelles. 65,97,109,110 More stringency can be achieved through rate-zonal UCF, which additionally separates particles based on their size. Both modalities are, however, laborious, time-consuming, and not scalable for commercial applications. 20,97,98 Good manufacturing practices (GMPs) for clinical-grade materials are moving toward gentler, faster, more robust, and, importantly, more scalable techniques.
Ultrafiltration
Ultrafiltration (UF, pore size 0.01–0.1 μm) retains larger particles on or in a porous membrane while smaller impurities flow through 97,98,100 and is at the basis of several commercial kits for exosome isolation. 20,112 Particle deformation and inactivation are preventable through optimization of flow rate, process time, and molecular weight (MW) cutoff 100 —usually chosen between 100 and 750 kDa. Larger MW cutoffs maximize particle yield but reduce sample purity, while smaller MW cutoffs maximize purity at the expense of yield. 94,99 The main limitation of UF is incomplete particle recovery due to membrane fouling—that is, the reduction of filtration efficiency due the accumulation of particles and contaminants within the membrane pores. This can be alleviated by performing sequential filtration on progressively smaller pore sizes 20,97 or by using tangential flow configurations. 94,97,109,113
Chromatography
Chromatography is widely used in the production of gene therapy products, with the combination of orthogonal separation principles ensuring the highest particle yield, purity, and potency. 97,99,100 Anion exchange (AEX) chromatography is a common choice for the purification of retroviral vectors, as these are typically negatively charged at neutral pH. 97 –99 Although less commonly used, AEX performs similarly to UCF for EV purification 98,114 and allows selective enrichment of VLPHIVs over cosecreted EVs. 115 However, AEX may copurify nucleic acids owing to their negative charge, and the pH and ionic strength changes required for desorption may damage fragile particles such as EVs and VLPs. 97,100 Another effective method that has been extensively used for VLP purification over the last several decades is affinity chromatography, 116 which enriches particles based on interactions between surface molecules, expressed physiologically or by genetic engineering, with ligands immobilized on the chromatographic support—such as heparin and biotin. 95,97,99,100 The most selective approach is immunoaffinity chromatography, which exploits antibody–antigen interactions and can potentially separate vesicle subtypes if specific markers are identified. 97,100 However, high costs, slow process times, and instability of antibodies along with the harsh elution conditions required to dissociate them from antigens—which can potentially cause toxicity in case of leaching—make immunoaffinity impractical for large-scale purification of fragile particles. 98,100,116
Precipitation
Precipitation with additives followed by low-speed centrifugation is a cost-effective and relatively effortless strategy used for the concentration of enveloped viral vectors, VLPs, and EVs, and it can accommodate large input volumes. 95,97,109,110 Currently, the crowding agent polyethylene glycol (PEG) is the most commonly used precipitation agent, 109 but its proinflammatory properties 117 and lack of selectivity—which leads to coprecipitation of lipoproteins and nucleoproteins 95,97,109 —reduce the potential for its use in clinical settings. 20 Lectin, which binds carbohydrates exposed on membranous envelopes, might offer more specificity. 20,110 Potential alternatives include highly concentrated salts, which could damage proteins and aggregate the particles due to changes in osmotic pressure, 97,100 and cationic polymers, whose irreversible bond to the vesicles' surface may interfere with the vesicles' functions or cause unintended issues. 100,110
Each technique has distinct strengths and drawbacks: precipitation provides high particle yield, but low purity; UCF removes most contaminants at the cost of particle recovery; chromatography offers flexible compromises between yield and purity, but its laboratory implementation can be challenging. The choice of purification method depends on the desired product and application, with optimal outcomes often achieved by combining techniques. Notably, each technique enriches vesicle subpopulations with slightly different size distribution profiles and molecular markers. 112,114,118,119
In addition, contaminants are not only found in the supernatant. Contaminating cellular RNAs 120,121 and proteins 122 –124 have been found in MLV and HIV particles as well as VLPs, 68 either as the result of stochastic encapsulation or by active enrichment during particle assembly. Similarly to how EVs preferentially package certain nucleic acids and proteins as cargos, 22 viruses can in fact selectively encapsidate certain host cell factors that they have evolved to co-opt for purposes such as promoting infectiousness and immune evasion. 120,121,124 There are currently no purification techniques capable of removing these contaminants, but their encapsulation may be addressed more upstream—for example, by knocking out the endogenous machinery for RNA loading in EVs, as proposed by Dubey et al. 125 Whether these host-derived elements are truly contaminants or function as cofactors involved in efficient cargo delivery is, however, yet to be determined.
Polishing
Lastly, polishing is the removal of remaining impurity traces, which is often performed through size-exclusion chromatography (SEC). This ensures high particle recovery and activity preservation but results in an undesirable dilution of the final product. Multimodal chromatography, which combines SEC and AEX, has also been applied successfully for polishing. 94,99,100 The final stage for all clinical-grade materials is sterile filtration, which reduces yields due to nonspecific particle-filter interactions, along with the addition of excipients to preserve biological activity and product stability through freezing. 94,95,99 The unavoidable particle loss is compensated by a reduced activation of proinflammatory pathways, which is likely to improve editing efficiencies and target cell viability. 103
Characterization and quality control of CDVs
Before CDV-based drug products can be introduced into clinical settings, a detailed characterization of their functional components and critical quality attributes is paramount (Fig. 3). However, due to the emerging nature of the field, significant gaps in both technology and expertise will need to be bridged before this goal can be achieved (reviewed by Adlerz et al. 126 ).
Nonfunctional titration of Cas9-CDVs
At the laboratory scale, the quality of CDVs is usually assessed through nonfunctional titration methods such as immunoblotting, enzyme-linked immunosorbent assay (ELISA), and mass spectrometry, which analyze the bulk population of produced particles and quantify them by measuring cargo proteins and envelope markers—or, in the case of VLPs, capsid components—as well as detecting contaminating proteins. These approaches have several limitations. First, they cannot distinguish between vesicle-associated and free proteins, thus not providing insight on the actual localization of either contaminants or cargo. Second, due to the lack of suitable CDV-specific markers, these techniques cannot analyze the entire secretome nor isolate a specific vesicle subpopulation. Finally, they do not provide accurate insight on the internal particle organization.
Nonetheless, ELISA is the most frequently used strategy to estimate the efficiency of Cas9 loading in VLP preparations. Following the downstream processing, antibodies directed either against Cas9 or an embedded peptide tag are used to estimate the concentration of gene editor proteins in a VLP sample. 36,69,70,127 –131 Similarly, the concentration of viral particles is derived from the concentration of the CA protein, the most abundant retroviral protein—p24 for VLPHIVs 36,38,67,84,132,133 and p30 for VLPMLVs. 69,70 Importantly, this is not a direct quantification but rather an estimation based on the assumption that individual VLPHIV and VLPMLV particles contain ∼2000 p24 molecules 134 and ∼1800 p30 molecules, 135 respectively. For VLPHIVs, commercial kits ensure the anti-p24 antibodies solely bind to particle-associated p24 molecules. For VLPMLVs, ∼20% of the total p30 molecules detected in solution are assumed to be particle associated. 135 Together, this allows to estimate the average number of Cas9 molecules per particle. PCR quantification of viral genomes, a key method for titrating viral vectors, is not suitable for Cas9-CDVs because a viral genome is usually lacking. However, reverse transcription quantitative PCR (RT-qPCR) and droplet digital PCR (ddPCR) can estimate the average number of sgRNAs per particle. 96,98,136
It is worth noting that all protein- and RNA-based approaches for CDVs titration are physical titration methods: they quantify total particle counts without assessing their integrity and potency and are therefore incapable of discriminating between functional and nonfunctional particles.
Functional titration of Cas9-CDVs
In contrast to physical titration, functional titration methods quantify only those particles capable of delivering complete and functional cargo. In the case of Cas9-CDVs, the readout for cargo functionality is the efficiency of genome editing. However, this measurement is heavily influenced by several factors that are not directly related to the CDV architecture, including the type of gene editor used, the targeted genomic location, and the type of target cell (either cultured or in vivo). These factors introduce cargo-specific variations in editing efficiencies, complicating direct comparisons between CDVs that carry different gene editor RNPs.
