
Abstract
Weaver syndrome is a rare neurodevelopmental disorder that encompasses macrocephaly, tall stature, obesity, brain anomalies, intellectual disability, and increased susceptibility to cancer. This dominant monogenic disorder is caused by germline variants in enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2), a key epigenetic writer. Unfortunately, there are no effective treatments for Weaver syndrome. However, preclinical results support the potential for therapeutic gains, despite the prenatal onset. Thus, for the first time, we tested whether CRISPR/Cas9 gene-editing strategies may be able to “correct” a Weaver syndrome variant at the DNA level. We initiated these preclinical studies by humanizing the region surrounding the most-common recurring patient-variant location in mouse embryonic stem cells (ESCs). Humanization ensures that DNA-binding strategies will be directly translatable to human cells and patients. We then introduced into ESCs the humanized region, but now carrying the Weaver syndrome EZH2 variant c.2035C>T p.Arg684Cys, and characterized the enzymatic properties of this missense variant. Our data showed a significant and dramatic reduction in EZH2-enzymatic activity, supporting previous cell-free studies of this variant as well as in vitro and in vivo mouse work by other teams. Intriguingly, this most-common variant does not create a complete loss-of-function, but rather is a hypomorphic allele. Together with prior reports describing hypomorphic effects of missense EZH2 variants, these results demonstrate that the etiology of Weaver syndrome does not require complete loss of EZH2 enzymatic activity. Toward therapy, we tested four CRISPR gene-editing strategies. We demonstrated that Streptococcus pyogenes Cas9 (SpCas9) showed the highest variant correction (70.5%), but unfortunately also the highest alteration of the nonvariant allele (21.1–26.2%), an important consideration for gene-editing treatment of a dominant syndrome. However, Staphylococcus aureus Cas9 (SaCas9) gave a variant correction (52.5%) that was not significantly different than SpCas9, and encouragingly the lowest alteration of the nonvariant allele (2.0%). Thus, the therapeutic strategy using the small SaCas9 enzyme, a size that allows flexibility in therapeutic delivery, was the most optimal for targeting the Weaver syndrome EZH2 variant c.2035C>T p.Arg684Cys.
INTRODUCTION
Weaver syndrome is a rare neurodevelopmental disorder that causes macrocephaly, tall stature, obesity, brain anomalies, intellectual disability, and increased susceptibility to cancer (OMIM #277590). It is a dominant monogenic disorder caused by germline variants in the gene “enhancer of zeste 2 polycomb repressive complex 2 subunit” (EZH2), the human ortholog of the mouse Ezh2 and Drosophila E(z) genes. 1 EZH2 is a key epigenetic writer, acting as a catalytic subunit of polycomb repressive complex 2 (PRC2). 2,3 PRC2 plays a role in transcriptional repression by adding up to three methyl groups onto histone H3 lysine residue 27 (H3K27). Methylation of H3K27, in particular to H3K27me3, has epigenome-wide effects across multiple developmental stages among many cell types and organ systems. 1 –3
Currently, there is no effective treatment or cure for Weaver syndrome. However, preclinical results support the potential for positive gains from therapy, despite the prenatal onset of this disorder. For example, adding back EZH2 to null human cells restored H3K27me3 levels and profiles, demonstrating that histone patterns are appropriately reestablished. 4 Furthermore, inhibition of H3K27 demethylases (a strategy predicted to increase the amount of H3K27 methylation) substantially reversed the excessive osteogenesis observed in Ezh2 mutant cells. 5 Thus, we hypothesize that treatment would improve neurodevelopmental trajectories as well as reducing the lifetime risk of common diseases, such as leukemia and diabetes, in these patients.
For the first time, we explore here the efficacy of CRISPR/Cas9 therapies as proof-of-principle to “correct” a Weaver syndrome pathogenic variant at the DNA level. Notably, for optimizing DNA- and RNA-binding therapies such as CRISPR, it is important to first “humanize” the region surrounding the variant location in the mouse ortholog with canonical human sequence. 6 This “minimally humanized” approach allows CRISPR guides and templates to be designed to bind human DNA and use human protospacer adjacent motifs (PAMs) such that treatment will be directly translatable from preclinical mouse cell or animal models to human cells and patients. For this study, we introduced the most common recurring missense pathogenic Weaver syndrome patient variant c.2035C>T p.Arg684Cys 7 –9 into minimally humanized Ezh2. As is typical, this is a loss-of-function variant. However, for missense variants such as this one, competing mechanistic hypotheses have been proposed based on the bias among clinical samples toward missense variants versus frameshift or gene deletions. 5,10 Thus, the aim of this project was to generate a series of mouse embryonic stem cell (ESC) lines minimally humanized around the variant site in exon 18, to further characterize the variant, and to test several CRISPR gene-editing strategies as potential therapeutic approaches for Weaver syndrome.
