
Abstract
Adeno-associated virus (AAV)-based therapeutics have the potential to transform the lives of patients by delivering one-time treatments for a variety of diseases. However, a critical challenge to their widespread adoption and distribution is the high cost of goods. Reducing manufacturing costs by developing AAV capsids with improved yield, or fitness, is key to making gene therapies more affordable. AAV fitness is largely determined by the amino acid sequence of the capsid, however, engineered AAVs are rarely optimized for manufacturability. Here, we report a state-of-the art machine learning (ML) model that predicts the fitness of AAV2 capsid mutants based on the amino acid sequence of the capsid monomer. By combining a protein language model (PLM) and classical ML techniques, our model achieved a significantly high prediction accuracy (Pearson correlation = 0.818) for capsid fitness. Importantly, tests on completely independent datasets showed robustness and generalizability of our model, even for multimutant AAV capsids. Our accurate ML-based model can be used as a surrogate for laborious in vitro experiments, thus saving time and resources, and can be deployed to increase the fitness of clinical AAV capsids to make gene therapies economically viable for patients.
INTRODUCTION
The adeno-associated viral (AAV) capsid is composed of three structural proteins: VP1 (87 kDa), VP2 (72 kDa), and VP3 (63 kDa), 1 all encoded by overlapping transcripts of the cap gene. Tropism, biodistribution, and capsid assembly are all determined by the capsid sequence. Capsid engineering efforts aim to modify the sequence to improve specificity, biodistribution, and safety. However, many capsid sequence changes are deleterious, reducing the fitness of the capsid and resulting in nonviable or low-yield capsids.
Despite its potential, the widespread adoption of AAV-based gene therapies faces notable challenges, among them, the high cost of goods (CoGS). While early AAV gene therapies 2 were based on naturally occurring AAV serotypes, current work aims to enhance the efficacy and safety, and to reduce costs by developing engineered capsids with improved tissue tropism and specificity. 3,4 AAV engineering studies frequently proceed by screening capsid libraries generated by methods such as peptide insertion or saturation mutagenesis of capsid regions. While this approach has yielded many improved capsids, an exhaustive search across the entire capsid sequence could uncover beneficial properties in previously uncharacterized regions of AAV. However, due to the vast search space—20735 possible combinations for AAV2 VP1—such a search is not feasible using traditional library screening techniques. Computational methods, including machine learning (ML) approaches, have been successfully applied in drug discovery. For example, virtual screening predicts interactions between small molecules and targets across the vast landscape of chemicals and proteins. 5 We reasoned that a similar computational approach could predict the fitness of novel capsid sequences, thereby unlocking new capsid regions for exploration.
ML comprises an ensemble of algorithms that can be trained on datasets to create models capable of generalizing to unseen data. One of the important early applications of ML was in natural language processing (NLP), where statistical models were developed for computers to understand and generate human languages. Protein language models (PLMs) were adapted from NLP and have had success in protein structure prediction and protein engineering. 6 – 9 PLMs treat protein sequences as sentences in which amino acid residues represent words. These models are trained on large protein sequence datasets, learning the “grammar” and “semantics” of protein. The resulting PLMs represent protein sequences as numerical representations or embeddings that capture functional and structural properties of the protein.
We used two models, both trained on the UniRef50 dataset. 10 These models have markedly different architectures. The first model (PLM) uses a transformer architecture based on a multihead attention mechanism. It has no recurrent units and is trained to predict the masked amino acids given the surrounding amino acids. In contrast, the second model (ESM2) uses a multiplicative short-term memory network based on a recurrent neural network architecture for sequence modeling, combining the long short-term memory and multiplicative recurrent neural network architectures and is trained to predict the next amino acid in the sequence. Both models have been shown to encode structural and biologically meaningful information about proteins but, because of their different architectures, they may capture different protein properties. 11 Previous studies have used classical ML to predict capsid fitness (a score that captures multiple facets of AAV quality) 12 and assembly, both of which can be used to reduce the number of nonviable AAV variants in libraries. 13 We reasoned that the addition of a PLM could complement other ML approaches, taking advantage of additional information about protein structure and function.
Here, we present a method for predicting the fitness of AAV2 mutants based on the amino acid sequence of the capsid monomer VP1, which contains all the sequence information for the VP1, VP2, and VP3 gene products. Our custom model architecture is trained on a publicly sourced dataset 14 and leverages an ensemble of models using both PLM and one-hot encoded embeddings. The result is a robust prediction model for AAV fitness. We validated our model on independent datasets and demonstrated that its predictions translate effectively to capsids with mutations across the AAV2 capsid sequence.