Cargo-specific biases in functional titration can be minimized by implementing standardized reporter systems, where each CDV shuttles the same gene editing machinery, targets the same genomic site, and is tested in the same cell type using a consistent methodology to measure editing efficiency. This approach ensures that observed differences in genome editing efficiencies between different CDV architectures are attributable solely to variations in delivery efficiency. Reporter systems can be established in increasingly complex models, ranging from cell lines to primary cells and even organoids and organ-on-chip platforms. In addition, this approach would provide a valuable tool to monitor the consistency between batches of the same Cas9-CDV product manufactured at different times, thereby supporting the scalability and reliability of these delivery vehicles and helping to narrow the gap between preclinical research and clinical applications of Cas9-CDVs as advanced therapeutic medicinal products. Specifically, genome-editing medicinal products (GEMPs) are an area where accurately estimating the numbers of functional versus nonfunctional particles in a sample is essential. 137 Potency assessment should directly relate to the intended biological function (defined as on-target efficiency). Establishing a suitable potency assay, predictive of biological activity, during the early stages of product development is key to ensure comparability between batches of the same product and between different product versions in preclinical studies, as well as to demonstrate product consistency during clinical testing. 138 The recent approval of Casgevy by the European Medical Agency is an important milestone that will provide further regulatory guidance for GEMPs. 139
Analytical techniques to assess Cas9-CDVs
The use of CDVs as delivery vehicles in gene therapy is limited by our incomplete understanding of their biology, the lack of uniform standards for their production, and the absence of straightforward, cost-effective methods to assess their functionality. As a result, CDVs are still a “black box” very far from meeting the rigorous quality standards imposed on clinical-grade materials. To advance the use of CDVs in GEMPs, we propose that advanced techniques for particle characterization can be imported from the fields of viral vector production, VLP vaccine manufacturing, and EV-based diagnostics. Although these approaches cannot address all currently unmet needs, they can undoubtedly serve as driving force in the clinical advancement of Cas9-CVD applications.
Microscopy-based techniques
Electron microscopy (EM) can characterize single particles in detail regardless of their size. Transmission EM (TEM) is the gold standard method for measuring the diameter of vesicles and characterizing their internal structures, whereas scanning EM (SEM) can map the particle surface. Cryogenic EM (CryoEM), due to sample preparation being based on rapid freezing rather than metal coating, can analyze particles in their physiological state. 96,98,136 CryoEM has, for example, been used to investigate VLP maturation 54,140 but does not have sufficient resolution to assess cargo loading. EM is, however, limited by the low-throughput and complexity of sample preparation, the technical expertise required to perform the experiments, and the high cost of the hardware. Furthermore, the similar morphology of most vesicle subtypes and the high background noise caused by nonvesicular contaminants may hinder the identification of the particles of interest. 98,136
Optical techniques
Optical-based methods for interrogating molecular particles enable a direct and relatively fast quantification and assessment of the size distributions of the particles in a sample. Dynamic light scattering (DLS) measures light intensity fluctuations due to particle movement in solution, and uses Brownian motion to estimate their hydrodynamic diameter. 96,141 The poor performance of DLS in heterogeneous samples is overcome by multiangle light scattering 98,142,143 and by nanoparticle tracking analysis (NTA), which captures the Brownian motion of individual particles. 96,136 NTA is a frequent choice for particle quantification because it is a rapid, sensitive, and high-throughput technique, although it can be hindered by very poor reproducibility, 136,143 a tendency to overestimate particle size and concentration, 96,143 and the inability to differentiate vesicles from contaminants or to resolve distinct vesicle populations. 96,98,144 All of these technologies depend heavily on the purity of the sample and on the concentration of the particles, and do not allow for accurate measurements of particles in crude lysates.
Flow cytometry-based techniques
Capture bead-based technologies can extend the capabilities of flow cytometry, which is typically calibrated for cells, to the detection of smaller particles while providing semiquantitative information on their surface epitopes. 96 Similarly, particle concentration, size, and surface composition can be characterized by single-EV flow cytometry and flow virometry, two related techniques that rely on fluorescence rather than light scattering to analyze particles as small as ∼40 nm. 96,98,136 Flow cytometry, however, may underestimate particle concentration due to simultaneous detection of multiple particles and to smaller particles falling within the background signal range. 136
Chromatography-based techniques
High-performance liquid chromatography, used either with AEX or SEC modalities, can also be used for particle quantification, provided that a calibration curve can be established using fully characterized materials. This has been achieved for retroviral and lentiviral vectors but poses a bigger challenge for newly developed CDVs with different cargos. Chromatography is very robust and can profile sample impurities but is usually unable to estimate particle size. When coupled to mass spectrometry, chromatographic techniques can provide insight on the bulk protein composition of CDV samples. 98,143,144
Limitations of current analytical techniques
Choosing the optimal method is challenging, as each excels for a different particle size range, and all have an unclear limit of detection. Technique-specific biases as well as varying abilities to resolve aggregates and exclude contaminants can cause significant discrepancies in particle concentration and size distribution—making the use of multiple orthogonal methods recommended. 96,136 Moreover, the uptake kinetics of any vesicle are influenced by both its size and surface charge 141,145,146 —an often overlooked parameter in quality assessments. The zeta potential can be assessed using electroacoustic 147 and electrophoretic 145 techniques, specialized NTA protocols, 148 or tunable resistive pulse sensing. 96,98