MATERIALS AND METHODS
Design and optimization of CRISPR components for developing humanized cell lines
The online tool Benchling (https://benchling.com/crispr) was used to design guide RNAs using the Streptococcus pyogenes Cas9 (SpCas9) nuclease, to introduce the “humanization” region, using two double-stranded breaks in Ezh2 surrounding exon 18. Guides were selected based on the predicted on-and off-target effects, and the corresponding sense and antisense templates designed for the humanization of exon 18 with and without the introduction of the pathogenic patient variant of interest, c.2035C>T p.Arg684Cys rs587783626 (from NM_001203247.2; c.2050C>T from MANE transcript NM_004456.5 for EZH2). Single-guide RNAs (sgRNAs) were all synthesized with chemical modifications (2’-O-methyl and phosphorothioate bonds at the first two 5′- and 3′-terminal RNA residues) (Synthego, Menlo Park, CA). Single-stranded oligodeoxynucleotide (ssODN) templates were synthesized with 100 bp mouse homology on each side of the 217 bp humanization region (Integrated DNA Technologies (IDT), Coralville, IA).
Male Ezh2 wild-type (Wt) ESCs (mEMS6131) 6,11 were derived as previously described, 12 from a C57BL/6NTac mouse (Taconic, Hudson, NY). Using the 10 µL Neon transfection system (Thermo Fisher Scientific, Waltham, MA) to deliver CRISPR components to ESCs as previously described, 6,13 the top two 5′ and 3′ sgRNAs were tested in combination with four ssODN templates. First, a ribonucleoprotein (RNP) complex was formed by combining SpCas9 (PNA Bio, Thousand Oaks, CA) with the sgRNAs, separately, by incubating at room temperature (RT) for 15 min. ssODN template was added to the RNP complex, and 3 × 105 ESCs grown on gelatin were used per reaction, using the Neon optimization setting 14 (1200V, 20 ms, 2 pulses). Final concentrations of reagents were as follows: sgRNA (each) 1.2228 µM, SpCas9 0.614 µM, and ssODN 100 nM. Electroporated ESCs were plated on a fresh 24-well plate on gelatin and incubated for 48 h, and cells were lysed in a tissue homogenization buffer (THB) as previously described. 13 The optimal combination of sgRNA and ssODN sequences is detailed in Supplementary Table S1, as determined by PCR using the primers detailed in Supplementary Table S2.
Picking and screening single clones to produce humanized nonvariant, humanized patient variant, and knockout ESC lines
ESCs (mEMS6131) grown on mouse embryonic fibroblasts (MEFs) were then electroporated with the optimized guides/templates (Supplementary Table S1), and directly plated onto 6 cm dishes. Individual clones were picked and plated onto 96-well plates on MEFs, until confluent. At that time clones were passaged onto two sister plates, one on MEFs for freezing and one on gelatin for PCR. Once ESCs were confluent, cells were either frozen using ESC-freeze media, 12 or lysed in THB as described above.
Individual ESC clones were screened using Taq1 polymerase-driven PCR and primers as listed in Supplementary Table S2. In addition, restriction fragment length polymorphism (RFLP) assays were developed to confirm the presence or absence of the variant, using enzymes AciI and BfuAI (New England Biolabs, Ipswich, MA) (Supplementary Table S3). Selected clones showed: no editing events (Wt/Wt), knockout (KO) deletion between the cut sites (KO/KO), or passed all screening assays for humanization (He18/He18). Cell lines were thawed from the 96-well freeze plate and expanded further on MEFs for freezing, and on gelatin for further sequencing, immunoblotting, and variant-correction experiments. DNA samples were purified using the PureLink PCR Purification Kit (ThermoFisher), and sent for bidirectional Sanger-sequencing to confirm Wt/Wt, the presence or absence of the variant in humanized clones (He18+/He18+ or He18−/He18−), respectively, or KO/KO. Humanization clones were then further sequenced at least 315 bp upstream and downstream of the homology arms to confirm no further editing events occurred surrounding the humanization region.
Immunoblotting
Immunoblotting was performed as per Mochizuki et al (2021), 14 with the following modifications: Cells were lysed in RIPA buffer containing 150 mM NaCl and protein extracts separated via 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Membranes were blocked with 5% bovine serum albumin (BSA) in tris buffered saline (TBS) for 1 h at RT, and probed with primary antibodies to both H3K27me3 (Cat#9733, Cell Signaling, Danvers, MA; 1:1,000) and total histone H3 (H3) (Cat#3638, Cell Signaling; 1:2,000) in 1% BSA in tris buffered saline with tween 20 (TBST) overnight at 4°C with agitation. Blots were probed with both IRDye 800CW goat anti-rabbit (Cat#925–32211, LI-COR; 1:10,000) and IRDye 680RD goat anti-mouse (Cat#925–68070, LI-COR; 1:10,000) secondary antibodies in 1% BSA in TBST for 1 h at RT with agitation. Protein band intensities were quantified using densitometric analysis in Image Lab Software (v6.1, Bio-Rad Laboratories, Inc.).
CRISPR therapeutic patient variant correction in ESCs
sgRNA and ssODN templates for the pathogenic patient variant c.2035C>T p.Arg684Cys correction were designed using Benchling as above. sgRNAs had the variant site toward the 3′ end, and ssODN templates had 40 bp flanking homology arms. The ssODNs were designed not only to correct the patient variant, but also with single-base “blocking” mutation(s) at and around the PAM site. Blocking mutations were selected to preserve the amino acid and match codon usage in mouse and human via the GenScript Codon Usage Frequency Table Tools (www.genscript.com).