METHODS
Predictive model development (Capsid-PLM)
We use the single-point mutant data to train an ML model to predict the fitness of AAV2 mutants utilizing custom Python scripts. 24,25 The fitness score of AAV2 mutants is described in the same method as the original publication for our training data. 14 Specific definitions can be found in Supplementary Equation S1.
After extracting the sequences and fitness scores from the training data, we embedded the sequences using PLM or ESM2 and used these embeddings as the inputs for top model training. We did a randomized 80–20 training-test set split, setting 20% of the embeddings aside for validation of the top model. Using Scikit-learn, we performed a grid search on K-nearest neighbor regression, support vector regression, and random forest regression to see which top model produces the highest Spearman/Pearson correlation with the experimental data on the set aside test set. This same 80–20 split was used across all the later hyperparameter tuning procedures, with k-fold cross-validation search across the training and test set showing little difference in the Pearson and Spearman correlation on the given test set. A summary of the steps taken to train capsid (CAP)-PLM can be found in Supplementary Figure S5.
Validation of CAP-PLM
We validated CAP-PLM using a library of 23 mutants generated by site-directed mutagenesis for which individual infectious titer assays were available. We selected Class A mutants, which had production and infectious titers similar to that of WT AAV2, and Class B nonproducing mutants, and manually parsed the mutations from the original article. We then used our model to predict their fitness scores and compared them using KS-test. Additional validation was performed using a library generated by saturation mutagenesis of positions 561–588 of the AAV2 VP1 sequence for which fitness was measured. 3 Individual variant validation was performed utilizing a random search for high and low fitness score sequences. We compared predicted fitness with experimental fitness using KS-test.
Biological validation of CAP-PLM
ITR capsid plasmids were synthesized by Twist Biosciences, reconstituted in TE buffer, pooled at equivolumetric ratios to create an ITR-capsid plasmid pool. A quadruple transfection method, followed by iodixanol-based density ultracentrifugation, was used to generate a pure AAV pool from the ITR-capsid plasmid pool. Unique barcodes corresponding to specific capsids were sequenced using next-generation sequence from plasmid and AAV pools and quantified using a custom Python-based pipeline. We used the same definition given in the training dataset for the fitness score (Supplementary Equation S1).
RESULTS
Model development and training
CAP-PLM is a discriminative model trained to predict the fitness of an AAV2 single-point mutant from the amino acid sequence alone (Fig. 1A). Building the CAP-PLM is broken down into five steps (Supplementary Table S1): reformatting the data, rebalancing the input data, embedding the sequences, training a regression model, and predicting fitness of a held-out validation set. For steps 2–4, we evaluated the effect of varying various hyperparameters on the model’s performance. To train and assess the performance of different capsid fitness models, we used publicly available AAV2 fitness data. 14 This library is the most comprehensive AAV capsid mutagenesis library available, comprising fitness scores from a pooled next-generation sequence library for all single-point mutations spanning the entire length of the AAV2 cap sequence and including all possible single-point insertions, deletions, substitutions, and stop codon mutations. Because this dataset spans the entire cap gene, 3 it allows us to predict the effect of mutations across the entire gene. We used the same definition given in the dataset for the fitness score (Supplementary Equation S1). We define the fitness score of a particular capsid as the log2 enrichment of the virus against the input plasmid normalized to parental AAV2 log2 enrichment. With this definition, the fitness score of the wild-type AAV2 is 0, and a fitness score of 1 means that a specific mutant produces twice more virus than AAV2, given the same amount of input plasmid.