The biomolecular corona of CDVs is perhaps as important as its size and charge, but it is not frequently considered when characterizing the molecular properties of vesicles. The corona is a dynamic layer of macromolecules that rapidly adsorb—that is, adhere to the surface—onto nanoparticles upon contact with biological fluids. This process is driven by biomolecule–nanoparticle and biomolecule–biomolecule interactions. The protein corona is a key determinant of a vesicle’s biological identity and tropism. 149 –151 Recent studies have used mass spectrometry to investigate the corona formed on inorganic and biomimetic nanoparticles, including liposomes, in human plasma. 149,152,153 Similar analyses have been conducted for enveloped viruses 154 and circulating EVs, where an additional challenge lies in distinguishing macromolecules that are part of the vesicle from those associated with its corona despite their shared biological origin. 151,155 To our knowledge, the corona of CDVs used in drug delivery following in vivo administration has not yet been characterized. Some evidence suggests that the corona starts forming during particle production, raising the possibility that some macromolecules currently considered contaminants may actually be parts of the vesicle’s identity. 96,151
Finally, another understudied aspect in the quality assessment of CDVs is the posttranslational modification status of their structural proteins. Glycosylation, in particular, is gaining recognition as a key determinant of protein function and stability in several biological processes. For example, N- and O-linked glycosylation of viral and host cell proteins is an important tool through which certain viruses, including influenza, HIVs, and coronaviruses, modulate protein folding and receptor binding. Analytical methods to examine and interrogate these modifications, however, have not yet been developed (Fig. 3). 156 –158
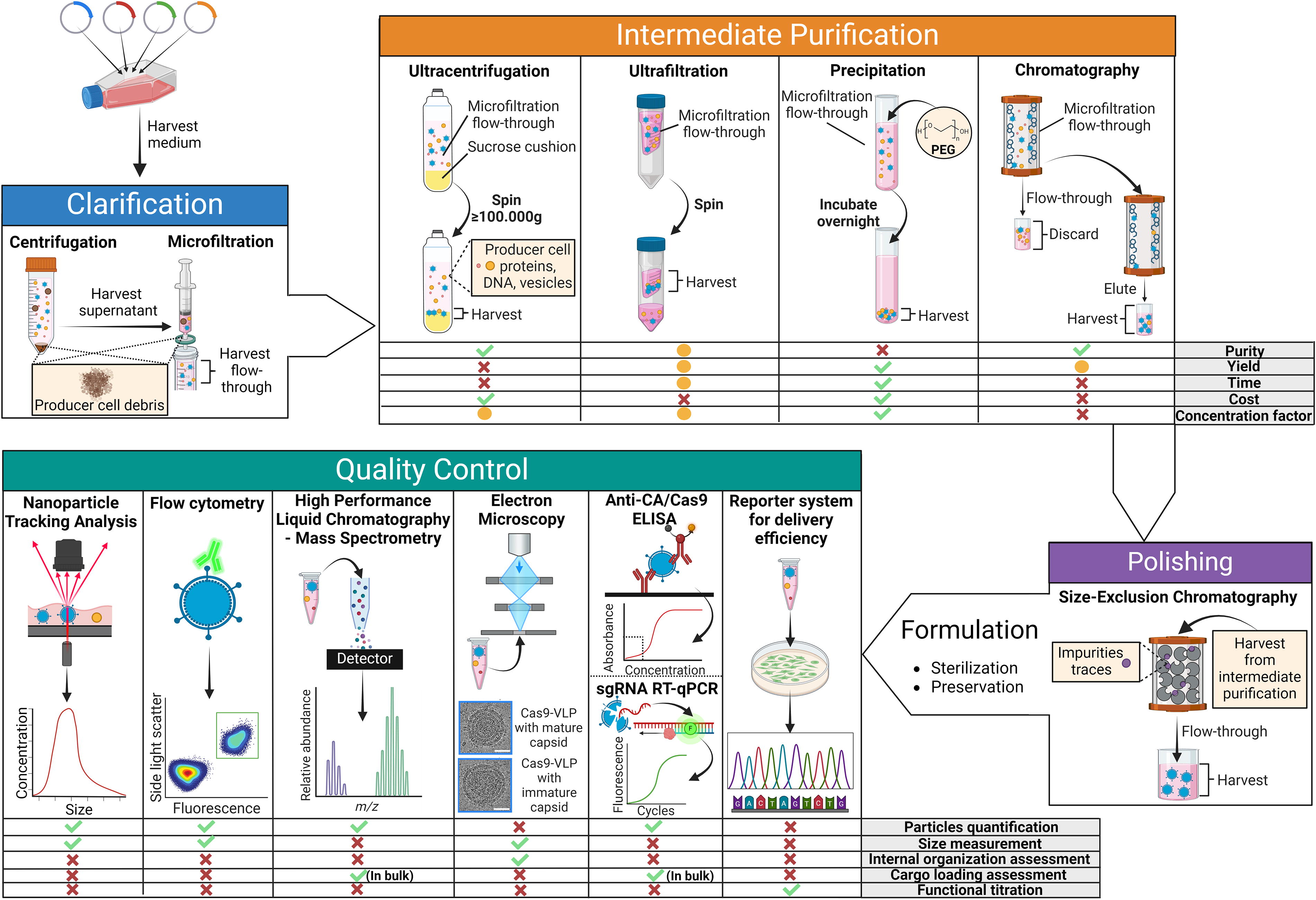
Downstream processing and quality control of Cas9-CDVs. The figure outlines the fundamental steps of the downstream processing of Cas9-CDVs, from harvesting the particle-enriched medium to the three purification phases (clarification, intermediate purification, and polishing), followed by final formulation for long-term storage. A final quality assessment step for the purified particles is included. For each step in the pipeline, the most used techniques are highlighted. Various alternative techniques for intermediate purification and quality control are presented, along with a schematic overview of their strength and limitations. Depending on the intended application, a single technique or a combination of methods may be used at each stage. The cryo-electron microscopy (EM) images are reproduced from Raguram et al. 140 and is used here without alterations under a Creative Commons license (http://creativecommons.org/licenses/by/4.0/).
HITTING THE MARK: CELL-TYPE-SPECIFIC TARGETING OF CDVS
EVs and retrovirus-based VLPs, collectively referred to as CDVs, have become essential tools for shuttling molecular cargo to cells in research settings, with significant therapeutic potential for delivering genetic medicines in clinical settings. Ensuring that CDVs selectively target the desired cell types is critical for advancing gene therapy applications by reducing off-target effects and enhancing therapeutic efficacy—which is particularly relevant for in vivo applications. Here, we explore the mechanisms of CDV uptake and discuss pseudotyping as a versatile strategy to achieve cell-type-specific delivery. For more detailed discussion on this topic, see Frank et al., 159 Duvergé et al., 160 and Stitz et al. 161
Mechanisms of cell targeting and uptake
Receptor binding, fusion routes, and cell entry
Similarly to how EVs and enveloped viruses—and, consequently, their derived vectors and VLPs—share similarities in their biogenesis pathways, they are also known to rely on similar mechanisms for cell entry.
The ability of a virus, along with its derived viral vectors and VLPs, to target specific cell types is defined as tropism. For enveloped viruses, this is determined by the affinity between host cell-type-specific receptors and the glycoproteins embedded in the viral envelope.
The binding of viral envelope proteins to cell receptors is a necessary first step in effective transduction of the target cells—meaning that only cells expressing these target receptors are permissive to viral entry. Once the viral particle is tethered to its cognate receptor, cell entry and thus cargo delivery may occur through one of two principal mechanisms: (i) direct fusion of the viral envelope with the target cell plasma membrane or (ii) internalization via endosomes and subsequent pH-dependent escape—the latter being more common than the former. In fact, the large majority of viruses—including VSVs, from which the most widely used envelope glycoprotein VSV-G is derived—rely on the endocytic machinery of the target cells to “hitch a ride” on the cytoskeleton from the plasma membrane to the nuclear periphery. This eases the passage through the crowded cytoplasm and minimizes the viral components presented at the plasma membrane, thereby delaying antibody or immune cell recognition. 162 –164 There are, however, notable exceptions to this general rule: Some members of the Retroviridae family, including HIV, as well as the Herpesviridae and Paramyxoviridae families, 165 –167 can directly fuse with the plasma membrane through a pH-independent entry mechanism that relies on specific conformational changes in the envelope protein upon receptor binding. This allows them to release their contents into the target cell cytoplasm, either as their primary entry strategy or as an additional route alongside endocytosis, depending on the target cell type.
While the molecular interactions of some viruses are well understood, they remain elusive for many others. However, as virology shifts from cultured cell models to in vivo experiments, it becomes evident that viruses exhibit significant redundancy and adaptability in receptor usage, endocytic pathways, penetration sites, and uncoating mechanisms. 163 Of note, receptor-independent, nonspecific uptake pathways have also been described for both MLV and HIV virions 168 —an important consideration when evaluating the specificity and safety of CDV-based strategies for the targeted delivery of gene editing tools.
Endosomal escape mechanisms
The endosomal–lysosomal system is a dynamic cellular network responsible for recycling, sorting, and processing internalized cargo. Following uptake via endocytosis, vesicles enter early endosomes—the cell’s primary sorting precinct. Here, depending on factors such as membrane composition, pH gradients within the early endosome, and the presence of molecular tags (e.g., ubiquitin), the vesicles will be either returned to the plasma membrane or directed toward the late endosomes, which subsequently fuse with lysosomes for degradation. To avoid being degraded in the host’s hydrolytic catabolic process, CDVs must escape from the endosome before its fusion with the lysosome. To achieve this, viruses and VLPs can co-opt some of the routes to prevent endosomal entrapment that are used by EVs, whose role as mediators of intercellular communication requires them to deliver intact molecular cargo to target cells and compartments.