The top sgRNA and corresponding ssODN sequences are shown in Supplementary Table S4 and were used in combination with SpCas9 or Staphylococcus aureus Cas9 (SaCas9; Aldevron, Fargo, ND). The sgRNA scaffold for the SaCas9 guide (cgWTG8) was from Ran et al (2015). 15 The four conditions were electroporated as described above, with He18−/He18− (mEMS6677) ESCs and He18+/He18+ (mEMS6713) ESCs, and a final concentration of reagents as follows: sgRNA 1.2228 µM, Cas9 0.614 µM, ssODN 6 µM. Electroporated ESCs were plated on a fresh 24-well plate on gelatin and incubated for 48 h. Cells were lysed using TBH before Sanger sequencing to quantify intended base changes and off-target edits.
Sanger sequence peak quantification
Relative peak height data were extracted from chromatograms using EditR (www.moriaritylab.shinyapps.io/editr_v10/). At sites of interest, the peak height data were normalized to the mock (electroporated, but with no reagents) to determine the efficiency of CRISPR editing.
Statistical analysis
Statistics were performed with GraphPad Prism (v10.2.0, 10.3.1, and 10.4.0 for Windows).
RESULTS
Generation of humanized Ezh2 exon-18 patient variant and knockout cell lines
To generate minimally humanized EZH2 cells, we used CRISPR editing to replace a single exon of mouse Ezh2 with human EZH2 in mouse ESCs (Fig. 1). More specifically, we edited mouse Ezh2 exon 18 and the adjacent-intronic regions to exactly match the canonical version of human EZH2 from nucleotides 74878 to 75094, based on the human EZH2 sequence (ENSG00000106462) (Fig. 1A). While humanization of exon 18 changes the nucleotide sequence, the amino acid sequence remains unaltered between human and mouse EZH2. In addition, we also generated exon 18 humanized mouse ESCs with the addition of the pathogenic Weaver syndrome variant c.2035C>T p.Arg684Cys (Fig. 1B). 8 To achieve humanization, optimized CRISPR sgRNAs and ssODNs (Supplementary Table S1), along with SpCas9, were electroporated into ESCs. Due to the complicated nature of the replacement event, a large number of clones were picked per electroporation to increase the likelihood of recovering clones with the desired genotypes. To obtain a clear understanding of the editing events in each ESC clone, we designed genotyping assays that included the target site and flanking genomic regions (Supplementary Table S2). In addition, RFLP assays were designed to distinguish cell lines with and without the variant (Fig. 2A, B, Supplementary Table S3), and sequencing confirmed the humanization and variant events (Fig. 2C), the lack of unwanted events surrounding the humanization, and deletions. Clones of four genotypes were obtained: Wt/Wt (unedited); He18+/He18+ (humanized exon 18 nonvariant); He18−/He18− (humanized exon 18 with the variant); and KO/KO (deletion between the two sgRNA cut sites). Table 1 details the nine cell lines used in this study (two–three of each genotype). All were independent, established from clones on different plates, except 6752 and 6754, which were thus not used in the same experiment. Sequencing results were as expected for all clones, except mEMS6713 (He18+/He18+ ), which included a low level of mouse sequence with abnormalities (∼12% based on peak height), presumably due to cellular contamination with an abnormal clone; and mEMS6687 (KO/KO) that contained an extra 10 bp of deletion at the cut sites.
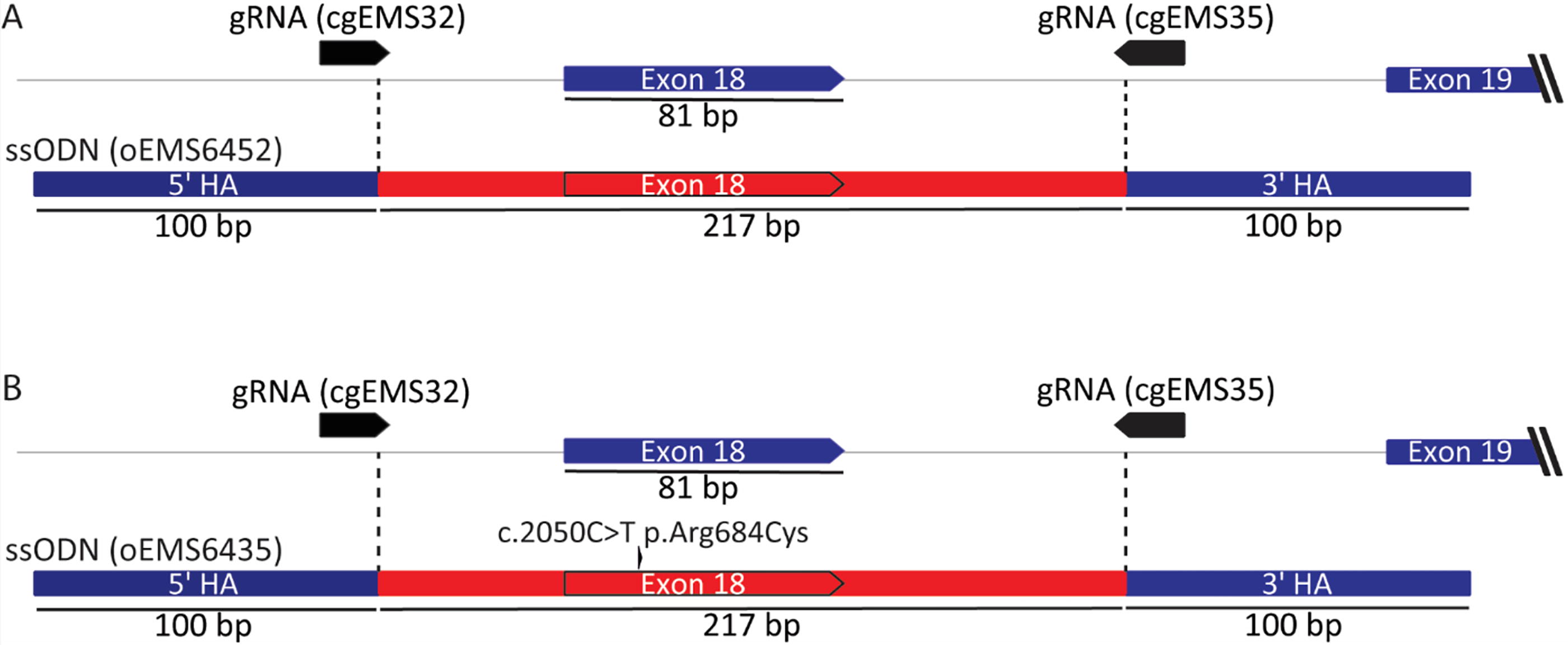
Strategy to derive humanized Ezh2 exon-18 mouse ESCs with and without the pathogenic patient variant c.2035C>T p.Arg684Cys.