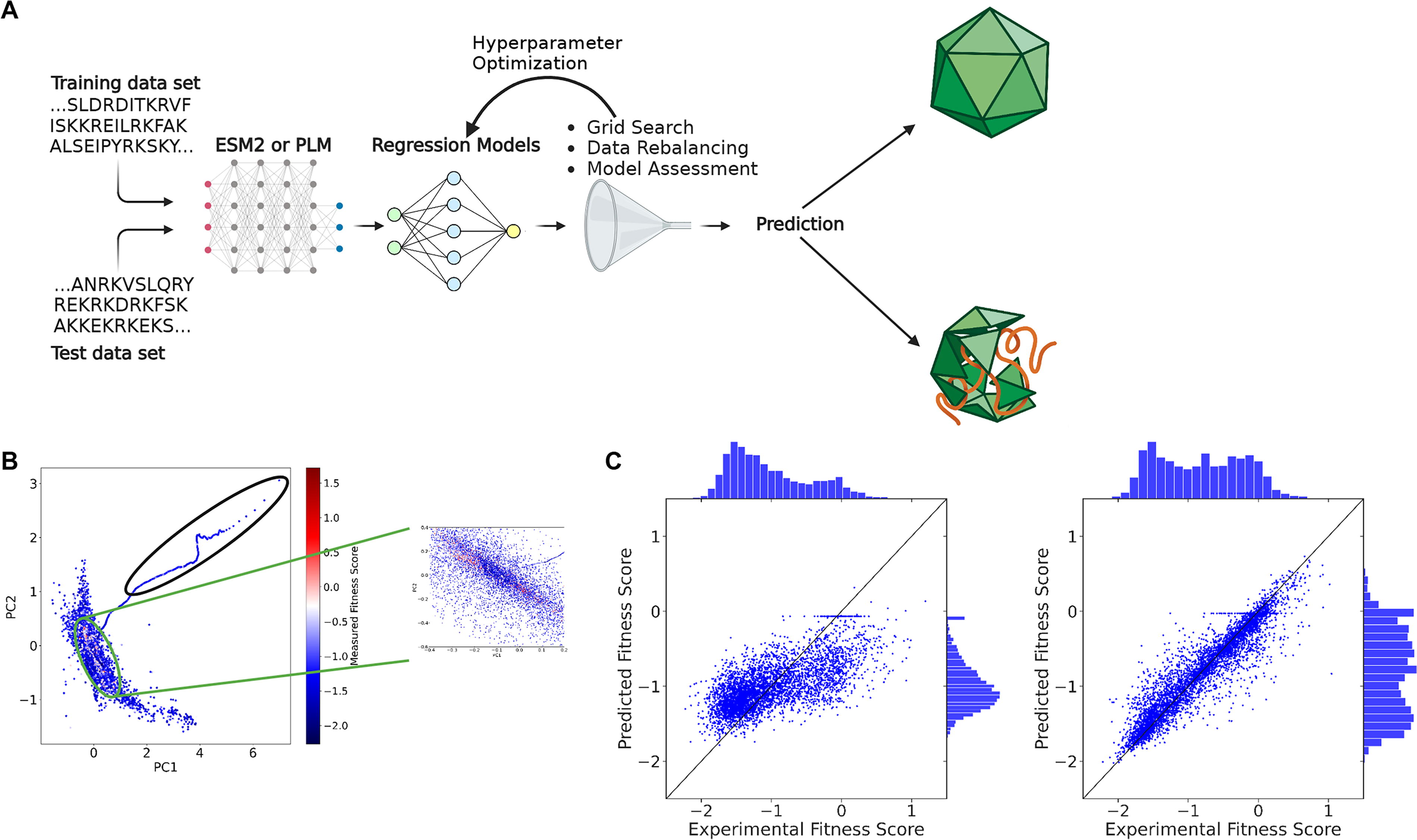
The capsid-protein language model (CAP-PLM) predicts fitness of AAV2 mutants.
Protein embeddings are vector representations encoding structural and functional properties of encodings. PLM’s embeddings have been shown to encode a variety of properties, including structural, physiochemical, and evolutionary information. 6 To show that PLM captures meaningful biological properties of the AAV2 sequence, we conducted principal component analysis (PCA) on the embeddings of AAV2 variants, generated by PLM. We found unsupervised clustering trends related to the fitness of the VP3 region of the AAV2 mutants (Fig. 1B, Supplementary Note S1). Specifically, we found that the PCA representations of the PLM embeddings cluster high-fitness score mutants together and display a distinctive arm containing stop-codon mutants near the beginning of VP3. Since PLM effectively captures unsupervised biological relationships in the AAV2 single-point mutation dataset, we chose to use PLM embeddings for our downstream model development.