Four distinct escape mechanisms have been proposed: membrane fusion, pore formation, back fusion, and pH buffering. 169 –173
Direct membrane fusion
EVs and some viruses can escape the endosomal compartment through direct fusion with the endosomal membrane, resulting in release of their cargo into the cytoplasm. This process is triggered by the unique composition of the CDV membrane: specific lipids, such as ceramide and cholesterol, induce curvature of the membrane to form inward-budding vesicles, whereas specialized proteins, such as soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs), tetraspanins (CD9, CD63, and CD81), and Rabs GTPases, promote fusion. The mildly acidic endosomal environment plays a key role in initiating the fusion process by prompting conformational changes in pH-responsive EV transmembrane proteins or VLP envelope glycoproteins. In some cases, additional binding of endosomal receptors, activation of pH-dependent proteases, and specific lipid compositions of the endosomal membrane can also be required for a successful fusion. 169,174,175
Pore formation
CDVs can escape endosome entrapment through the formation of transient pores in the endosomal membrane, which serve as channels for the cytoplasmic release of the CDV cargo. 169,173 The mechanism of pore formation is not yet fully unraveled, but it is thought to be triggered by particular lipids, such as phosphatidylethanolamine, within the EV membrane, as well as by certain cell-penetrating peptides (CPPs) of viral origin. These include peptides derived from the HIV protein TAT and the influenza protein HA-2, which undergo pH-mediated conformational changes enabling them to insert into the endosomal membrane and disrupt its integrity. 176 CPPs, whether isolated from viruses or engineered, have been shown to enhance the endosomal escape of EVs when chemically tethered to their membrane. 177 However, further studies will be required to understand the full extent of their effects, as they have been linked to aberrant transcriptional responses and morphological alterations in target cells. 178 Finally, it should be noted that this model fails to explain how larger cargos, which can easily reach a diameter comparable with the thickness of the phospholipid bilayer, could transit through the pores, which should be too small to allow them to pass. This suggests that there are mechanisms of this process that are yet to be fully elucidated. 179
pH buffering or “proton-sponge effect”
The mildly acidic endosomal environment that is essential for the sorting and degradation processes is maintained through the action of proton pumps. Studies suggest that certain ionizable CDV proteins, including CPPs, and membrane lipids may act as pH buffers by capturing or transporting protons. This mechanism is thought to safeguard the cargo from the pH-activated catabolic lysosomal enzymes and to induce the overactivation of the proton pumps, resulting in increased osmotic pressure to cause endosomal rupture. However, this model remains hypothetical and is still the subject of ongoing debate. 35,173,179
Back-fusion
EVs can utilize a process known as “back-fusion” to escape the endosomal compartment. MVBs are specialized structures within the endosomal system that contain smaller ILVs. MVBs can fuse with the plasma membrane to release ILVs as exosomes into the extracellular space. Similarly, MVBs are thought to “back-fuse” with the endosomal membrane to release their contents, such as embedded EVs, into the cytoplasm, thus preventing their lysosomal degradation. This mechanism, similar to other membrane fusion events, relies on specialized proteins such as SNAREs and Rabs. 173,180,181
Nuclear import of cargo
It is currently unclear whether enveloped vehicles with a structured virus-like capsid, such as VLPs, can more effectively support directed transport of RNPs through the cytoplasm and into the nucleus compared with enveloped vehicles without a viral core, such as EVs. Nuclear entry of the viral capsid core, for example, in the case of HIV, has been debated for years, and current findings suggest that the core, at least under some circumstances, is transported through the nuclear pores before its disassembly within the nucleoplasm. 182,183 Docking of the core at the nuclear pore complex is mediated by interactions between the capsid and cellular factors, leading to nuclear entry of an intact core. Hence, recent findings suggest that HIV nuclear capsids are intact or nearly intact until they uncoat immediately before integration of viral DNA. 184 A certain structural elasticity of the core is thought to enable its nuclear entry. 185
Whether such mechanisms also support delivery of Cas9-sgRNA RNPs by VLPHIVs, such as LVNPs, and may promote unloading of genome-engineering proteins near open chromatin is currently unknown and needs further scrutiny. Likewise, further studies are needed to identify the cellular factors on which VLPs may rely to successfully deliver proteins—potentially uncovering variations between cell types and delivery systems that could impact efficacy.
Interestingly, the potential nuclear translocation of soluble and possibly even membranous EV components has also been suggested, for example, by the presence of the nuclear transport receptor karyopherin within EV proteome analyses. 186
Cell-type-specific retargeting of CDVs
Tropism and pseudotyping
Pseudotyping involves altering the surface proteins of a viral particle or VLP by replacing its native envelope protein with a different targeting moiety. This changes the tropism of the particle, expanding or narrowing its ability to target specific cells—making this a crucial strategy for enhancing targeting specificity. 187
One of the most widely adopted envelope proteins is VSV-G, which is characterized by high structural stability and very broad tropism. VSV-G, in fact, targets the members of the low-density lipoprotein receptor family, which are expressed in most human cells and tissues. 188 To achieve targeted delivery to specific cells, naturally occurring envelope glycoproteins from other enveloped viruses with distinct tropisms can be leveraged. 127 These include the envelope proteins of gamma-retroviruses (e.g., MLV), 189 baboon retroviruses, 190 and paramyxoviruses. 191,192 Dual pseudotyping—that is, adding two envelope proteins to a particle—has also been demonstrated to provide specific advantages. For example, dual pseudotyping of LVs with VSV-G and Sendai virus hemagglutinin-neuraminidase (SeV-HN) glycoprotein significantly improved transduction of human cells, including hematopoietic stem cells. The unexpected synergy between these two elements stemmed from VSV-G facilitating the incorporation of wild-type SeV-HN into LVs, while SeV-HN cleaved the sialic acid on the VSV-G structure, ultimately enhancing transduction efficiency. 193
Similarly, EVs must first bind to their target cells to deliver their cargo. A significant challenge hindering their therapeutic application is ensuring precise delivery to the desired tissues while avoiding accumulation at off-target sites. EVs are known to exhibit preferential affinity toward certain cell types, depending on a plethora of factors such as their surface protein profile, lipid and glycan composition, and charge (reviewed in Murphy et al. 194 ), as well as cell type of origin. 195 These factors can be harnessed and optimized toward cell-type-specific delivery of gene editor RNPs. In addition, similarly to viral vectors and VLPs, EVs can be customized by expressing viral glycoproteins or other recombinant fusion proteins on their surface via genetic modification of producer cells. 196,197 VSV-G, for example, was found to enhance the delivery efficiency of EVs without causing significant changes to their size, morphology, and composition. 198 Despite recent progress, many aspects of EV biology remain unclear. While the biodistribution and cell targeting of EVs can be modified, the uptake pathways that internalize them into cells are diverse and the mechanism can vary by cell and EV type. As is the case for viral particles, nonspecific uptake routes can limit the safety and efficacy of EV-based therapies. For both EVs and VLPs, endosomal entrapment remains a significant bottleneck, highlighting the need for new tools to enhance cargo release.
It is important to underscore that pseudotyping does not solely alter the tropism of a particle but also influences its biodistribution kinetics and mechanisms for entry and cargo release. The internalization dynamics and turnover rates of the target receptor can also impact these processes 199 —making careful selection of the target receptor essential when designing a retargeting strategy.
Novel retargeting strategies
In addition to using naturally occurring viral envelopes, envelope proteins can be engineered or replaced with synthetic constructs to enhance cell-type specificity and thus improve safety by minimizing binding to off-target cells. The use of specific ligands to redirect delivery vehicles toward a specific target is known as ligand-directed targeting display.
To achieve this, the particles are first modified with a targeting ligand that binds with high affinity to a chosen receptor on the target cell surface. This binding mediates attachment but is not sufficient to initiate entry. Then, proteins that promote cell entry by facilitating membrane fusion, such as viral glycoproteins, are displayed on the surface of the particle. However, the intrinsic affinity of these proteins for their own canonical receptor could mediate cargo delivery to nontherapeutically relevant cell types. To prevent this, the natural receptor-binding properties of fusion protein should be inactivated via mutagenesis. 199 –201 This strategy has proven to be particularly difficult for viral glycoproteins that combine receptor binding and membrane fusion features in a single polypeptide, as is the case for VSV-G. Initial experiments using a membrane-anchored targeting moiety combined with a truncated VSV-G in LVs showed efficient delivery to the intended target cells, 202 but nontarget cells were also transduced. 203 In contrast, some viruses, such as alpha-viruses (e.g., SINV) and paramyxoviruses (e.g., MV, TPMV, NiV), have separate polypeptides for receptor attachment and membrane fusion (reviewed by Perrin et al. 181 ). Engineering or replacing the receptor attachment polypeptide with specific targeting moieties, while maintaining the fusion protein unmodified, could provide a more straightforward approach to achieve cell-type-specific cargo delivery—although the existence of complex interactions between the receptor-binding and fusion glycoproteins may add a different layer of complexity to this strategy. 165,204,205
One strategy for glycoprotein engineering is the production of tailored chimeric proteins that combine targeting domains from different viral proteins, creating hybrid envelopes that can enable more efficient and selective pseudotyping. For example, fusing the internal domains of the MLV glycoprotein to the target-binding domain of a codon optimized variant of the gibbon ape leukemia virus envelope protein enabled transduction of human primary B and T lymphocytes. 206
A different approach, which can further increase targeting specificity, leverages single-chain variable fragment antibodies (scFvs) against specific cell-associated antigens fused to the membrane-anchoring domain of an envelope protein. 207 For example, Hamilton et al. used scFvs against several CD antigens, fused to the transmembrane domain of CD8a and coexpressed with a mutant VSV-G, to selectively and specifically retarget their VLPHIV design (termed EDVs) toward distinct subpopulations of human T cells. 108 Recently, Strebinger and colleagues developed DIRECTED (Delivery to Intended REcipient Cells Through Envelope Design), a modular platform that offers several immobilization strategies (i.e., protein A/G and SNAP-tag®) to stably display scFvs or full-length antibodies on the surface of LVs and VLPMLVs, which is compatible with in vivo applications. 199 In parallel, Stranford et al. developed GEMINI (Genetically Encoded Multifunctional Integrated Nanovesicles), a suite of methods for the production of genetically engineered EVs that display an anti-CD2 scFv fused to the C1C2 lactadherin domain scaffold for membrane tethering, in combination with the measles H/F glycoproteins to promote fusion. 80
An alternative to scFvs is designed ankyrin repeat proteins (DARPins). Large DARPins libraries can be easily produced and screened to identify high-affinity binding partners for any given target molecule of interest. DARPins are less prone to aggregation compared with certain scFvs and enabled higher transduction efficiencies compared with scFvs directed toward the same target when fused to the measles H glycoprotein. 208 The use of the SpyCatcher-SpyTag chemistry—where a SpyTag inserted into an engineered Sindbis virus envelope protein serves as a covalent anchoring site for a targeting moiety fused to a truncated SpyCatcher 209 —as well as split-intein systems that exploit the extremely rapid, high-affinity binding between the two halves of an intein catalytic site, 210 has been described for tethering cell-targeting ligands to the envelope of delivery vehicles.