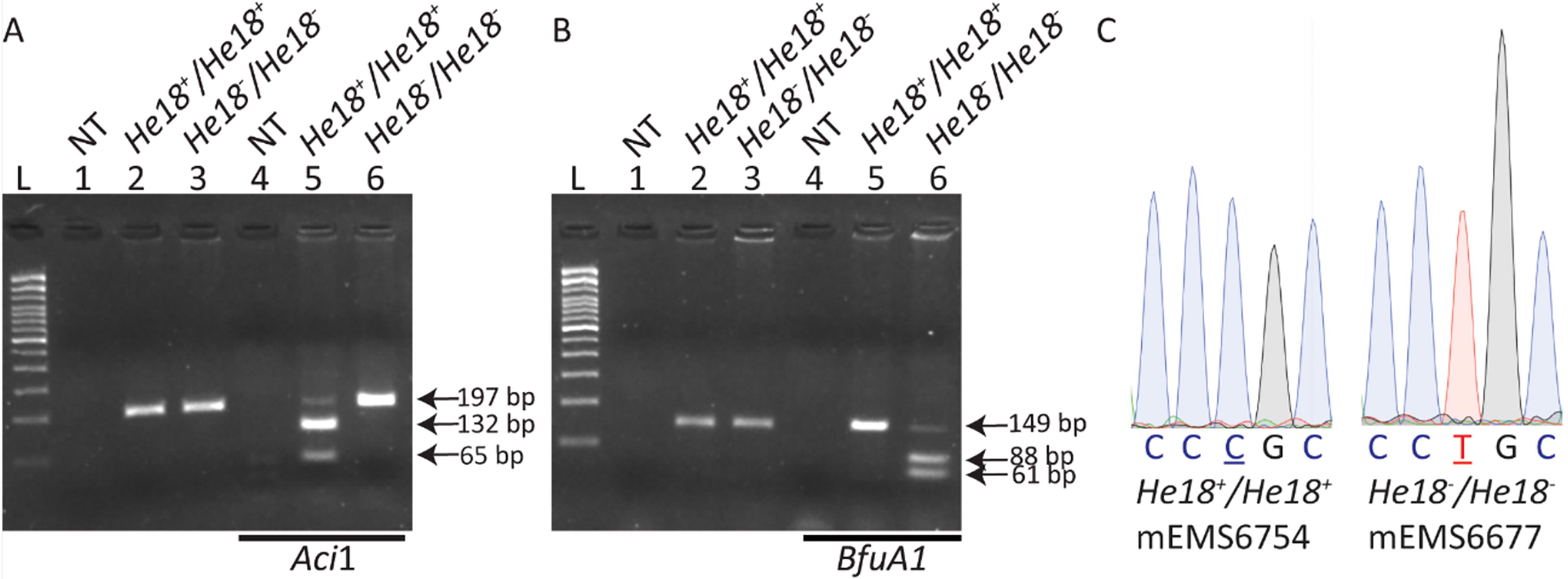
Identification of homozygous humanized Ezh2 exon-18 mouse ESCs with and without the pathogenic patient variant c.2035C>T p.Arg684Cys.
Embryonic Stem Cell Lines Generated
mEMS, mammalian cell lines; Wt, wild type; He18 + , humanized exon 18 nonvariant; He18 − , humanized exon 18 variant.