To train our model, we randomly set aside 20% of the input dataset as the validation data and used the remaining 80% as training data. After training, we tested the model’s ability to predict the fitness of the held-aside test set by calculating Pearson correlation between the prediction and the experimental data (Fig. 1C), testing the effect of varying hyperparameters on model performance. Hyperparameters that improved the model were selected and incorporated stepwise into subsequent model iterations (Supplementary Fig. S1, Supplementary Table S2). Based on the results of our PCA (Fig. 1B), we began by exploring removing stop codon sequences from the input data for model training, but this did not improve model performance (Supplementary Fig. S1). To determine whether the VP3 sequence, which comprises a subsection of the VP1 sequence and roughly 5/6 of the proteins in a full capsid, 15 encodes comparable information with the VP1 sequence, we compared the model performance trained by using the full VP1 sequences with that of a model trained using only the VP3 sequences. The result showed 6% improvement on Pearson correlation of the predicted fitness against the experimental fitness by using VP1 sequences (Supplementary Fig. S1; Pearson 0.721 vs. 0.678). As 55% of the fitness scores of the input data fell between −0.5 and −1.5, we reasoned that the model would be likely to overfit on low fitness scores. To combat this, we rebalanced the input data by randomly removing 5,095 sequences with scores between −0.5 and −1.5 (about 1/6th of the total) (Supplementary Fig. S2, Supplementary Table S3, Supplementary Note S2). This further improved the model by 5% (Supplementary Fig. S1; Pearson correlation of 0.755 vs. 0.721). Then we tested different regression models, including ridge, K-nearest neighbors, radius nearest neighbors, support vector regression, nu support vector regression, random forest regression, and decision tree regression, and found that a random forest regression model outperforms others (Supplementary Fig. S3).
One-hot encoding is commonly used to encode protein sequences and has previously been combined with evolutionary data to improve protein fitness prediction. 16 One-hot encoding represents each amino acid as a binary vector, thereby representing the categorical variable of an amino acid as a numerical value. While PLM embeddings can encode information about functional and structural properties of a protein, one-hot encoding provides only positional properties. We hypothesized that combining one-hot encoding with PLM embeddings might improve the representation. To determine if a composite model trained on both PLM and one-hot encoded embeddings could outperform the one trained on PLM embeddings alone, we used the best-performing random forest model to predict the fitness scores for the training set and used the differences between this prediction and the ground truth as labels for a second training scheme using one-hot encoded sequences as input. This improved the Pearson correlation by another 8% (Supplementary Fig. S1; Pearson correlation of 0.755 to 0.818). Overall, we saw a 20% increase in Pearson correlation over the course of these hyperparameter improvements.
Finally, we evaluated the performance of two pretrained PLMs, PLM and ESM2 with a downstream model on the fitness prediction task. Interestingly, although transformers in principle capture global dependencies better through self-attention mechanisms, we found that the PLM-trained model had a slight advantage in Pearson correlation of 2%, in line with observations from other use cases 17 (Supplementary Fig. S1; Pearson correlation of 0.818 vs. 0.805). Therefore, our final model (Fig. 1D) comprises two random forest models trained on the PLM and one-hot encoded embeddings, respectively. To allow for large insertions into the VP1 sequences, we permitted an input of up to 755 amino acids.
Validation of model on additional datasets
To test the robustness and determine if the predictive power of the CAP-PLM extends beyond the single-point mutant data on which it was trained, we tested the method against three additional datasets, a library of 93 AAV2 mutants generated by site-directed mutagenesis of 48 approximately evenly spaced sites in the cap gene, 18 a library of capsids generated by saturation mutagenesis of positions 561–588 of the AAV2 VP1 sequence, 3 and an internally randomly generated library of AAV2 mutants with 2–3 mutations. These three datasets contain multiple-point mutations; therefore, we chose to validate CAP-PLM with these multiple-point mutations that are not contained within the single amino acid mutations in our original training/test dataset.
For the first dataset, we analyzed two types of mutants: class A (n = 13) mutants were fully competent and had titers comparable with wild-type AAV2, while class B mutants (n = 8) were nonviable. We found that the predicted fitness scores of the class B (nonviable) mutants were significantly (KS-test, p < 0.005) lower than those of the class A (viable) mutants (Fig. 2A). Therefore, despite being trained on a single mutation dataset, CAP-PLM is robust enough to extrapolate to capsids harboring more than one mutation.