Most of the research on CDV pseudotyping has focused on protein-based strategies. However, other innovative approaches are emerging, such as the use of nucleic acid aptamers, which can be designed to bind to cell-specific target molecules and can be tethered to membranes through fusion with cholesterol or other membrane lipids. 211 –213
Finally, the introduction of click-chemistry—a group of reactions involving the conjugation of molecules in a modular manner—has opened a wealth of options for the postisolation surface functionalization of vesicles. This method has proven to be robust, rapid, and easily scalable, 194,214,215 and it has been reported not to induce significant changes in vesicle size and properties. 216,217
This overview provides a general summary of available strategies for CVD retargeting; for a more comprehensive exploration, refer to He et al. 201 and Gutierrez-Guerrero et al. 127 (Fig. 4).
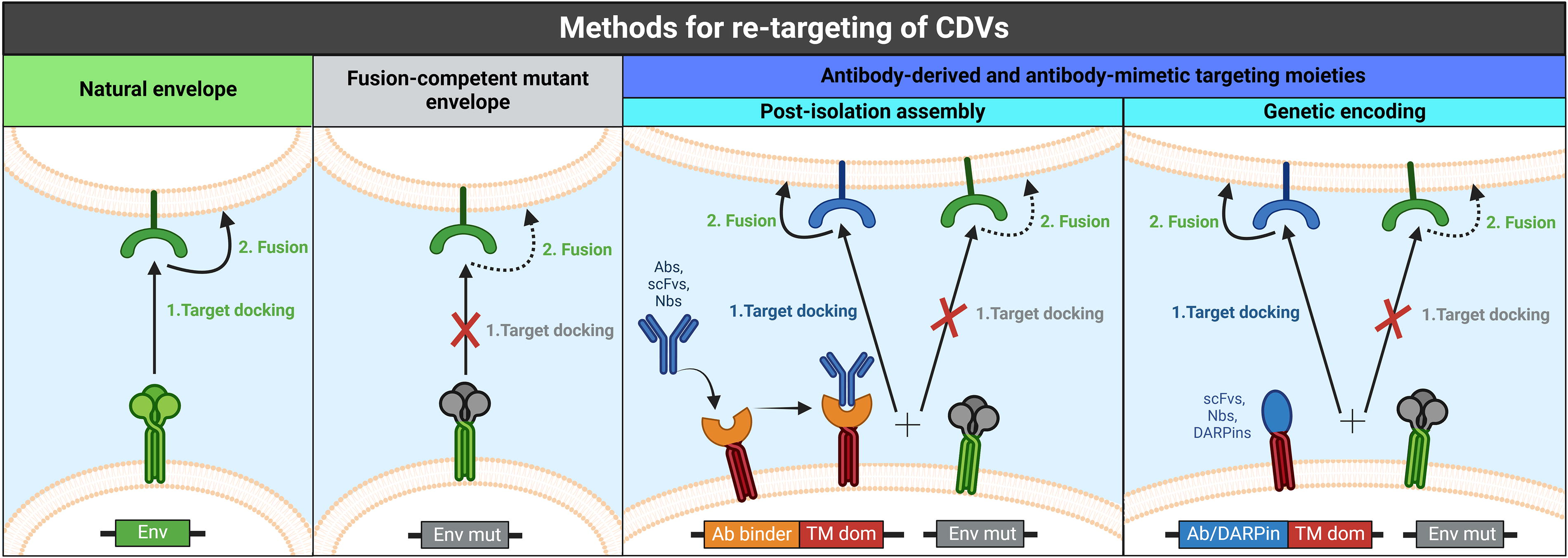
Methods for retargeting of CDVs. The figure depicts the most common retargeting strategies used to define the tropism of CDVs, as discussed in Chapter 4. In this illustration, a viral glycoprotein that combines receptor binding and membrane fusion features in a single polypeptide is shown—as is the case for the most commonly used viral envelope, vesicular stomatitis virus glycoprotein (VSV-G). Native envelope:
Immune responses and host restriction factors affecting delivery and editing efficiency
Host immune responses play a pivotal role in determining both the safety and efficacy of gene therapies. Cas9-CDVs, as complex and multifaceted medicinal products, require a comprehensive evaluation of their potential immunogenicity from various perspectives.
Immune responses to the vector components
Due to their structure and origin, VLPs are expected to share at least some of the immunogenic features of the retroviral vectors from which they originate. For example, certain interferon-induced transmembrane proteins located on the plasma membrane as well as the endosomal membrane are known to restrict the cell entry of several viruses and viral vectors, including the ones of retroviral origin, presumably by reducing membrane fluidity upon recognition of several viral envelope glycoproteins 218,219 —including VSV-G. 220 In addition, certain members of the Toll-like receptor (TLR) family have also been shown to restrict retroviral entry upon envelope glycoprotein sensing via activation of the NF-kß-dependent transcriptional program and consequent release of proinflammatory cytokines that stimulate the chemotaxis of phagocytes toward the site of infection. 218,221,222 This suggests that concerns related to innate antiviral immunity may extend to EVs decorated with viral glycoproteins as well.
If the retroviral vectors overcome this first line of defense and reach the cellular cytosol, another array of innate immune responses can then be elicited by the retroviral capsid protein components. For instance, binding of the E3 ubiquitin ligase TRIM5α onto the retroviral capsid has a destabilizing effect on the latter, resulting in dysregulation of its uncoating and promotion of its proteasomal degradation—thus preventing reverse transcription of the retroviral vector-encoded transgene—as well as release of inflammatory mediators through the activation of the NF-kß and AP-1 transcription factors. 218,223 In parallel, retroviral capsid proteins can also be sensed by certain TLRs, eliciting robust NF-kß activation and secretion of type 1 interferons (IFNs-1). 218,222
The RNA-encoded transgenes packaged by retroviral vectors are shielded from the host immune system by the capsid and nucleocapsid structures until the reverse transcription step, during which they are first exposed to the cellular cytoplasm as uncoating occurs—although, as previously discussed, direct nuclear import of the assembled capsid has also been reported in certain conditions. 182,183 The resulting double-stranded DNA is imported into the nucleus, where it integrates into the host genome. While for native retroviruses the integrated viral DNA is transcribed into new viral RNAs, which serve as retroviral genomes and as templates for the synthesis of retroviral proteins, 43 in the case of single-round retroviral vectors, expression of the integrated transgene only gives rise to the product of interest. Retroviral nucleic acids are a major source of pathogen-associated molecular patterns that can be recognized by cellular pattern recognition receptors to stimulate innate immune responses. The viral RNAs, as well as retroviral vector-encoded transgenes, can be sensed both by TLRs located in the endosomal compartment and by cytosolic RNA helicases belonging to the retinoic acid-inducible gene I-like (RIG-I-like) receptor family, resulting in the activation of NF-kß and in the secretion of IFNs-1 and proinflammatory cytokines. A wide range of specialized receptors, including members of the TLR family, IFN-inducible proteins, and helicases, are able to specifically detect the reverse-transcribed viral DNA or retroviral vector-encoded transgene, along with all the intermediates formed during reverse transcription (e.g., RNA-DNA hybrids). Particularly notable among these receptors is cGAS, a cytosolic cGMP-AMP synthase that acts as the major sensor of retroviral DNA: the products of its enzymatic activity can activate the cGAS/STING signaling cascade, resulting in the induction of IFNs and IFN-stimulated genes—which include antiretroviral restriction factors such as APOBEC3G, a cytidine deaminase that mutates nascent viral DNAs during reverse transcription, TRIM5α, the aforementioned capsid proteins receptor, tetherin, a lipid raft-associated protein that can prevent the egress of newly assembled virions, and SAMHD1, which inhibits reverse transcription by depleting the dNTP pool. Such responses may affect cell function and, in some cases, even trigger apoptosis.