EZH2 variant led to significantly decreased but detectable H3K27me3 levels
To determine whether the genetic changes introduced at the EZH2 locus impacted its histone methyltransferase activity, we measured H3K27me3 levels by Western blot in eight ESC lines described above (Table 1, Fig. 3). Three replica Western blots were performed (Fig. 3A). H3K27me3 and total H3 protein levels were determined via 2-color fluorescence, quantified using densitometric analysis, and H3K27me3 levels normalized to total H3 protein. Statistical analysis was used to address three questions (Fig. 3B). We were surprised that a comparison of Wt versus humanized cell lines revealed that humanization alone reduced EZH2-protein function (Wt/Wt vs. He18+/He18+ ). A possible explanation is that to maintain a canonical human sequence for CRISPR studies we could not codon optimize the human sequence, which may have led to reduced translation efficiency of the mRNA. We then compared humanized cell lines with and without the pathogenic patient variant c.2035C>T p.Arg684Cys, which resulted in a significant reduction in protein function due to the variant (He18+/He18+ vs. He18−/He18− ). The final comparison revealed significantly greater protein function with the variant versus the knockout cells (He18−/He18− vs. KO/KO). Although faint, the H3K27me3 band observed from the variant cell lines is evidence of a hypomorphic protein with detectable enzymatic activity, in contrast to the undetectable activity within the KO cells.
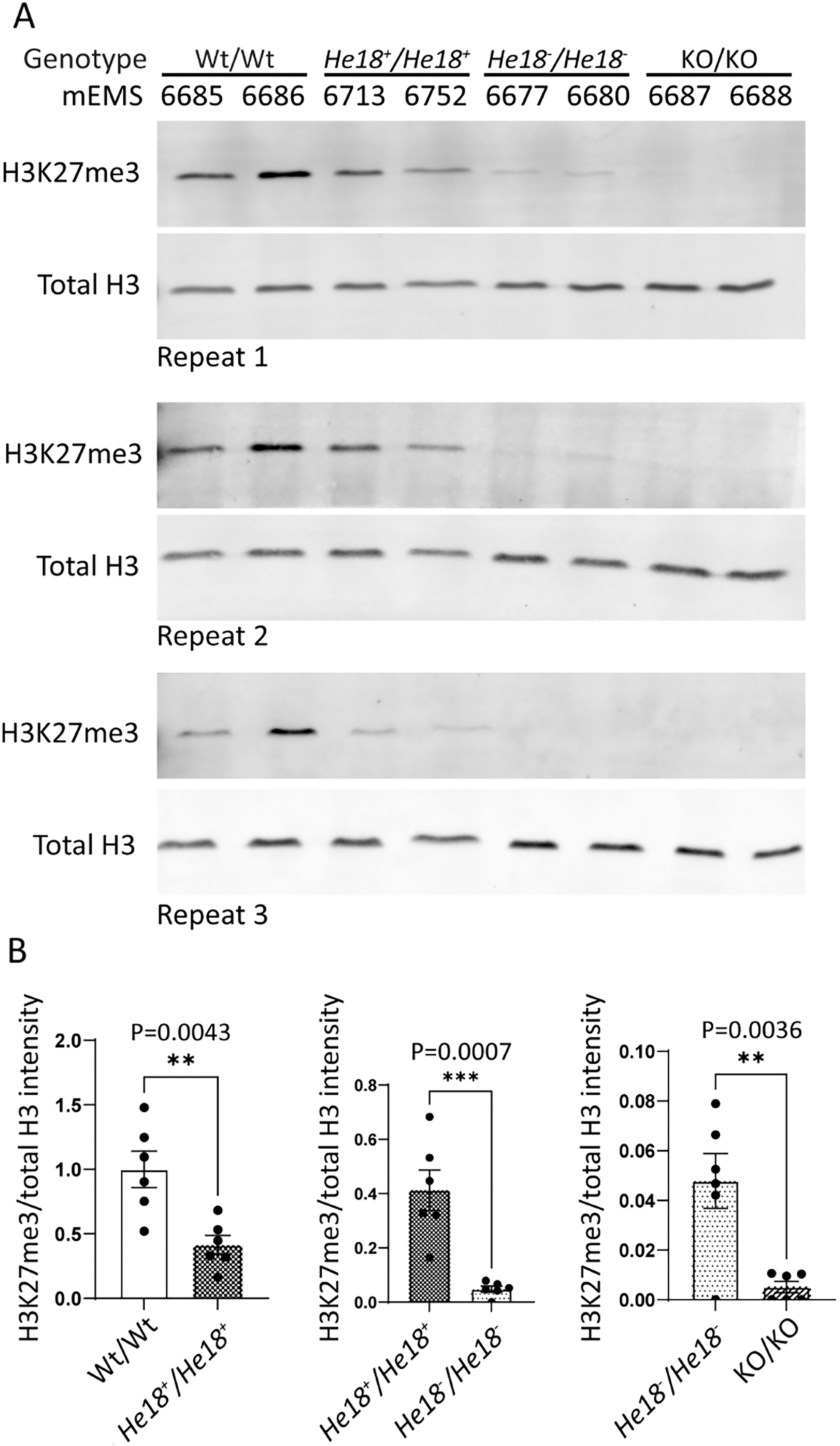
H3K27me3 was significantly impacted by Ezh2 humanization, and by the pathogenic variant, yet demonstrated some residual activity (i.e., hypomorphism).