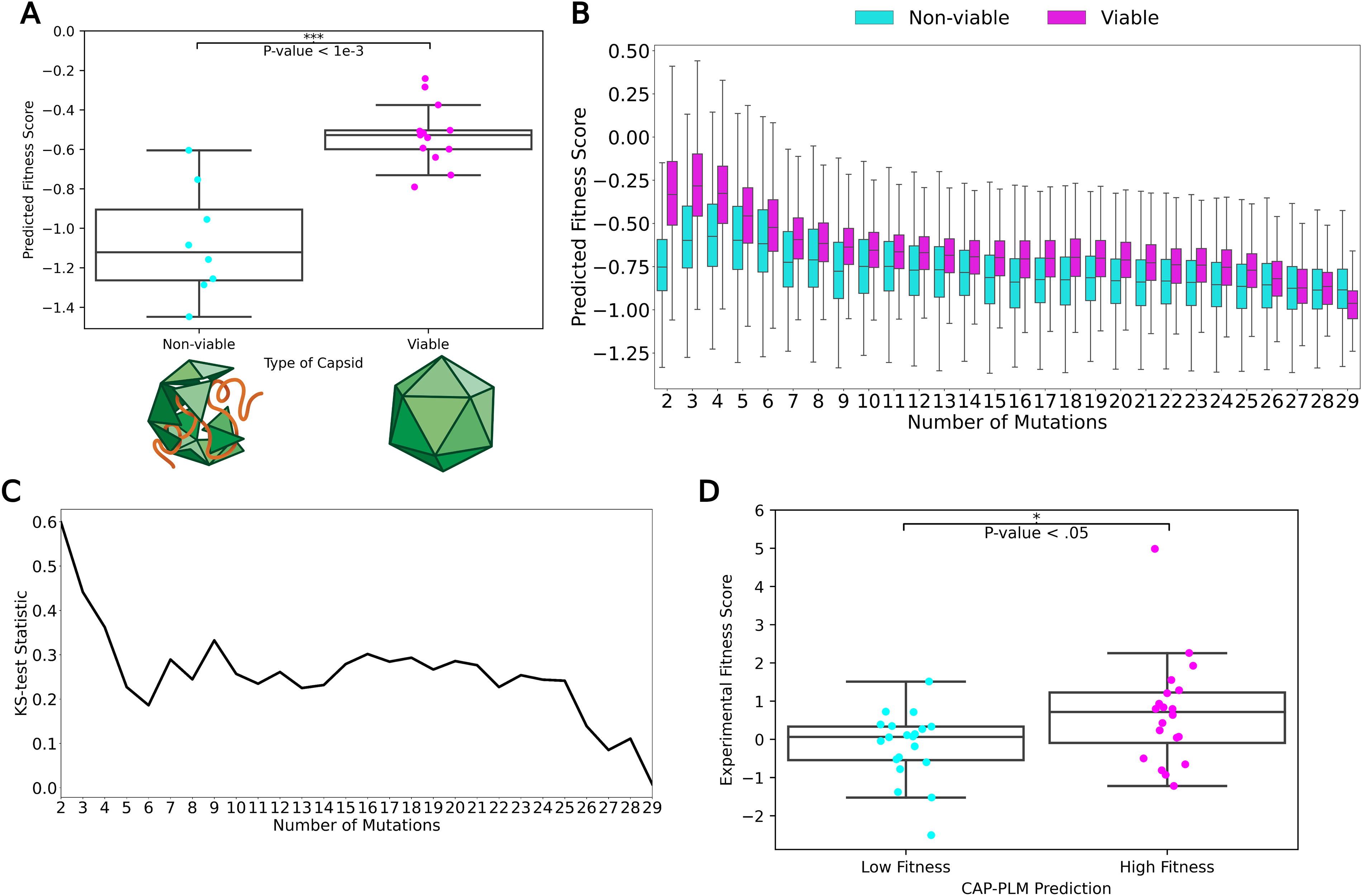
CAP-PLM generalizes to predictions on fitness of AAV2 multiple-mutation mutants.
Next, we tested CAP-PLM on a more comprehensive dataset, one containing 201,426 mutants between 2 and 39 mutations away from the wild-type AAV2 capsid sequence. We divided these mutants by number of mutations into viable or nonviable mutants as assigned by the original publication and compared the two classes by their predicted fitness scores (Fig. 2B). Predicted fitness scores of nonviable mutants with fewer than 25 mutations were significantly lower than for viable mutants by KS-test (FDR-corrected KS-test, p < 0.05), but the test statistic of the comparison decreases as the number of mutations increased (Fig. 2C). Prediction power drops and stabilizes after four mutations, becoming unreliable after 25 mutations suggesting that the prediction power of CAP-PLM decreases with increasing numbers of mutations. Thus, CAP-PLM can make accurate predictions on a variety of mutants that are at a considerable distance from those in the training dataset.