It is worth noting that, while native retroviruses are equipped with accessory proteins that can counteract or mitigate the effect of host restriction factors, these are generally omitted in the architectures of retroviral vectors and VLPs. 218,224 –226 Another important consideration is that CDV-based platforms that transiently deliver protein cargo are unlikely to be hampered by the same nucleic acid-induced native immune responses that restrict retroviral vectors.
For a more extensive characterization of the immune responses triggered by various components of retroviral vectors, refer to Coroadinha 218 and Shirley et al. 224
Adding onto the landscape of innate immune responses potentially triggered by CDVs, it is relevant to mention that recent studies have revealed complex interactions between EVs and the complement system, highlighting their role in immune regulation. In addition, EVs are known to carry key complement factors as well as complement regulators that can influence inflammation. 227,228 The exact molecular mechanisms driving these effects, and how they could impact the use of EV-based delivery vehicles for in vivo therapies, remain poorly understood and require further investigation.
Humans exhibit low preexisting immunity against retroviral vectors due to low infection rates with the wild-type viruses they are derived from. This implies that adaptive immune responses, such as production of neutralizing antibodies, are generally less likely to restrict the activity of retroviral vectors—and, by extension, of CDVs. This is in contrast with AAVs, where prior exposure and associated adaptive immunity are widespread in the human population. Adaptive responses may, however, be elicited when the retroviral vectors or CDVs are pseudotyped with envelope glycoproteins derived from viruses that more commonly infect humans, such as hemagglutinin (HA) from influenza viruses, 229 or with envelope glycoproteins derived from viruses against which vaccination is standard, such as the hemagglutinin (H) and fusion (F) peptides from the measles paramyxoviruses. 230
It is worth highlighting that, regardless of preexisting adaptive immunity, all viral fusogens are inherently immunogenic and likely to trigger innate immune responses, as well as being susceptible to inactivation via opsonization by the complement when circulating in human blood—which can undermine the safety profile and significantly hamper the efficacy of pseudotyped CDVs. Evading immune surveillance is not only paramount to ensure the safety of Cas9-CDVs but is also crucial for enhancing in vivo targeting. Modifying envelope proteins to avoid detection by neutralizing antibodies and complement, as well as to evade triggering innate immune responses, is therefore expected to significantly improve delivery efficiency. 218 VSV-G-pseudotyped particles, for instance, are known to be inactivated by human serum. 231 By contrast, alternative pseudotypes show different immune resistance profiles: LVs pseudotyped with Cocal virus envelope proteins, for example, are more resistant to the complement system, which opens possibilities for incorporating these components for in vivo applications. 232 Another strategy is to replace viral glycoproteins with nonviral proteins with fusogenic potential. For instance, Myomaker and Myomerger are two key mammalian proteins that govern myoblast fusion and muscle formation, but are structurally and functionally divergent from traditional, viral fusogenic proteins. Hindi et al. recently pseudotyped LVs with these proteins, enabling specific delivery of therapeutic cargo to the skeletal muscle. 233 Similarly, the fusogenic coiled-coil peptides CPE4/CPK4, 234 the envelope protein of human endogenous retroviruses, 235 and the gamete fusion protein HAP2/GCS1 of Arabidopsis thaliana, 236 as well as human-derived CPPs, 237,238 have been investigated for their potential to promote cell entry and cytosolic cargo release.
Importantly, a common trigger of adaptive immune responses following retroviral vector administration is the transgene they carry. Successfully transduced cells expressing a retroviral vector-encoded transgene, in fact, may be flagged as non-self by the immune system and selectively eliminated. This is due to transgene-derived peptides being presented by the major histocompatibility complex class I, which is displayed on the surface of all cells, and class II, which is exclusively expressed by professional antigen presenting cells. This triggers a signaling cascade that can produce cytotoxic T cells targeting the foreign antigen, besides promoting the synthesis of antigen-specific antibodies. 224,239 –241 As for other nucleic acid-related immune responses, this adaptive response is not likely to cause concern for the majority of Cas9-CDV architectures—with the exception of those that include a retroviral expression vector encoding the sgRNA or a therapeutic transgene.
Finally, viral components are not the only potential trigger of immune responses present in retroviral vectors and CDVs. For instance, HLA-I proteins, which are displayed on the plasma membrane of the CDV-producer cells, have been shown to be incorporated into the LV membrane envelope during the budding process—a mechanism also shared by CDVs. These vector-associated HLA-I proteins can elicit T cell-mediated alloimmune reactivity. As a solution, genetic ablation of the HLA-I complex in producer cells yields substantially less immunogenic LVs. 242
On top of the ubiquitous immune pathways discussed here, cell-type-specific mechanisms of antiviral defense have also been described 218 —warranting a careful, case-by-case evaluation of the immunogenicity of CDVs redirected toward different tissues. Severe systemic immune reactions are recognized as potential complications arising from the administration of gene therapeutic products—and, in the case of high-dose AAV vectors, have sometimes proven fatal. 243,244
LVs are the vehicle of choice for ex vivo gene therapy—more specifically, for gene addition strategies, whereas electroporation is favored for the delivery of gene editing cargo. As a consequence, data on the immunogenicity of the latest generation LVs following in vivo administration remain limited. However, the few clinical trials that have recently utilized these vectors for in vivo gene therapy have reported favorable safety profiles. 245,246
Immune responses to the CRISPR-Cas cargo
A second concern associated with Cas9-CDVs, and common to all gene editing strategies, is the risk of immunogenicity against Cas nucleases and any derived editing tools.
Due to their bacterial origin, Cas nucleases may in fact act as pathogen-associated molecular patterns and trigger intracellular innate immune responses. Furthermore, it has been observed that—strikingly—up to 80% of individuals show preexisting humoral and cellular immunity against proteins derived from Staphylococcus aureus and Streptococcus pyogenes—including Cas nucleases. 247 –252 This has fueled ongoing efforts to rationally engineer minimally immunogenic nucleases for gene editing applications. 253 Anti-Cas antibodies are very likely unable to neutralize either Cas9-LVs, as they do not directly shuttle the Cas9 protein, or Cas9-CDVs, as their vesicle structure would shield the Cas9 protein cargo from immune sensing. However, the preexisting, anti-Cas immunity may lead to cytotoxic T cell responses directed against transduced cells that express Cas9, thus rendering the gene editing therapy ineffective. 247,254 It can be hypothesized that retroviral vectors, which promote stable, long-term transgene expression, may trigger such a response more robustly than CDVs, which only transiently deliver the Cas9-sgRNA RNP.
This consideration underscores the importance of not only evaluating how the Cas9 cargo may trigger immune responses, but also how these responses may influence the success of gene editing strategies. Depending on the format of the Cas9 cargo components (e.g., retroviral expression vector or DNA encoding Cas9 and sgRNA, mRNA encoding Cas9 codelivered with chemically synthesized sgRNA, Cas9-sgRNA RNP complexes) and on the longevity of their presence in target cells, their uptake may elicit distinct cellular defense mechanisms, in turn affecting editing efficacy and cellular functions at different degrees. For example, as discussed previously, innate immune sensing mediated by activation of IFNs-1 may be triggered as a cellular protective response to retroviral infection or retroviral vector transduction. This initiates a plethora of defense mechanisms that include inhibition of reverse transcription, which can potentially impact retroviral vector-based gene transfer 255 but could also affect gene editing technologies that rely on the process of reverse transcription, such as prime editing. 256
Further investigation will be required to gain more comprehensive insights into whether delivery of Cas9-sgRNA RNP complexes via CDVs is restricted by specific cellular defense mechanisms—and, if so, how these may be circumvented.