CRISPR therapy strategies corrected the EZH2 exon-18 patient variant at rates ranging from 43.3% to 70.5%
To determine the best strategy for therapeutic correction of the EZH2 pathogenic patient variant c.2035C>T p.Arg684Cys in humanized ESCs, four CRISPR strategies were designed and implemented (Fig. 4A). We considered a variety of CRISPR options. Notably, the adenine base editor could not be used due to the unsuitable location of the nearest PAM. 16 Therefore, we used the canonical SpCas9, which is already being used in clinical therapies, 17 as well as the smaller SaCas9, 15 which allows more flexibility of therapeutic delivery methods. sgRNAs and ssODNs were designed to correct the variant. In addition, single-base “blocking” mutations that did not change the amino acid were introduced to reduce guide rebinding (and thus retargeting) by Cas9. 18 The blocking mutations also allowed for comparative quantification of base-pair alterations of the variant allele in He18−/He18− cells and the nonvariant allele in He18+/He18+ cells. Blocking mutations were introduced either at the PAM site or within 2–5 bp of the variant mutation site (Supplementary Table S4).
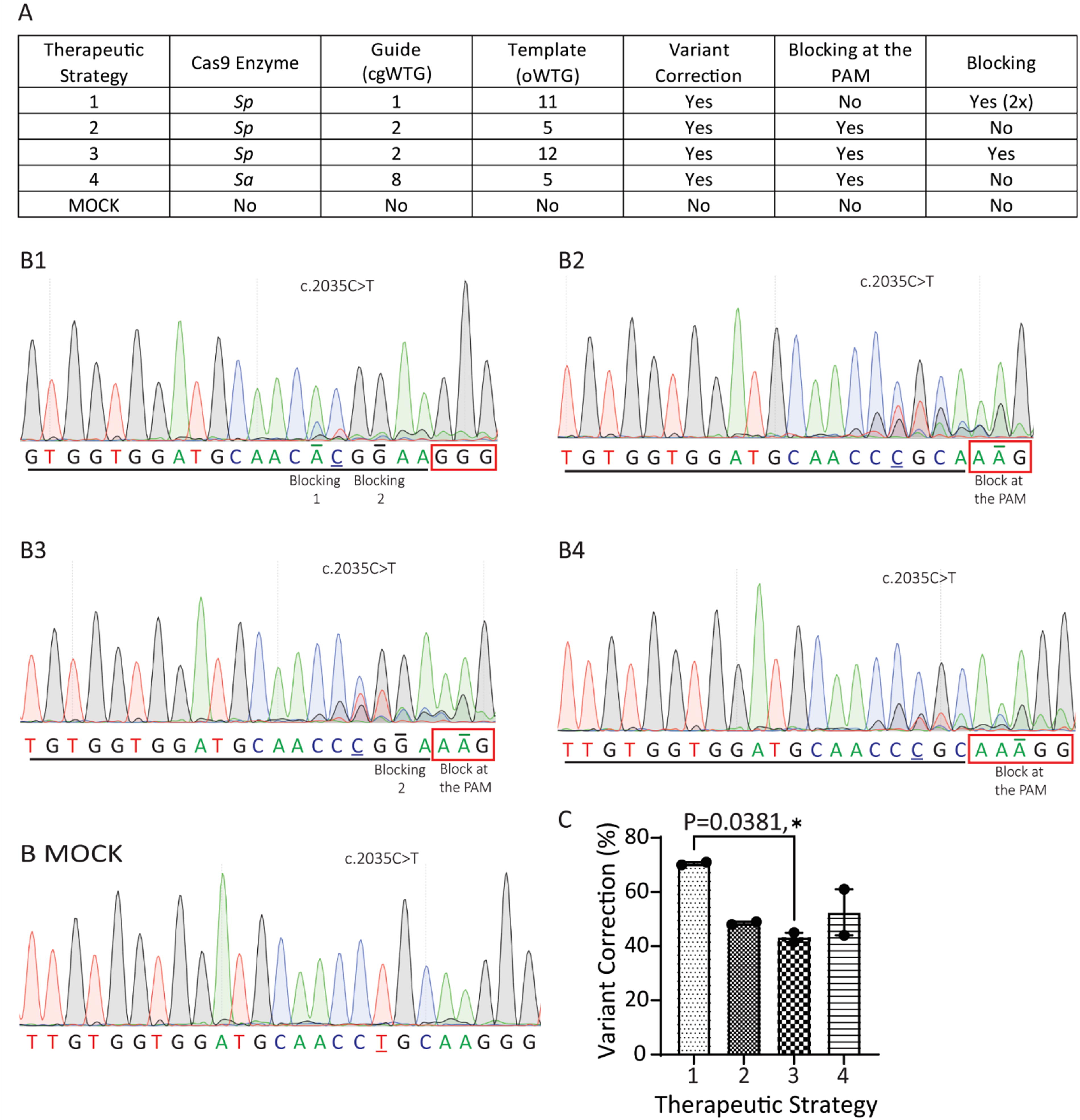
CRISPR editing using SpCas9 and SaCas9 corrected EZH2 exon-18 pathogenic patient variant c.2035C>T p.Arg684Cys in ESCs.