Finally, we tested CAP-PLM on an internally generated set of AAV2 sequences. We generated 100 sequences each 2–3 mutations away from the wild-type sequences. As the predicted fitness of these sequences cluster tightly around 0 (equivalent to wild-type AAV2), we needed to create two distinct classes; therefore, we took the top and bottom 20% of in silico generated sequences and separated them into high and low predicted fitness classes. Then, we manufactured the sequences and compared the experimental fitness scores of the two classes. Experimental fitness scores of the low and high fitness classes were significantly different by KS-test (p < 0.05), confirming the real-world applicability of our approach to fitness prediction. (Fig. 2D) In conclusion, CAP-PLM is a flexible and extensible framework, which is a strong predictor even on multimutant AAV2 fitness. Furthermore, CAP-PLM is generalizable to make predictions across the entire AAV VP1 sequence.
DISCUSSION
We have established a state-of-the-art ML model that accurately predicts the fitness of AAV2 capsid mutants from the amino acid sequence of VP1. Using this model, we can rapidly analyze far more sequences than can be screened using biological assays, filtering out inviable sequences, reducing costs, and improving efficiency in capsid library designs. As these dead mutants represent wasted library space, this increases the chance that a given library will contain variants with biological properties of interest.
We trained our model on a library of AAV2 mutants, the most comprehensive AAV2 capsid mutagenesis library publicly available at the time. However, the model should be flexible enough to provide a framework that could be extended to other clinically relevant AAV serotypes. This would only require an initial investment in a single-mutant library for training and testing of the model. Such a library could be leveraged to explore additional capsid properties, making the investment potentially valuable. In addition, regions or mutations that are associated with higher manufacturability in AAV2 might be grafted onto other AAV serotypes as has previously been shown for other properties. 19 Finally, with additional tuning, optimization, and inputs, the model might be extended to predict additional biological properties.
Our choice of model was driven by the application of NLP to biological problems. PLM has been shown to perform well on stability and function prediction, while ESM2 performs well on structure prediction. However, other PLMs, such as Deep SequenceVAE 20 or EVmutation, 21 have been shown to outperform PLM and ESM1b, 8 an earlier version of ESM2, on fitness prediction for a variety of different proteins. 16 It is likely that different models encode different information and have their own inductive biases, 22 which are a function of which proteins are used for training and how they are represented in the model. These biases can affect performance on specific downstream tasks. In particular, they can be biased toward sequences that are close to their training data. Therefore, it is possible that a PLM trained on viral sequences might perform better on this task. However, our results suggest that this is not necessary.
In developing this model, we prioritized a lightweight model with reduced potential for overfitting. Therefore, we used a classical ML top model rather than a neural network. This architecture is suitable for our relatively small dataset. However, it may not be scalable to larger training datasets, for prediction of more complicated capsid properties, or for prediction of multiple properties. A neural network-based architecture could potentially improve the scalability of the model and allow it to predict more complex capsid properties. Indeed, neural network-based architectures have been used to predict capsid assembly and to generate diverse viable sequence variants, 3,12 but these methods did not incorporate PLMs. The combination of a PLM that is trained off implicit fitness constraints present in known protein sequences, with site-specific fitness, can outperform either method alone.
While this predictive model is useful on its own, combining it with a generative model would be a consequential leap forward in AAV engineering. At present, most AAV library designs focus on known regions of the AAV capsid sequence. However, a generative model could be used to design a capsid library agnostic to known AAV biology, expanding the sequence space that could be explored for potentially therapeutically useful capsids while ensuring that libraries are not dominated by nonviable capsids. Variational autoencoders have been used to a degree of success in the past for optimizing AAV production. 23 Furthermore, capsids generated by such a model might exceed the parental capsid in fitness, which, in the long term, could contribute to lowering the CoGS for AAV gene therapies, making them more affordable and accessible. Finally, capsids generated by such a model might elucidate the functions of previously unexplored regions of capsid genomes. The predictive model described herein is a critical first step toward solving key issues in AAV engineering.
Footnotes
ACKNOWLEDGMENTS
AUTHORS’ CONTRIBUTIONS
J.W., Y.Q., E.L., and S.R.C.: Conceptualized the study. J.W.: Curated the datasets, designed and implemented the models, and designed the figures. J.W., Y.Q., E.L., and S.R.C.: Contributed to data interpretations. M.J.R. and T.T.: Performed the biological validation and next-generation sequencing. J.W. and E.L.P.: Drafted the article, contributed feedback to figure generation, and revised and edited the article. C.M. and S.R.C.: Supervised the project and provided feedback on the article with all the authors.
AUTHOR DISCLOSURE
All authors are present or past employees of Sanofi and may hold shares and/or stock options in the company.
FUNDING INFORMATION
This study was funded by Sanofi.
SUPPLEMENTARY MATERIAL
Supplementary Equation S1
Supplementary Note S1
Supplementary Note S2
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.