Immune responses to the contaminants
A final concern is related to the immunogenicity risk associated with host cell contaminants present in Cas9-CDV preparations—whether they are copurified with the particles or packaged within them. The World Health Organization poses clear guidelines for the acceptable limit of residual DNA in biological products—which is fixed at 10 ng/dose, with individual fragments no longer than 200 base pairs. 257 To our knowledge, the residual host cell DNA encapsulated by Cas9-CDVs has never been quantified nor comprehensively characterized. However, Banskota et al. verified that less than 0.03 molecules of plasmid DNA encoding the Gag-ABE fusion protein were packaged within their Cas9-VLPMLVs. 69 Mangeot et al., instead, characterized the full RNA content of their Cas9-VLPMLVs, revealing stochastic encapsulation of cellular mRNAs, rRNAs, and tRNAs, with less than 0.4% of the total RNA content being ascribable to the viral component-encoding constructs that are expressed in producer cells. 68 A more thorough investigation of the host cell DNA copurified and packaged within Cas9-CDVs would be highly advisable, as it has been shown that a significant portion of the innate immune responses triggered following the administration of VSV-G-pseudotyped retroviral vectors are actually due to tubulovesicular structures that contaminate retroviral vector preparations and contain nucleic acids—including the plasmids used to transfect producer cells. 258
In contrast to DNA, there is no clear consensus on the maximum accepted content of residual host cell proteins in viral vector preparations. By the early 2000s, residual proteins derived from the producer cells had already been identified as one of the major triggers of immune reactions following LV administration, and more stringent purification methods were shown to yield markedly less immunogenic vector preparations. 259 However, the favorable safety profiles highlighted by recent clinical trials involving viral vectors seem to suggest that following GMPs for particle synthesis and downstream purification minimizes the risk of host cell contaminant-induced immunogenicity. 260 –263 Furthermore, risk-assessment studies conducted on monoclonal antibody biotherapeutics—whose purification pipelines share several similarities with those of CDVs 264 —encouragingly suggest low levels of host cell protein-related immunogenicity. 265,266
The protein content of physiologically secreted EVs has been extensively characterized, and the resulting data have been compiled in publicly accessible databases such as Vesiclepedia. 267 However, given that the molecular profile of EVs exhibits significant variability based on vesicle type, cell type of origin, and healthy versus diseased state, it is unlikely that insights gained from these proteomic studies would be directly applicable to CDVs in general, and Cas9-CDVs in particular. This notion is further supported by the observation that distinct producer cell lines secrete EVs with unique molecular compositions, and that transfection of these cells—for example, to introduce the Cas9-encoding transgene—can alter the composition of their secretome. 105 –107 Mangeot et al. reported that a wide variety of host cell proteins are stochastically packaged in their Cas9-VLPMLV preparations. The majority of these were, as expected, membrane-associated proteins and could not be detected in target cells following transduction. Conversely, cytosol-derived proteins, which were presumably stochastically packaged within the particles, could be detected in the transduced cells—although transiently. 68
Given the novelty of CDV-based platforms for the delivery of gene editing tools, regulatory bodies will likely need to rely on the precedents established in LV regulation. Presumably, the same quality guidelines applied to LVs and other existing, similar gene therapy products may be extended to Cas9-CDVs as well. In any case, rigorous safety and efficacy assessment of Cas9-CDVs will be necessary before future clinical trials.
In conclusion, the regulatory landscape for CDVs harboring CRISPR tools is rapidly evolving, and we can anticipate that the guidelines for their manufacturing and downstream processing will be updated to account for the complexity of CDVs as therapeutic products—following the example set by AAVs produced as vectors for gene therapy. 268 Indeed, the field is hampered by the limitations of current analytical techniques, which are still not capable of providing detailed and in-depth characterization of all product-associated impurities. To bridge this gap, the European Medical Agency and the U.S. Food and Drug Administration published guidelines that promote the adoption of risk-based approaches balancing purity concerns with demonstrated in vivo safety and efficacy. 269 Moving forward, further studies, continuous monitoring of data from ongoing clinical trials, and development of more sensitive analytical techniques will be paramount to establish specific quality parameters and ensure robust safety profiles.
Challenges and future directions
Despite the advantages of pseudotyping for CDV retargeting, significant challenges remain in achieving complete specificity and efficiency. Immune responses to viral proteins, off-target effects, and low target receptor expression can limit the efficacy of pseudotyped vectors. In silico simulations have shown that nanotopography—the detailed nanoscale structure of a molecule, including the number, density, and size of the exposed region of envelope proteins—impacts cellular internalization and could be used in the rational design of future CDVs for biomedical applications. 270
LUNG IN SIGHT: PRESENT AND FUTURE OF TARGETED DELIVERY FOR CF
When the cystic fibrosis transmembrane conductance regulator (CFTR) gene was identified in 1989, 271 –273 the concept of a CFTR gene replacement strategy to treat CF lung disease seemed to be a “low-hanging fruit,” in particular for a target organ as conceptually easy to reach as the lung, which can be accessed directly through aerosol delivery. Unfortunately, more than 30 years later no clinical gene therapy product has reached market approval, and the lung has proven far more challenging to target than originally anticipated. However, the past three decades have witnessed enormous progress in molecular biology and medical science related to CF, which have again brought the possibility of CFTR gene replacement and editing within reach. We now have a greater understanding of CFTR’s biological function throughout the body and of the minimal levels of CFTR expression that are needed to reduce disease severity in the lung. 274,275 Technologically, there have been advances in lung-targeted gene therapy delivery vehicles, as well as major breakthroughs in mRNA manufacturing and CRISPR gene editing. The current technological limitation for CFTR gene therapy appears to be delivering the molecular cargo in sufficient amounts and to the right cell types in the lung.
Present landscape of lung gene editing for CF
Currently, several gene therapy products have reached clinical testing, 276 which represents leading proof-of-concept for lung-cell targeting success and CFTR correction efficacy in preclinical models of CF disease. In general, these gene therapy clinical trials focus on people with CF (pwCF) carrying mutations that are unresponsive or ineligible to market-approved CFTR modulators, and hence, focus on ∼15% of the CF patient population. 277 One first in-human trial is investigating SPL84, an antisense oligonucleotide (ASO) of 19 nucleotides in length that is delivered “naked” (i.e., without a delivery vehicle) to promote splice switching of a deep intronic CFTR splicing mutation, 3849+10 kb C→T. Early preclinical studies showed that the fluorescently labeled ASOs could penetrate the viscous mucus of CF disease lungs, transfect proximal and distal airway epithelial cells in a muco-obstructive mouse model, resist lysosomal degradation, and lead to high functional restoration of CFTR-dependent ion transport. 278,279 Also in Phase II clinical testing is an inhaled mRNA therapy, VX-522, which delivers a full-length CFTR mRNA transcript to lung cells using an LNP. Given the large payload, a delivery vehicle is indeed necessary to shuttle the cargo into the cytoplasm of airway epithelial cells. Likewise, ARCT-032 and RCT2100 are inhaled, LNP-encapsulated CFTR mRNA therapies currently being investigated in a Phase I trial. 276 LNP-mRNA formulations have gained traction in particular owing to their suitability for repeated administrations. However, the CFTR mRNA transcript only serves as a transient template for gene expression, so the drug must be readministered at regular intervals. Delivery of a DNA payload by means of viral vectors, in particular AAVs, holds promise to reduce this need for regular treatment intervals due to the persistence of AAV genomes as episomal elements in the host nucleus throughout the cell lifespan. However, the loss of the viral genome due to cell death, and its dilution due to division of episomes among daughter cells during cell replication—which is necessary for airway epithelium renewal—may still make readministration of the vector necessary. This is challenging, because the first administration elicits production of neutralizing antibodies against the AAV capsid—although strategies to prevent this are being explored. 280 Importantly, several AAV serotypes have proven able to migrate through viscous mucus and efficiently transduce a differentiated air–liquid interface airway epithelial culture from the apical side—the gold standard in therapeutic testing for CF. Viral vector-based candidates currently in clinical testing include SP-101, which uses an AAV2.5T capsid serotype, and 4D-710, which is based on a proprietary, evolved AAV capsid developed by 4D Molecular Therapeutics. 281 –283 Key amino acid substitutions in the AAV capsid structure seem to dictate the disruption of specific mucoadhesive interactions of the AAV serotype — a property that has been extensively studied in airway mucus both in vitro and ex vivo. 282 Another important determinant of AAV transduction efficiency is the prevention of proteasomal degradation of the capsid in the cytoplasm, for example, by cotreatment with doxorubicin. 281 A final consideration put forward is that the delivery vehicle size should be smaller than the predicted pore sizes of the mucus mesh lining CF airways. This is estimated to be around 150 nm (100–400 nm), 284 –286 which can be traversed by the small AAV particles (∼25 nm) and LNPs (∼90–130 nm). 10 However, pretreatment of pwCF airways with mucolytics or mucus-hydrating agents, such as 4.5% NaCl, 287 is expected to remove or loosen the CF mucus mesh, thereby also supporting delivery of particles with larger diameters, such as LVs or CDVs. In that light, a Phase I/II trial is recruiting adult pwCF who are ineligible to receive CFTR modulator treatment with an inhaled Sendai HN/F pseudotyped LV, Lenticlair 1 (BI 3720931). This LV introduces a normal copy of the human CFTR cDNA driven by an exogenous promoter (NCT06515002). Similarly, a clinical trial is planned to nebulize a replication-defective, nonintegrating herpes simplex virus type 1-derived vector engineered to deliver functional, full-length human CFTR to the airways of pwCF (NCT05095246).