Therapeutic CRISPR reagents were electroporated into He18−/He18− ESCs (mEMS6677), and cell lysates harvested. Sequencing peak quantification was used to assess the success of each strategy to correct the pathogenic patient variant c.2035C>T p.Arg684Cys (Fig. 4B). Statistical ANOVA was applied (Fig. 4C). Strategy 1 using SpCas9 (cg (CRISPR guide) WTG1 and oligodeoxynucleotide (o) (WTG11)) showed the highest average of correction at 70.50 ± 0.50% (Fig. 4C). However, this high percentage was not significantly different from strategy 4 using the smaller SaCas9 (cgWTG8, oWTG5) at 52.50 ± 8.50% correction, nor from strategy 2 using SpCas9 (cgWTG2 and oWTG5) at 48.50 ± 0.50% correction. In contrast, strategy 3 using SpCas9 (cgWTG2, oWTG12) showed the lowest average level of correction at 43.27 ± 1.73%, and was significantly different from strategy 1.
CRISPR therapy strategies altered the EZH2 nonvariant allele at rates ranging from 2.0% to 26.2%
To determine the level of blocking base-pair alteration each therapeutic strategy showed on the nonvariant He18+ allele, optimized CRISPR reagents (Supplementary Table S4) were also electroporated into He18+/He18+ ESCs (mEMS6713), and cell lysates harvested. Sequencing peak quantification was used to assess the level of base-pair alteration by each therapeutic strategy (Fig. 5A). The levels of alteration at the blocking sites were then compared between the He18+/He18+ (Fig. 4B) and He18−/He18− ESCs (Fig. 5B). Importantly, for all strategies, the alteration of the blocking mutations with the pathogenic patient variant c.2035C>T p.Arg684Cys allele was significantly greater than with the nonvariant allele. Notably, among the results for the nonvariant allele, strategy 4, the smaller SaCas9 with one blocking mutation at the PAM, showed the lowest level of nonvariant allele alteration at 2.00 ± 2.00%. The remaining SpCas9 strategies all showed higher levels of alteration of the nonvariant allele: strategy 2, with one blocking mutation at the PAM, showed 9.53 ± 3.47%; strategy 3, with two blocking mutations (the latter at the PAM), at the first blocking mutation showed 13.50 ± 1.50% (He18+ ) and the second showed 14.07 ± 0.93% (He18+ ); and the highest of all was strategy 1, with two blocking mutations (none at the PAM), at the first blocking mutation showed 21.00 ± 10.00% (He18+ ) and the second showed 26.20 ± 11.20% (He18+ ).
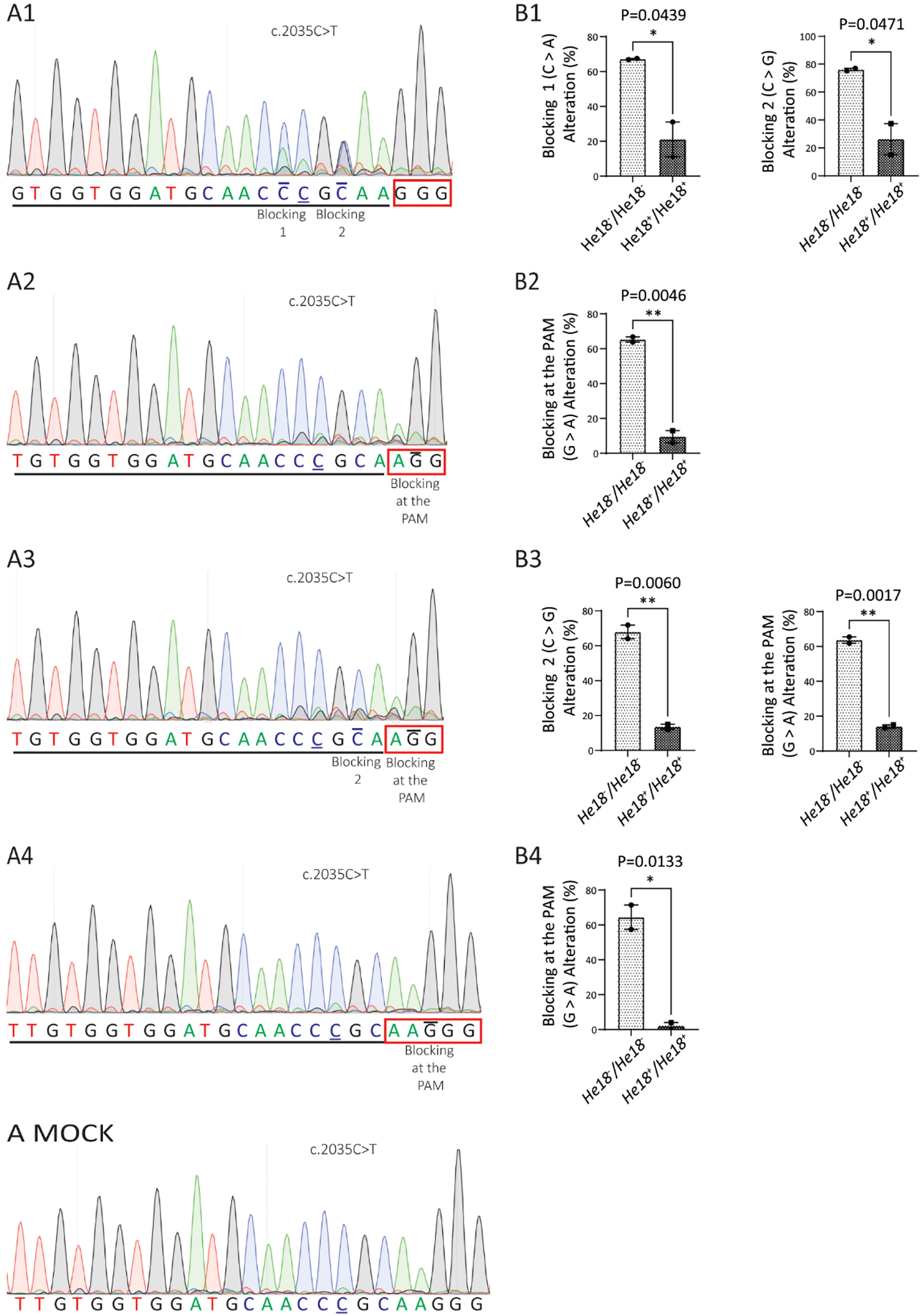
CRISPR editing using SaCas9 showed the lowest level of alteration of EZH2 exon-18 nonvariant location in ESCs.