To date, no gene editing strategies have yet reached clinical testing for CF, although there are numerous clinical trials ongoing for gene editing therapies to treat cancer, blood, eye, metabolic disorders, and other genetic conditions. 288 However, accumulating evidence in preclinical studies demonstrates that genomic CFTR mutations can be precisely edited. 289 –291 Two important considerations for packaging CRISPR-Cas cargo are (i) the typically large size of the transgene encoding the gene editing enzymes and (ii) the multicomponent nature of the functional gene editing machinery, which is minimally composed by an editing enzyme and an sgRNA. These factors often necessitate packaging and codelivery in different vectors, such as the dual AAV system. 292
RNP-formulated gene editors offer a ready-to-go solution for all-in-one packaging, leveraging the inherent ability of the Cas nucleases to recruit the sgRNA. This supports efficient loading of gene editor RNPs into VLPs during their assembly in producer cells, both for canonical Cas9 and ABEs. 67 –69 For prime editing, instead, enhanced recruitment of the pegRNA is required owing to the lower intrinsic affinity between the pegRNA and the prime editor protein—which is achieved by the use of aptamer-binding proteins. 70
VLP-mediated RNP delivery has not yet been demonstrated for lung delivery, but has been achieved in animal models for delivery to the eye, liver, and brain. 67 –69 Strategies to optimize targeting of the lungs, such as engineering novel pseudotype VLPs with ligands that support directed and preferential uptake into lung cell types of interest for CF (e.g., basal progenitor cells, secretory cells, and ionocytes), are being developed. 293 Given that VLPs have been engineered from retroviral vectors, and thus share with them a near-identical envelope structure, important lessons learned from successful lung-targeting strategies that were developed for retroviral vectors are being applied for VLPs. For example, LVs pseudotyped with Sendai virus (SeV) envelope proteins F and HN were delivered to mouse lung by tracheal instillation and were shown to transduce all major surface airway cell types, including ciliated, club, and goblet cells. Interestingly, basal cell transduction was obtained despite these cells not being in direct contact with the airway lumen, and being instead located at the basement membrane. 294 How airway delivery mechanistically leads to basal cell targeting remains elusive to date. LV pseudotyping with baculovirus glycoprotein gp64 led to apical airway epithelial cell transduction and functional CFTR recovery, although only when the airway epithelial cells were pretreated with 4.5% NaCl, pointing toward receptor location at the basolateral side for this viral envelope. 287
Another important consideration for appointing putative target cells for a given disease, besides being reachable and targetable by a given delivery vehicle, is the nature of the respective target cell. It is quite well possible, in fact, that features such as their terminal differentiation or noncycling nature might impact edibility. In particular, for gene editing techniques that require de novo DNA synthesis, such as prime editing, dNTP levels might be rate limiting, as was already shown for hematopoietic stem cells. 256 These findings might thus be extended to the editing of the progenitor cells in the CF airways. In vitro prime editing of proliferating basal cells has been achieved after both LV delivery and through RNA electroporation, reaching 1–25% of CFTR correction, respectively. 290,291 Also, in vivo, proof-of-concept targeting and editing of basal cells have been shown after systemic delivery of SORT-LNPs loaded with Cre mRNA to transgenic tdTomato reporter mice. 10 Although this route encompasses several extracellular barriers between the blood vessel lumen and the basolateral side of the airway epithelium, these data underscore that—at least in mice—this migration route seems to support access to and editing of basal cells.
Future perspectives for safe and efficient in vivo gene editing of the lung
In the coming years, the ongoing clinical trials aiming to introduce a functional copy of the CFTR mRNA or cDNA or aiming to restore CFTR protein function through ASO-mediated exon skipping will generate invaluable insights for the next generation of gene therapy products. We expect those trials to provide a deeper understanding of the relationship between the number and identity of the corrected lung cells and their level of functional correction ex vivo, and the improvements in clinically relevant endpoints. In addition, we expect to gain insights into the development of innate or adaptive immune responses against delivery vehicles and cargos.
Given the vast surface area of the lung, codelivery of multiple vectors appears impractical. Consequently, it will likely be crucial for gene editing strategies to deliver all-in-one particles that contain the necessary machinery to edit CFTR in a sufficient proportion of the envisioned target cells. These delivery vehicles should be minimally immunogenic, capable of supporting large payloads, and transient to minimize off-targets; moreover, their production pipeline should be easily scalable. While GMPs are well-established for delivery vehicles that have already reached market authorization, such as retroviral vectors, rAAVs, and LNPs, VLP-based systems as well as EV-based ones will require to pave a similar roadmap, based on identification and implementation of crucial steps in their up- and downstream manufacturing process. Several key aspects will need to be considered in the future development of CDVs for the in vivo delivery of gene editors to human tissues, including but not limited to the lung: (i) The reduction of the CDV size to ease migration through the tissues of the body, which can be achieved by engineering smaller CRISPR editors that are packaged into them, 295 or through elimination of redundant components of their cargo recruiting strategies. 4 (ii) The further refinement of the cell-targeting properties of CDVs using viral or synthetic ligands such as nanobodies or single-chain antibodies, 192,199 which should additionally take into account the role of the protein corona as a potential factor influencing cellular tropism in vivo—as has already been demonstrated for lung SORT-LNPs. 10 (iii) The reduction of immune responses induced by vector delivery. As previously discussed, delivering foreign nucleic acid or protein sequences into mammalian cells is anticipated to elicit innate and/or adaptive immune responses. Minimizing their presence in the host cell by using short-lived delivery systems can diminish these responses. Concurrently, stepwise advances to reduce the immunogenic components of both CRISPR therapy delivery vehicles and their cargo are an ongoing endeavor, for example, by rationally engineering CRISPR nucleases to display less immunogenic epitopes. 253
While we ultimately seek to develop an all-in-one, single-application, in vivo gene therapy product to treat CF patients, progress is being made on all aspects of the ideal therapy. The first major milestone will be to demonstrate efficient and safe gene correction in a sufficient number of target cells in the lung. Several clinical trials are currently being conducted for gene therapy products, and meaningfully improved outcomes would represent a major advancement of gene therapy, lung disease, and CF fields. This has the promise to improve the quality of life for the people living with a devastating disease such as CF, and to develop molecular tools that will benefit the advancement of therapies for other lung diseases, genetic disorders, and acquired conditions.
Footnotes
ACKNOWLEDGMENTS
The authors sincerely thank Dr. Thomas Gallagher for his expert editing and valuable suggestions that helped refine this article.
All figures were created in BioRender. The individual figures can be cited as follows: Figure 1 (https://BioRender.com/n38p529), Figure 2 (https://BioRender.com/wg7bbdd), Figure 3 (https://BioRender.com/p62z411), and ![]() (https://BioRender.com/otkda2b).
(https://BioRender.com/otkda2b).
AUTHOR DISCLOSURE
J.G.M. is a member of the scientific advisory board of nChromaBio. The company was not involved in the present work. M.S.C. report speaker fees from Vertex Pharmaceuticals. All other authors declare that they do not have any competing interests.
FUNDING INFORMATION
This research was funded by grants from the