DISCUSSION
Here, we document the development of mouse ESC lines that support preclinical approaches to understanding and treating Weaver syndrome. Critically, these cells have been humanized using the minimal humanization approach, giving them high translational value for optimization of DNA or RNA binding therapies. Given that ESCs are pluripotent, they can be differentiated to many different cell types, allowing for a broad exploration of Ezh2 function in a variety of cell types representing multiple organs, such as the brain. In this work, we studied the most frequent Weaver syndrome pathogenic patient variant, c.2035C>T p.Arg684Cys. Building on this work, others may now wish to use these cell lines and methods to introduce other EZH2 exon-18 amino acid changes to study protein function, or additional exon 18 pathogenic variants. 1,19,20 Finally, the methods may also be used to repeat this approach for any exon of interest in this or other genes.
The exact mechanisms by which EZH2 variants cause Weaver syndrome remain unclear. Using these humanized cell lines we show that the pathogenic patient variant c.2035C>T p.Arg684Cys results in a significant and dramatic reduction in Ezh2 catalytic activity. This result supports previous studies with this variant using a cell-free system, 7 and nonhumanized MEFs. 5 However, this most-common variant does not create a complete loss-of-function allele, but rather a hypomorphic allele, retaining a low but significant level of H3K27me3 activity. This result is consistent with a previous study by Lui et al. describing a different hypomorphic Ezh2 allele (c.1876G>A p.Val626Met) in a Weaver syndrome patient, as well as with those of Gao et al modeling p.Arg684Cys. 5,10 Together, these results show that the etiology of Weaver syndrome does not require a complete loss of EZH2 enzymatic activity.
In studies investigating the use of CRISPR for human therapies, gene humanization is critical, since then the guide RNAs and templates can be designed to bind and use PAMs in the human sequence—thereby facilitating translation to human cells and patients. Here, we compared four different CRISPR strategies and were thrilled to find corrections ranging from 43.3% to 70.5%. There was no significant difference in the correction percentage between the highest performing SpCas9 (70.5%) and SaCas9 (52.5%), supporting the use of either enzyme. Since Weaver syndrome patients are heterozygous at the EZH2 mutation site, a viable therapy must balance high-efficiency mutation correction with minimal alteration of the nonvariant allele. Comparing the alteration of blocking mutations present in the CRISPR templates, scored on both the humanized variant and nonvariant alleles, allowed us to discover that the SaCas9 enzyme gave the lowest level of alteration of the nonvariant allele at an impressive 2.0%.
CONCLUSIONS
CRISPR therapeutic approaches to Weaver syndrome were undertaken using minimally humanized mouse ESCs, and the pathogenic patient variant c.2035C>T p.Arg684Cys. We demonstrated that SpCas9 gave the highest variant correction (70.5%), but unfortunately also the highest alteration of the nonvariant allele (21.1–26.2%). However, SaCas9 gave a variant correction (52.5%) that was not significantly different than SpCas9, and importantly the lowest alteration of the nonvariant allele (2.0%). Thus, among the approaches tested here, the SaCas9 enzyme is the most suitable for correction of the Weaver syndrome variant. In addition, the SaCas9 enzyme is small, which allows flexibility in therapeutic delivery.
Footnotes
ACKNOWLEDGMENTS
The authors would like to thank Ms. Marketa Hlavon, Mr. Cillein Thorne, and Ms. Aida Sobhani of the CMMT DNA Sequencing and Bioanalyzer Core Facilities, for Sanger sequencing support. In addition, they also thank Ms. Melissa Braschel of the Clinical Research Support Unit, BC Children’s Hospital Research Institute, for statistical consultation.
DATA AVAILABILITY STATEMENT
AUTHORS’ CONTRIBUTIONS
Conceptualization, funding acquisition, and project administration: W.T.G. and E.M.S. Supervision: W.T.G., M.C.L., and E.M.S. Formal analysis: T.C.L. and S.M.J. Methodology: E.M.S. Resources: W.T.G. Investigation: T.C.L., A.J.K., S.M.J., B.A.A., and D.G. Visualization: T.C.L. and S.M.J. Writing—original draft: W.T.G., T.C.L., A.J.K., S.M.J., and E.M.S.
AUTHOR DISCLOSURE STATEMENT
No competing financial interests exist.
FUNDING INFORMATION
This work was funded by a
SUPPLEMENTARY MATERIAL
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.